Experimental Charge Density Analysis and Electrostatic Properties of Crystalline 1,3-Bis(Dimethylamino)Squaraine and Its Dihydrate from Low Temperature (T = 18 and 20 K) XRD Data


 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
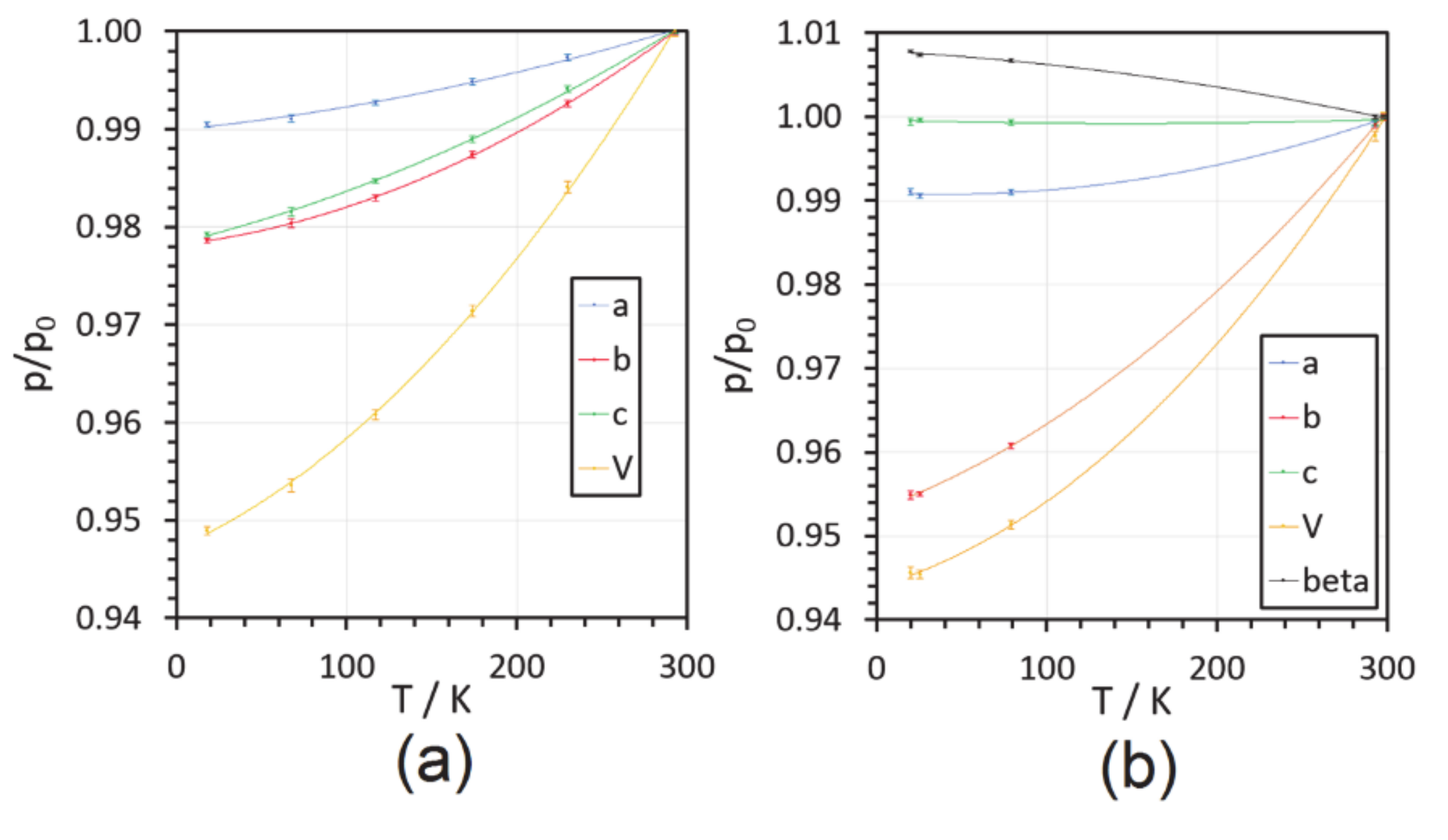
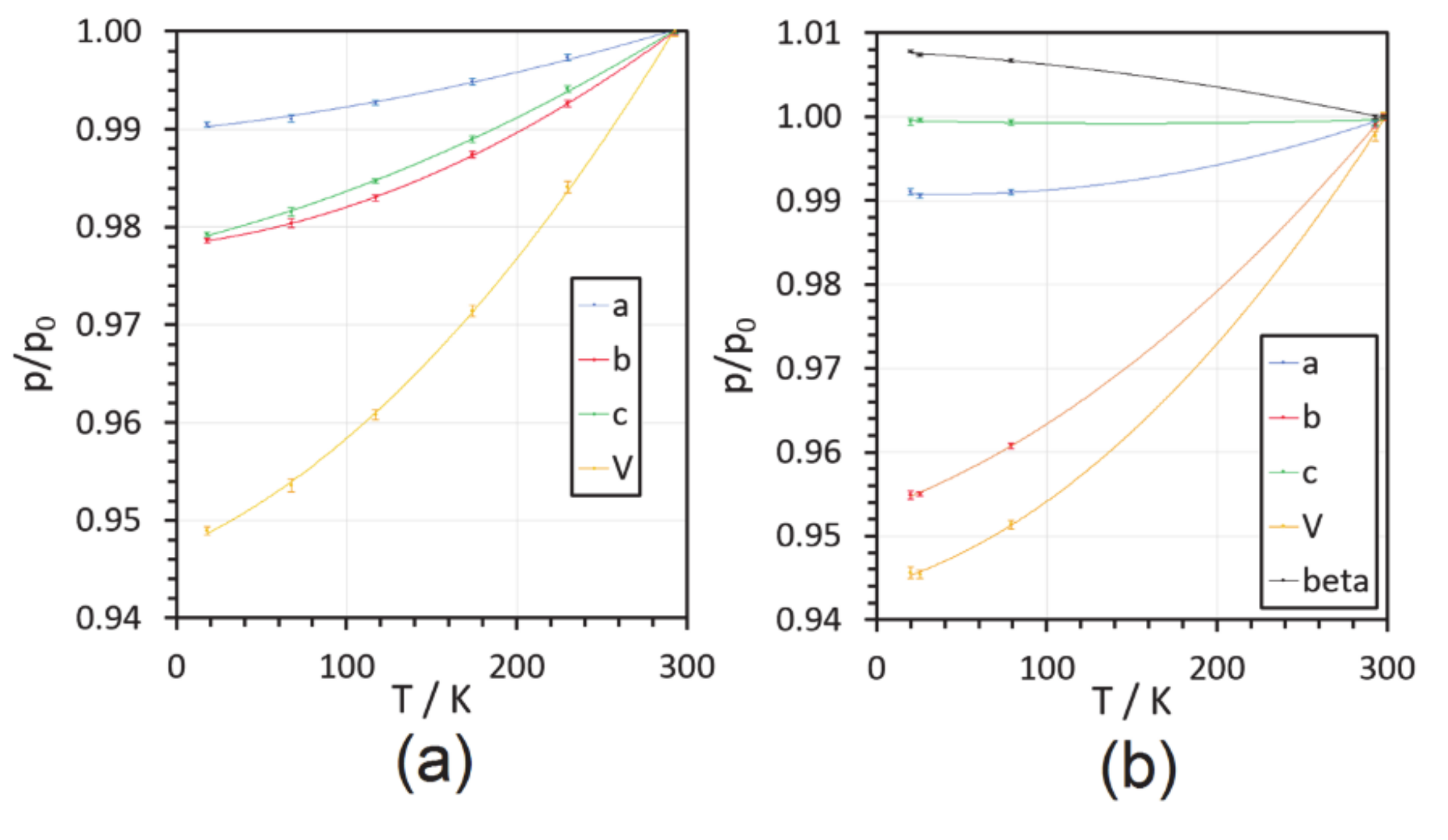
2.1. Cell Dimensions
2.2. Data Collection
2.3. Multipolar Refinement
3. Results and Discussion
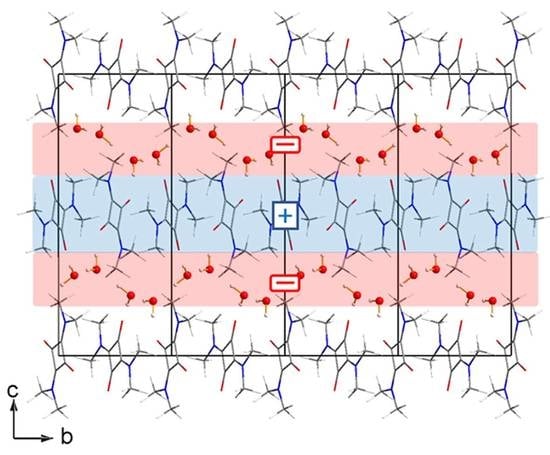
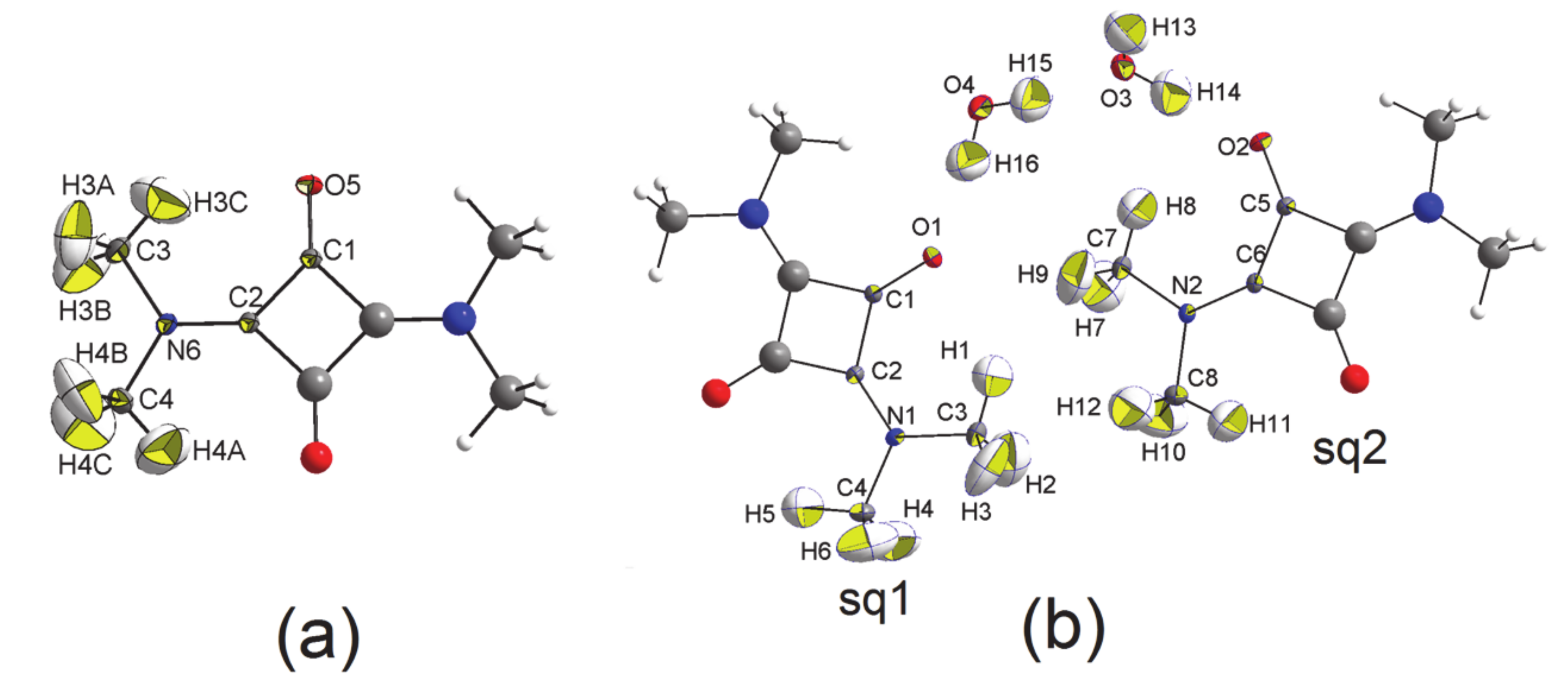
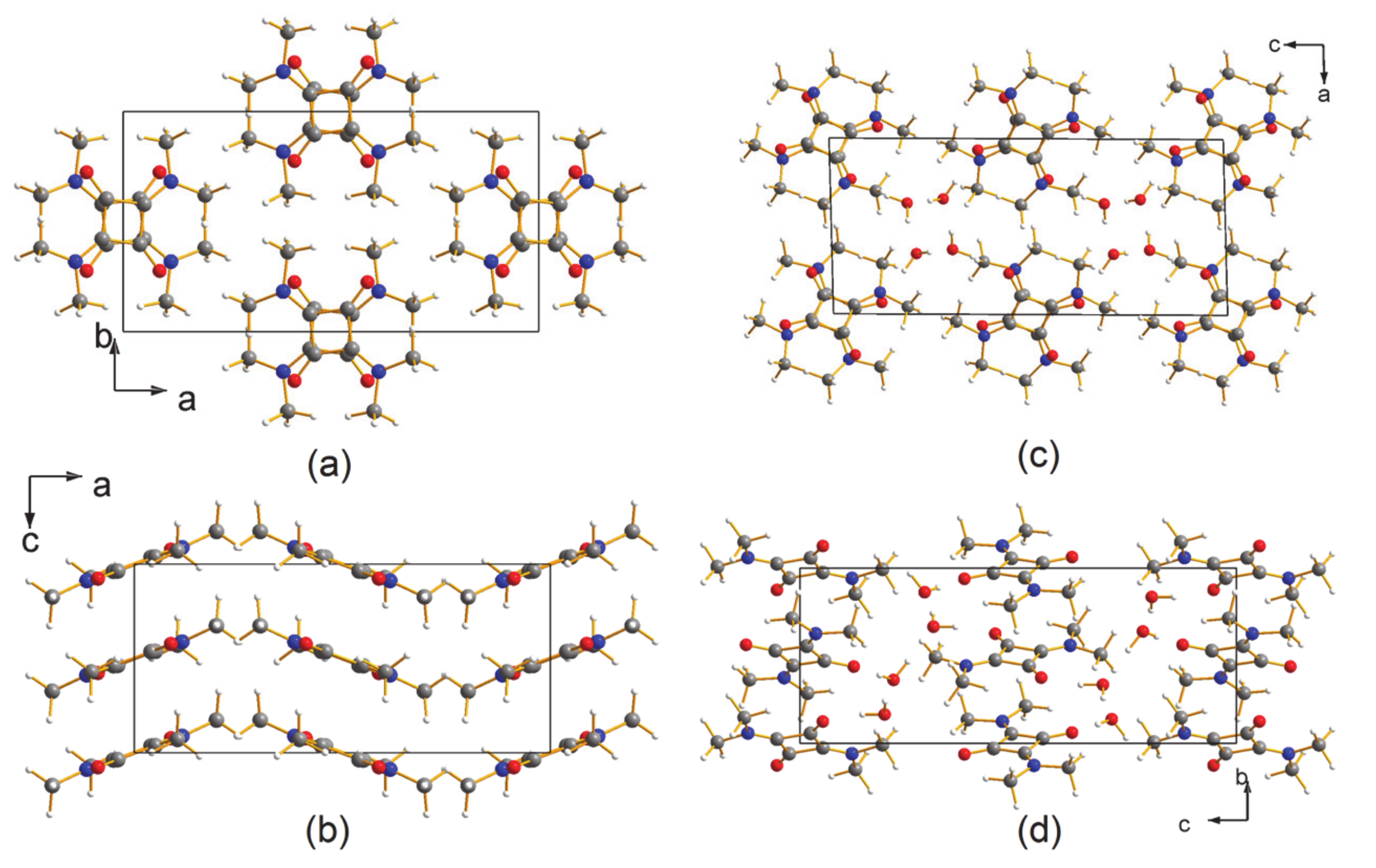
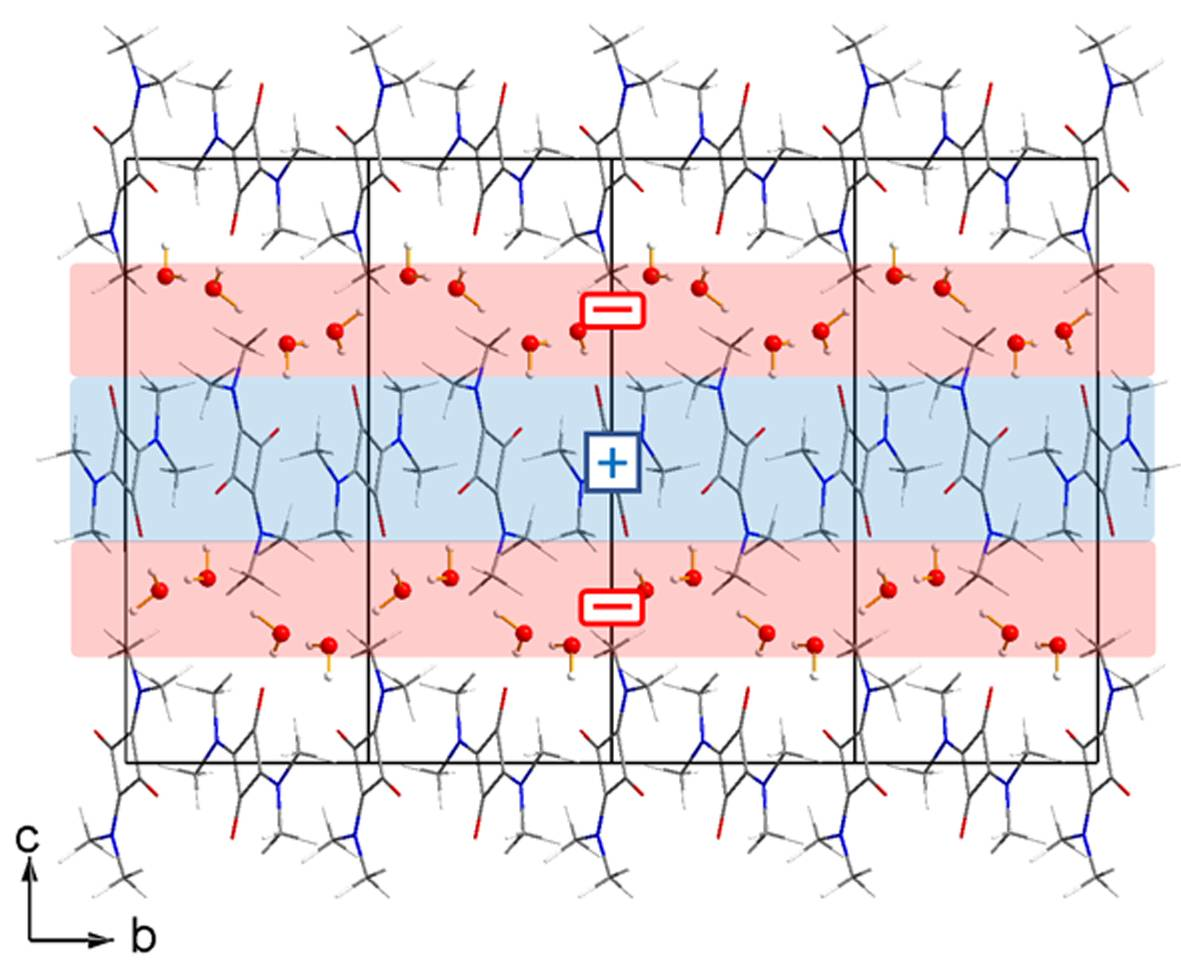
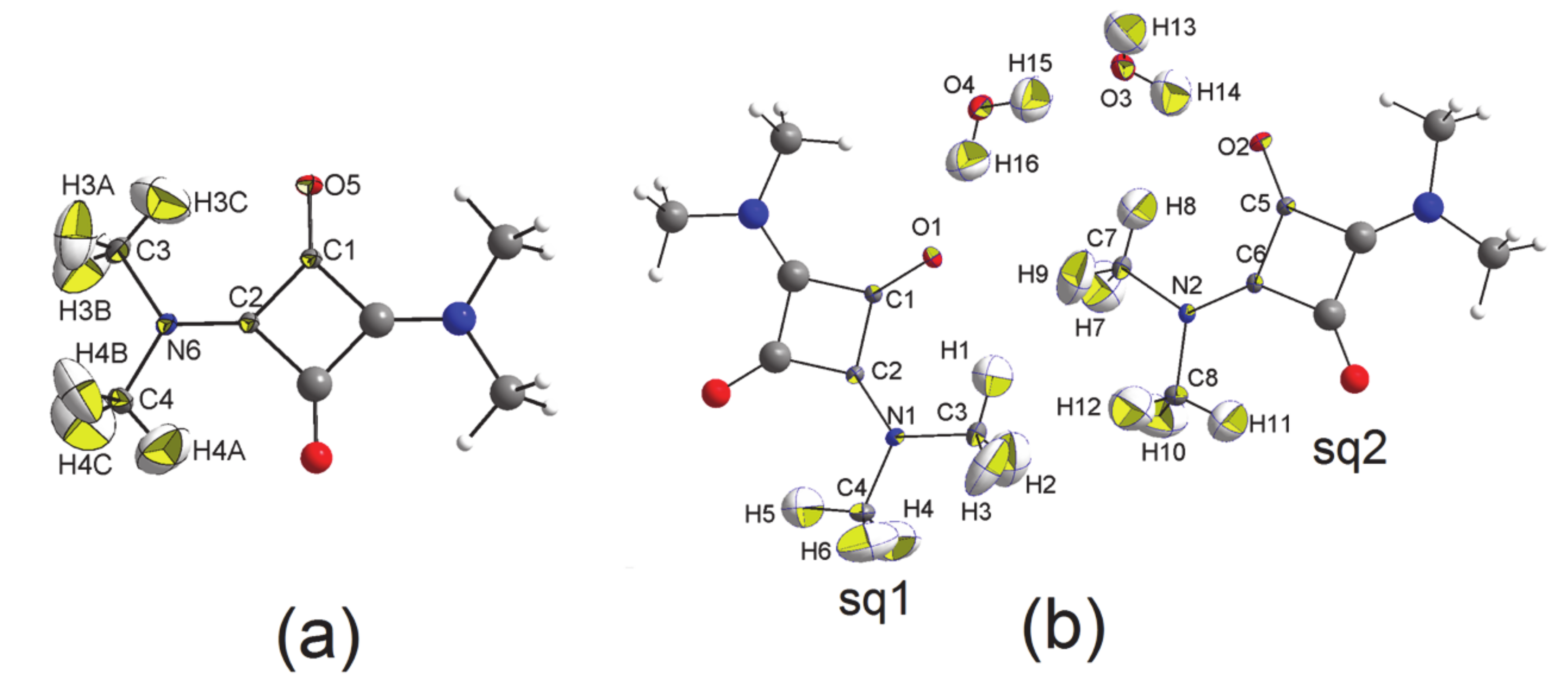
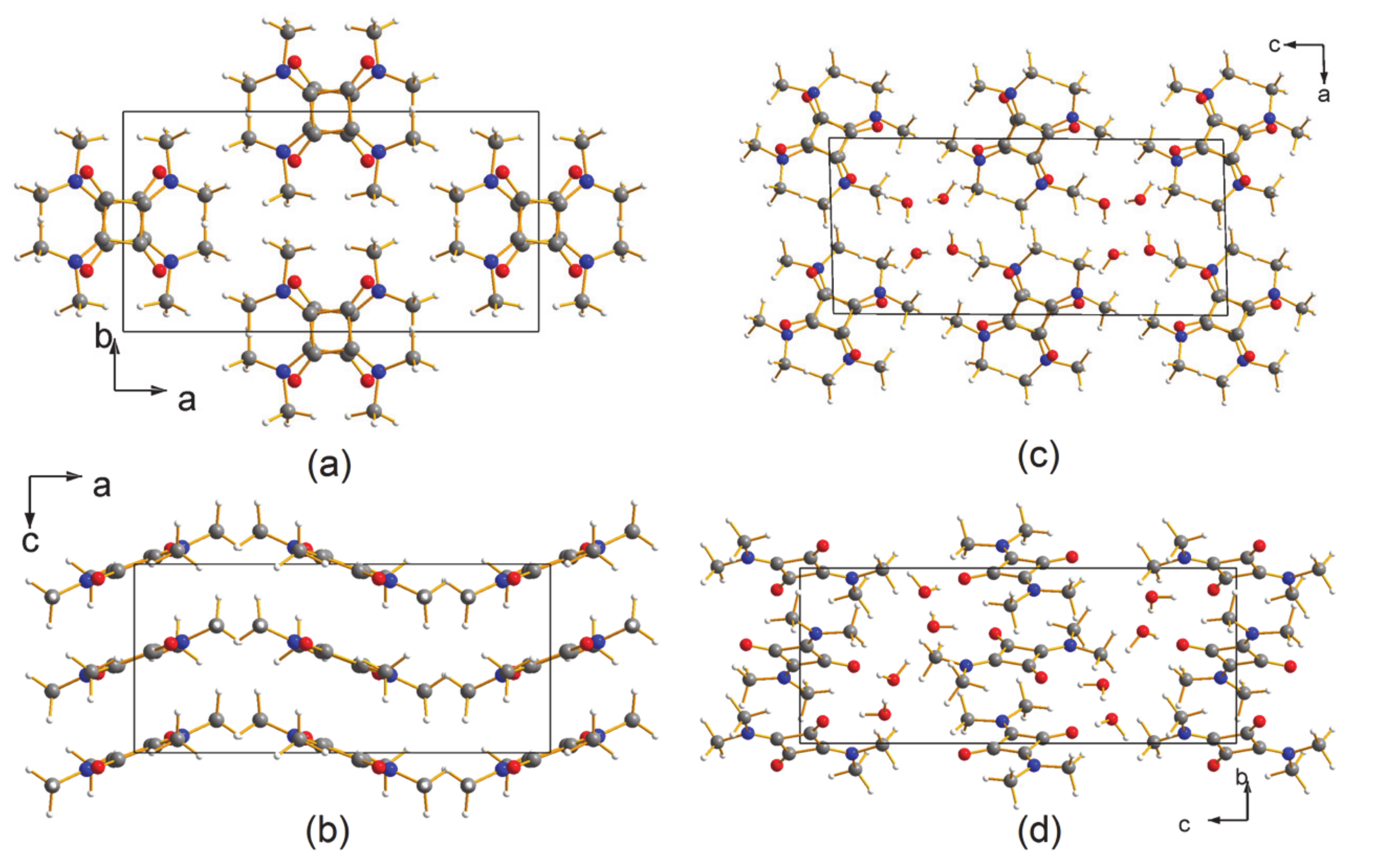
3.1. The Structures
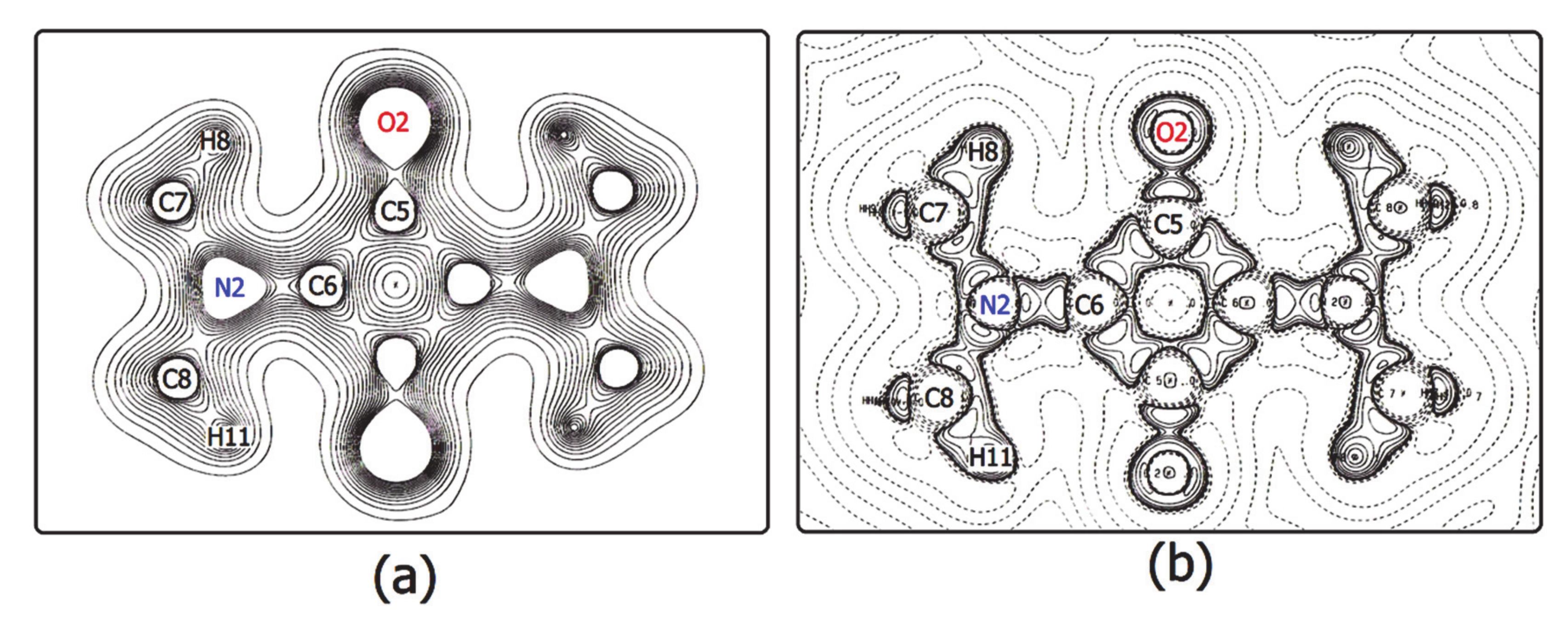
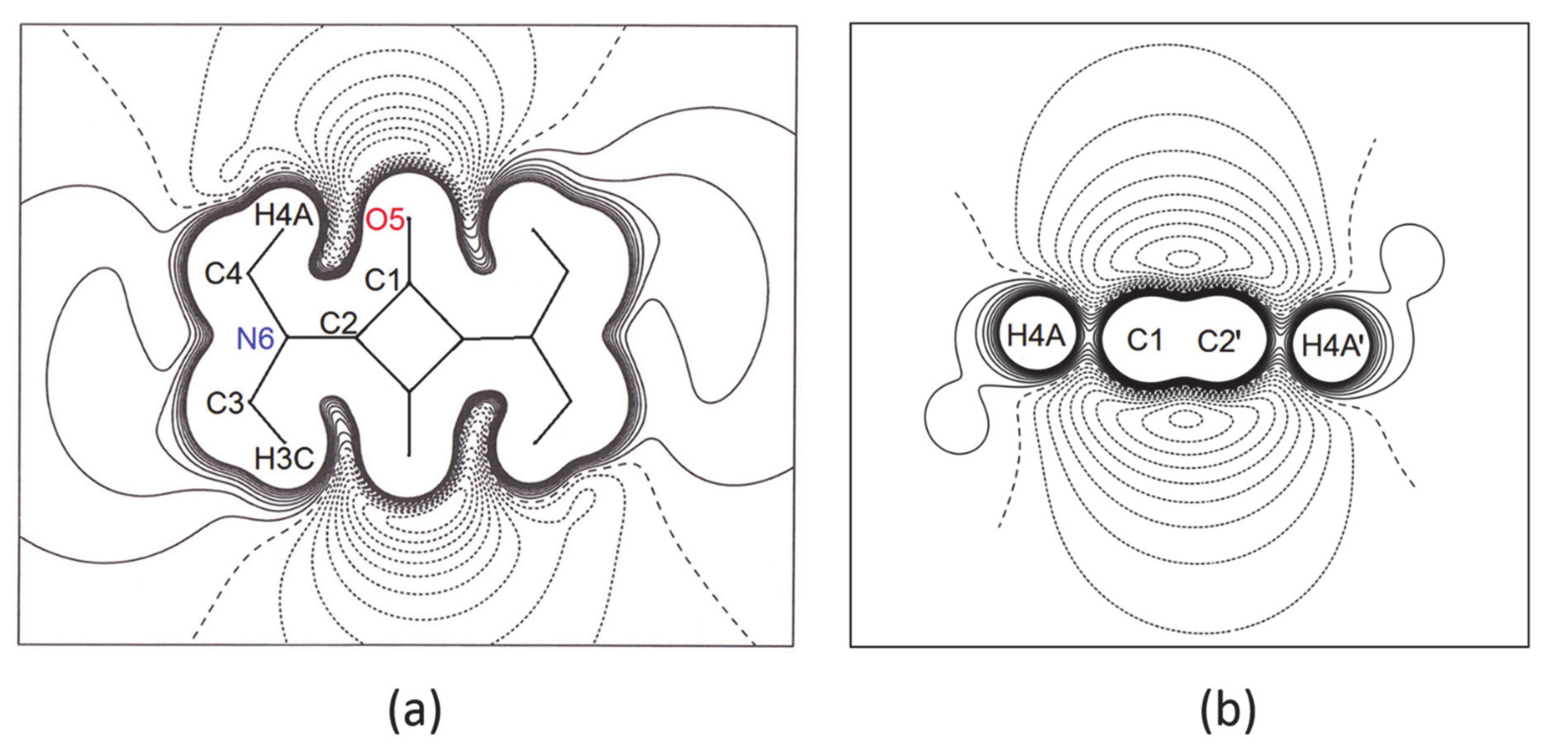
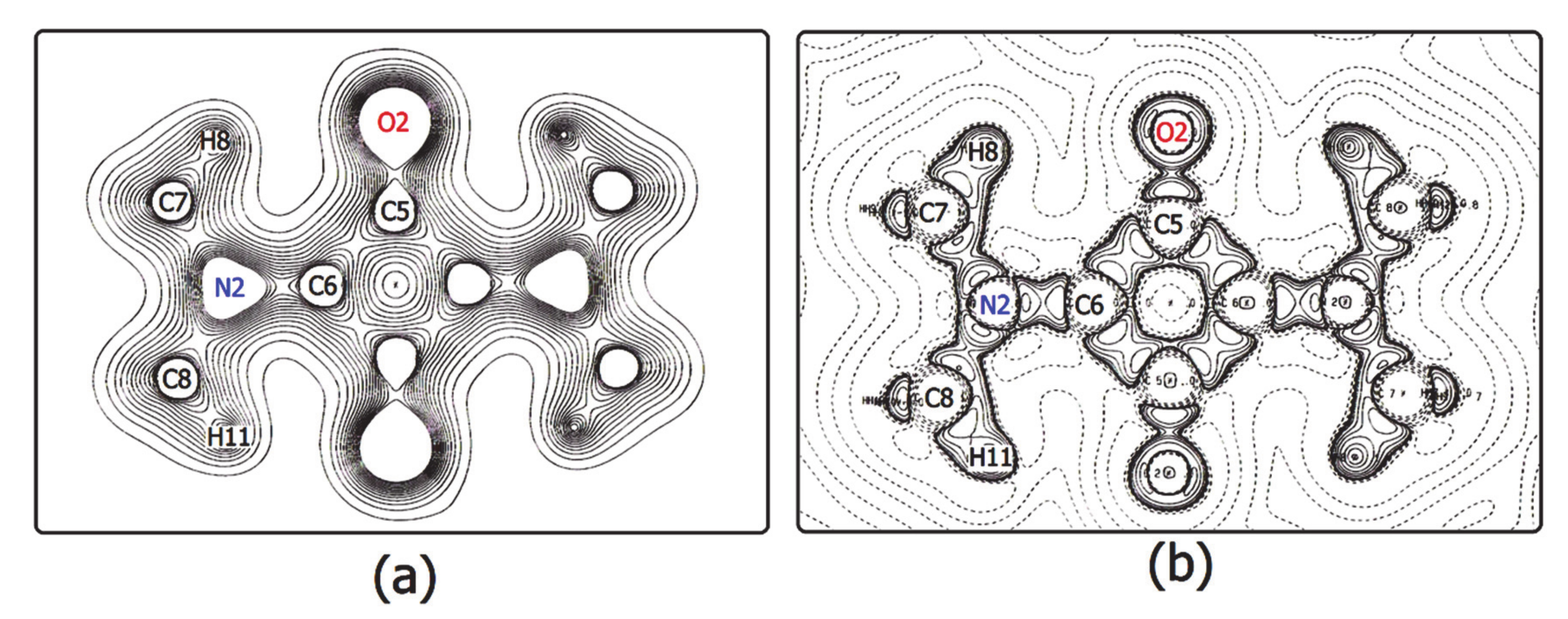
3.2. The Electron Density Distribution (EDD)
- (i)
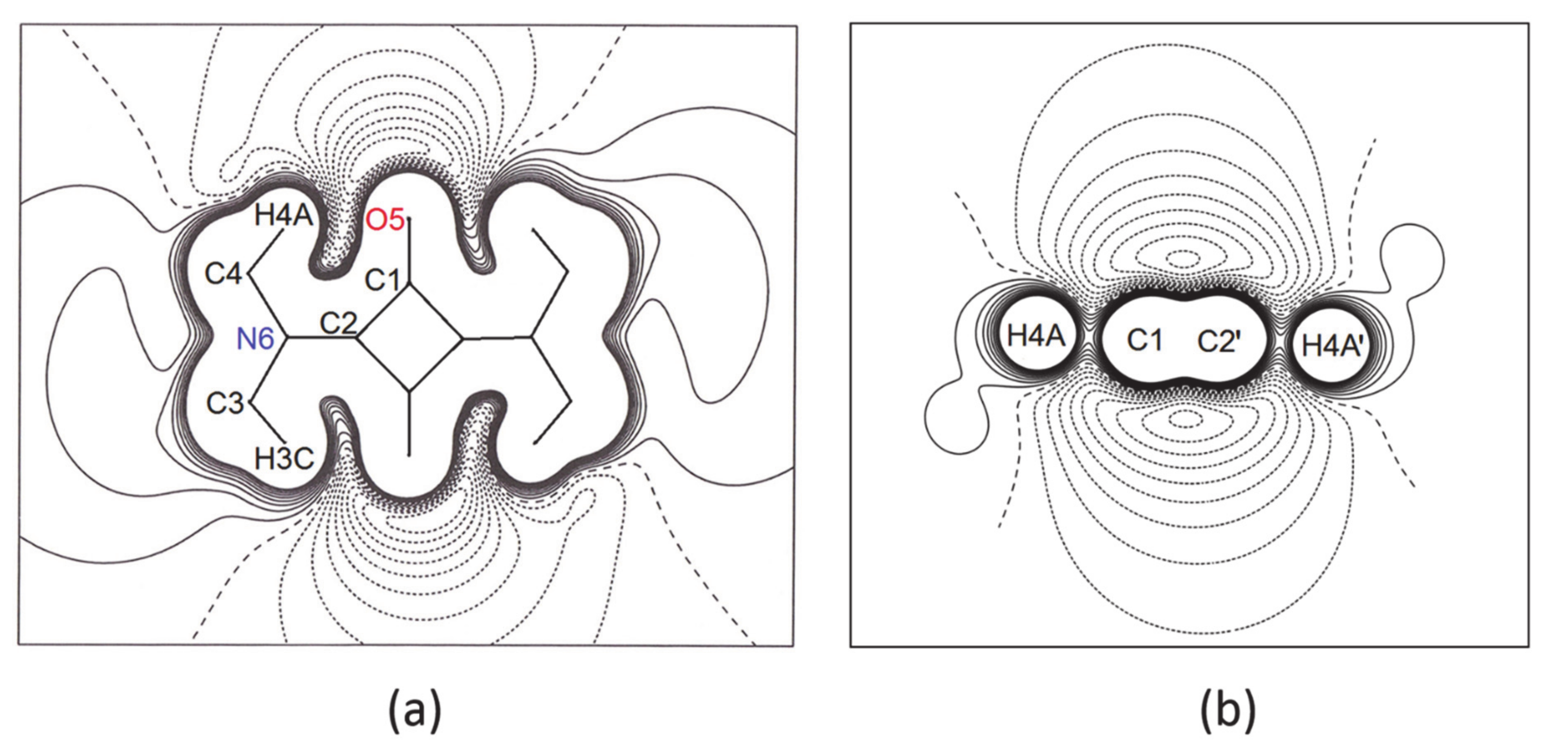
- In all three squaraine molecules, the lengths of the bond paths are very close to the related interatomic distances, except for the two independent bent bonds of the squarylium rings, where the numerical values of the bond paths differ significantly from the geometric distances (e.g., Δs = +0.0054 Å and +0.0051 Å in SQ, see Table 6). Longer bond path lengths are due to bond bending, as found in other 4-membred rings. In SQ, the two lines (bond path and bond length) make angles of 4.5 deg and 4.9 deg at the C atom bonded to the O atom [40,41], and of 5.7 deg and 5.9 deg at the other C atom, bonded to the N atom. Similar values—in the range 3.5 deg–6.7 deg for sq1 and 4.0 deg–5.5 deg for sq2—are observed at the corresponding angles of the squaraine rings of SQDH.
- (ii)
- There is a larger accumulation of charge on the ring C-C bonds of SQ than in those of sq1 and sq2 in SQDH: the values of ρ(rb) at the bcps of the former bonds are greater than those in the dihydrated crystal by 2.4–4.5 times the larger esds, and the values of the corresponding Laplacians differ by more than 5–6 times the esds of sq1 and sq2.
- (iii)
- The same features are shown by the N-Cmethyl bonds: the values of ρ(rb) at the bcps of these bonds in SQ are larger, on average, by 0.045 eÅ−3 (i.e., 4.5 esds) than those of sq1 and sq2, and their ∇2ρ(rb) quantities are more negative by ca. 2.4 eÅ−5 (again more than 4 esds). Differences of 4.1 and 4.6 eÅ−5 are observed also at the Laplacian of the (3, −1) bcp of the other N-C bond, whose length indicates a significant amount of double bond character in all three squaraine molecules. This statement is confirmed also by the value of ρ(rb) at the latter bond, 2.48 eÅ−3, only marginally smaller than that (2.51 eÅ−3) we found [42] at the bcp of the formally pure N = C double bond, 1.332 Å long, of a thiazete compound at T = 100(2) K.
- (iv)
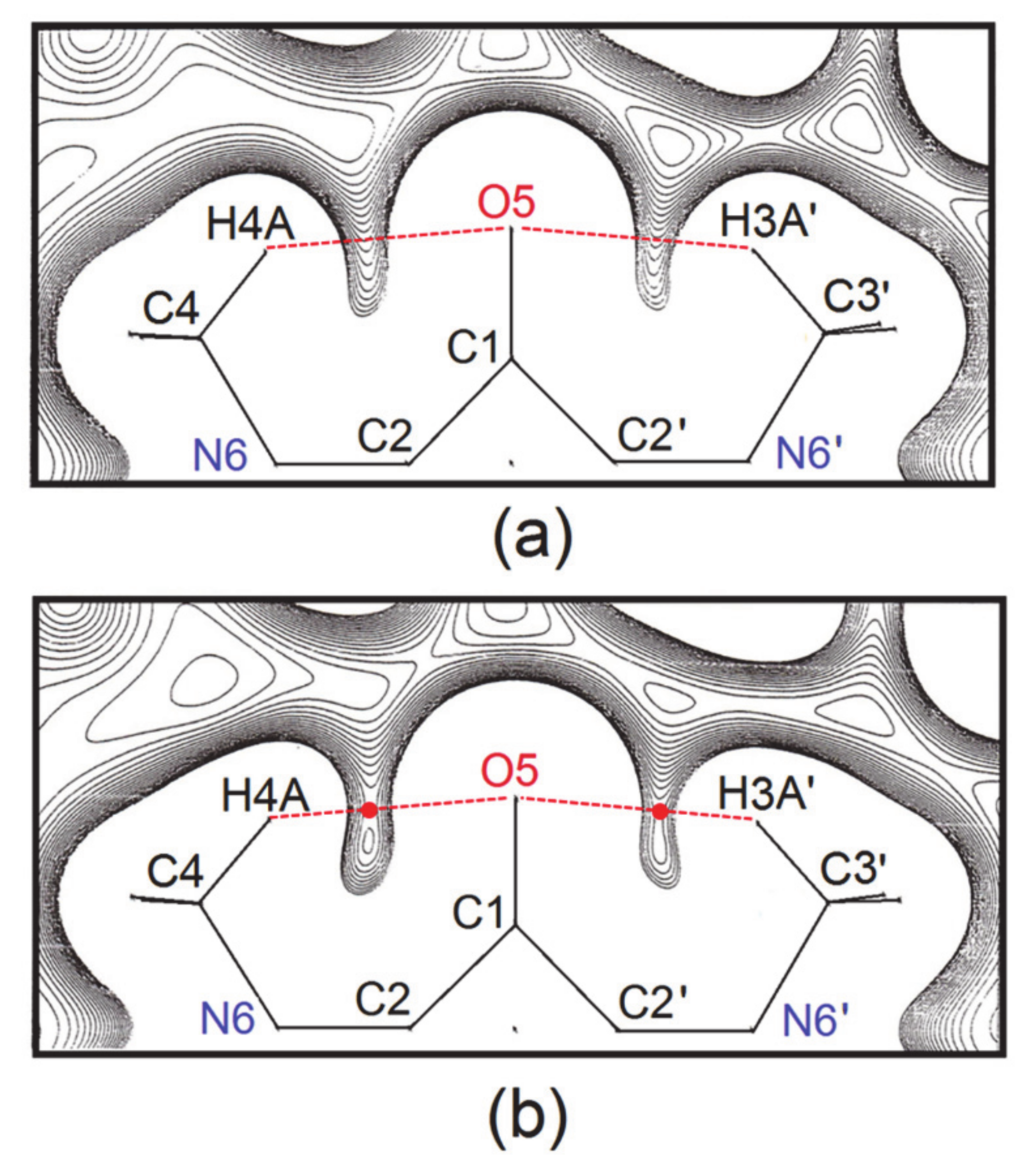
- For a complete isolated molecule of SQ (hence for a whole of two asymmetric units) the total number of (3, −1) bond critical points we found is 28, including 24 points for the 24 chemical bonds and 4 points for the four intra-molecular O···H interactions (Table 6). There are five (3, +1) ring critical points (rcps), corresponding to the squarylium ring plus the four six-membered rings formed by the sequence O-Cα-Cβ-N-Cmethyl-Hmethyl. With 24 attractors (the 24 atoms of the molecule) and no (3, +3) cage critical points, the Morse relation [43] is satisfied. The full corresponding set of atoms in SQDH includes two complete squaraine molecules (sq1 and sq2) plus two water molecules, for a total of 54 attractors. For this system, the Morse condition is also fulfilled: we found 61 bcps and 8 rcps. However, at odds with SQ, there are only two intramolecular O···H bond paths per sq molecule instead of four. As reported at the bottom of Table 6, no interaction lines were found for two of the O···H contacts and their centrosymmetric mates. Rather, each of the two H atoms no longer bonded to the squaraine O atom is now connected by a bond path to a water O atom (see Figure 4), and two eight-membered rings are formed, since each water molecule has one of its H atoms connected to the squaraine O atom. As a result, SQ and SQDH show different topologies of the squaraine molecules, in spite of the substantial geometric similarities described above.
3.3. The Electrostatic Potential
3.4. Atomic Volumes
3.5. Atomic Charges
3.6. Molecular Electrostatic Moments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schmidt, A.H. Reaktionen von Quadratsäure und Quadratsäure-Derivaten. Synthesis 1980, 1980, 961–994. [Google Scholar] [CrossRef]
- Schmidt, A.H. The Chemistry of Squaraines. In Oxocarbons; Elsevier: Amsterdam, The Netherlands, 1980; pp. 185–231. [Google Scholar]
- Ajayaghosh, A. Chemistry of Squaraine-Derived Materials: Near-IR Dyes, Low Band Gap Systems, and Cation Sensors. Acc. Chem. Res. 2005, 38, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Ilina, K.; MacCuaig, W.M.; Laramie, M.; Jeouty, J.N.; McNally, L.R.; Henary, M. Squaraine Dyes: Molecular Design for Different Applications and Remaining Challenges. Bioconjugate Chem. 2020, 31, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Khopkar, S.; Shankarling, G. Synthesis, photophysical properties and applications of NIR absorbing unsymmetrical squaraines: A review. Dye. Pigment. 2019, 170, 107645. [Google Scholar] [CrossRef]
- Meier, H. Extended conjugation in stilbenoid squaraines. Z. Nat. B 2019, 74, 241–254. [Google Scholar] [CrossRef]
- Law, K.Y. Squaraine chemistry. A study of the solute-solvent complexation of squaraine in solvents by proton NMR spectroscopy. J. Phys. Chem. 1989, 93, 5925–5930. [Google Scholar] [CrossRef]
- SciFinder. Chemical Abstracts Service, Columbus (OH). Available online: https://scifinder.cas.org (accessed on 1 September 2020).
- Lunelli, B.; Roversi, P.; Ortoleva, E.; Destro, R. Geometry and molecular parameters of 3,4-bis(dimethylamino)-3-cyclobutene-1,2-dione and its isomer bis(dimethylamino)squaraine. Combined study by IR spectroscopy, XRD and ab initio MO calculations. J. Chem. Soc. Faraday Trans. 1996, 92, 3611. [Google Scholar] [CrossRef]
- Lunelli, B.; Roversi, P.; Ortoleva, E.; Destro, R. Corrigendum to Geometry and molecular parameters of 3,4-bis(dimethylamino)-3-cyclobutene-1,2-dione and its isomer bis(dimethylamino)squaraine: Combined study by IR spectroscopy, XRD and ab initio MO calculations. J. Chem. Soc. Faraday Trans. 1997, 93, 513. [Google Scholar]
- Lunelli, B.; Soave, R.; Destro, R. Structure and stability of bis(dimethylamino)squaraine and its hydrates: A study using XRD, IR spectroscopy, and thermodynamic measurements. Phys. Chem. Chem. Phys. 1999, 1, 1469–1477. [Google Scholar] [CrossRef]
- Chen, Z.; Lohr, A.; Saha-Möller, C.R.; Würthner, F. Self-assembled π-stacks of functional dyes in solution: structural and thermodynamic features. Chem. Soc. Rev. 2009, 38, 564–584. [Google Scholar] [CrossRef]
- Ashwell, G.J.; Jefferies, G.; Hamilton, D.G.; Lynch, D.E.; Roberts, M.P.S.; Bahra, G.S.; Brown, C.R. Strong second-harmonic generation from centrosymmetric dyes. Nature 1995, 375, 385–388. [Google Scholar] [CrossRef]
- Honeybourne, C.L. Charge distortion by sparkles can explain strong SHG by centrosymmetric squaraine dyes. J. Mater. Chem. 1999, 9, 2241–2244. [Google Scholar] [CrossRef]
- Sissa, C.; Terenziani, F.; Painelli, A.; Siram, R.B.K.; Patil, S. Spectroscopic Characterization and Modeling of Quadrupolar Charge-Transfer Dyes with Bulky Substituents. J. Phys. Chem. B 2012, 116, 4959–4966. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Painelli, A.; Pati, S.K.; Terenziani, F.; Sissa, C. Aggregates of quadrupolar dyes for two-photon absorption: the role of intermolecular interactions. Phys. Chem. Chem. Phys. 2016, 18, 28198–28208. [Google Scholar] [CrossRef]
- Orian, L.; Pilot, R.; Bozio, R. In Silico Stark Effect: Determination of Excited-State Polarizabilities of Squaraine Dyes. J. Phys. Chem. A 2017, 121, 1587–1596. [Google Scholar] [CrossRef]
- Divya, V.V.; Suresh, C.H. Electronic Structure of Bis(4-dimethylaminophenyl)squaraine. ChemistrySelect 2019, 4, 3387–3394. [Google Scholar] [CrossRef]
- Nazarov, A.E.; Ivanov, A.I.; Vauthey, E. Modeling Infrared Spectral Dynamics upon Symmetry Breaking of a Photo-Excited Quadrupolar Dye. J. Phys. Chem. C 2020, 124, 2357–2369. [Google Scholar] [CrossRef] [Green Version]
- Destro, R. Detection and characterization of a reversible solid-solid phase transition at 147 K in crystalline 3,4-bis(dimethylamino)-3-cyclobutene-1,2-dione. Chem. Phys. Lett. 1997, 275, 463–468. [Google Scholar] [CrossRef]
- Stewart, R.F.; Spackman, M.A. VALRAY User’s Manual; Carnegie Mellon University: Pittsburgh, PA, USA; University of Copenhagen: Copenhagen, Denmark, 2000. [Google Scholar]
- Samson, S.; Goldish, E.; Dick, C.J. A novel low-temperature X-ray goniometer with closed-cycle cooling to about 18 K. J. Appl. Crystallogr. 1980, 13, 425–432. [Google Scholar] [CrossRef]
- Destro, R. Experimental Determination of Scan-truncation Losses from Low-temperature (16 K) Single-crystal X-ray Measurements. Aust. J. Phys. 1988, 41, 503. [Google Scholar] [CrossRef] [Green Version]
- Destro, R.; Roversi, P.; Barzaghi, M.; Marsh, R.E. Experimental Charge Density of α-Glycine at 23 K. J. Phys. Chem. A 2000, 104, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Destro, R.; Marsh, R.E. Scan-truncation corrections in single-crystal diffractometry: an empirical method. Acta Crystallogr. Sect. A Found. Crystallogr. 1987, 43, 711–718. [Google Scholar] [CrossRef]
- Destro, R.; Marsh, R.E. On predicting scan profiles: the nature of the ‘aberration function’. Acta Crystallogr. Sect. A Found. Crystallogr. 1993, 49, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.F. Generalized X-Ray Scattering Factors. J. Chem. Phys. 1969, 51, 4569–4577. [Google Scholar] [CrossRef]
- Stewart, R.F. Electron population analysis with rigid pseudoatoms. Acta Crystallogr. Sect. A: Cryst. Physics, Diffr. Theor. Gen. Crystallogr. 1976, 32, 565–574. [Google Scholar] [CrossRef]
- NATO Advanced Study Institute. The Application of Charge Density Research to Chemistry and Drug Design; Jeffrey, G.A., Piniella, J.F., Eds.; Springer: Boston, MA, USA, 1991; Volume 250, ISBN 978-0-306-43880-6. [Google Scholar]
- Roversi, P.; Destro, R. Approximate anisotropic displacement parameters for H atoms in molecular crystals. Chem. Phys. Lett. 2004, 386, 472–478. [Google Scholar] [CrossRef]
- Barzaghi, M. PAMoC (version 2002.0) online User’s Manual 2002. Available online: https://www.pamoc.it/ (accessed on 6 June 2020).
- Steiner, T. C–H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Gatti, C.; May, E.; Destro, A.R.; Cargnoni, F. Fundamental Properties and Nature of CH··O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(dimethylamino)-3-cyclobutene-1,2-dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Destro, R.; Sartirana, E.; Loconte, L.; Soave, R.; Colombo, P.; Destro, C.; Lo Presti, L. Competing C═O···C═O, C–H···O, Cl···O, and Cl···Cl Interactions Governing the Structural Phase Transition of 2,6-Dichloro-p-benzoquinone at Tc = 122.6 K. Cryst. Growth Des. 2013, 13, 4571–4582. [Google Scholar] [CrossRef]
- Lo Presti, L.; Soave, R.; Destro, R. On the Interplay between CH···O and OH···O Interactions in Determining Crystal Packing and Molecular Conformation: An Experimental and Theoretical Charge Density Study of the Fungal Secondary Metabolite Austdiol (C12H12O5). J. Phys. Chem. B 2006, 110, 6405–6414. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory. In International Series of Monographs on Chemistry 22; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Gajda, R.; Stachowicz, M.; Makal, A.; Sutuła, S.; Parafiniuk, J.; Fertey, P.; Woźniak, K. Experimental charge density of grossular under pressure—a feasibility study. IUCrJ 2020, 7, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Destro, R.; Ruffo, R.; Roversi, P.; Soave, R.; Loconte, L.; Lo Presti, L. Anharmonic motions versus dynamic disorder at the Mg ion from the charge densities in pyrope (Mg3Al2Si3O12) crystals at 30 K: Six of one, half a dozen of the other. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Destro, R.; Ortoleva, E.; Soave, R.; Loconte, L.; Lo Presti, L. Detection and kinetics of the single-crystal to single-crystal complete transformation of a thiiranium ion into thietanium ion. Phys. Chem. Chem. Phys. 2009, 11, 7181. [Google Scholar] [CrossRef]
- Lo Presti, L.; Ellern, A.; Destro, R.; Soave, R.; Lunelli, B. Rationalizing the Effect of Halogenation on the Molecular Structure of Simple Cyclobutene Derivatives by Topological Real-Space Analysis of Their Electron Density. J. Phys. Chem. A 2011, 115, 12695–12707. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, L.; Orlando, A.M.; Loconte, L.; Destro, R.; Ortoleva, E.; Soave, R.; Gatti, C. Single N-C Bond Becomes Shorter than a Formally Double N=C Bond in a Thiazete-1,1-dioxide Crystal: An Experimental and Theoretical Study of Strong Crystal Field Effects. Cryst. Growth Des. 2014, 14, 4418–4429. [Google Scholar] [CrossRef]
- Collard, K.; Hall, G.G. Orthogonal trajectories of the electron density. Int. J. Quantum Chem. 1977, 12, 623–637. [Google Scholar] [CrossRef]
- Roversi, P.; Merati, F.; Destro, R.; Barzaghi, M. Charge density in crystalline citrinin from X-ray diffraction at 19 K. Can. J. Chem. 1996, 74, 1145–1161. [Google Scholar] [CrossRef]
- Saleh, G.; Soave, R.; Lo Presti, L.; Destro, R. Progress in the Understanding of the Key Pharmacophoric Features of the Antimalarial Drug Dihydroartemisinin: An Experimental and Theoretical Charge Density Study. Chem. A Eur. J. 2013, 19, 3490–3503. [Google Scholar] [CrossRef]
- Finocchio, G.; Rizzato, S.; Macetti, G.; Tusha, G.; Lo Presti, L. Unravelling the Chemistry of the [Cu(4,7-Dichloroquinoline)2Br2]2 Dimeric Complex through Structural Analysis: A Borderline Ligand Field Case. Crystals 2020, 10, 477. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M.; Dietrich, H.; Macheleid, J.; Weiss, R.; Schleyer, P.V.R. The molecular and electronic structure of dipiperidinosquaraine. Eur. J. Inorg. Chem. 1985, 118, 2118–2126. [Google Scholar] [CrossRef]
- Bahtiar, A.; Tusaddiah, S.H.; Safriani, L. Improved charge carrier transfer in squaraine-capped ZnO nanoparticles layer for electron transport of hybrid solar cells. J. Physics: Conf. Ser. 2018, 1080, 012001. [Google Scholar] [CrossRef] [Green Version]
- Hestand, N.J.; Zheng, C.; Penmetcha, A.R.; Cona, B.; Cody, J.A.; Spano, F.C.; Collison, C.J. Confirmation of the Origins of Panchromatic Spectra in Squaraine Thin Films Targeted for Organic Photovoltaic Devices. J. Phys. Chem. C 2015, 119, 18964–18974. [Google Scholar] [CrossRef]
- Spackman, M.A. Molecular electric moments from x-ray diffraction data. Chem. Rev. 1992, 92, 1769–1797. [Google Scholar] [CrossRef]
- Koritsanszky, T.S.; Coppens, P. Chemical applications of X-ray charge-density analysis. Chem. Rev. 2001, 101, 1583–1628. [Google Scholar] [CrossRef]
- Bohorquez, H.J.; Obregon, M.; Cárdenas, C.; Llanos, E.; Suárez, C.; Villaveces, J.L.; Patarroyo, M.E. Electronic Energy and Multipolar Moments Characterize Amino Acid Side Chains into Chemically Related Groups. J. Phys. Chem. A 2003, 107, 10090–10097. [Google Scholar] [CrossRef]
- Destro, R.; Soave, R.; Barzaghi, M. Physicochemical Properties of Zwitterionic L - and DL -Alanine Crystals from Their Experimental and Theoretical Charge Densities. J. Phys. Chem. B 2008, 112, 5163–5174. [Google Scholar] [CrossRef]
- Cardamone, S.; Hughes, T.J.; Popelier, P.L.A. Multipolar electrostatics. Phys. Chem. Chem. Phys. 2014, 16, 10367. [Google Scholar] [CrossRef]
- Coppens, P. X-Ray Charge Densities and Chemical Bonding; Oxford University Press: New York, NY, USA, 1997; ISBN 0-19-509823-4. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T/K b | a/Å | b/Å | c/Å | β/° | V/Å3 |
|---|---|---|---|---|---|
| SQ | |||||
| 293 | 15.217(3) | 8.162(2) | 6.986(1) | 90. c | 867.8(3) |
| 230 | 15.177(4) | 8.102(2) | 6.945(2) | 90. c | 854.0(4) |
| 174 | 15.139(4) | 8.059(2) | 6.909(2) | 90. c | 843.0(4) |
| 117 | 15.106(3) | 8.023(1) | 6.879(1) | 90. c | 833.8(3) |
| 67.5 | 15.081(5) | 8.002(3) | 6.857(3) | 90. c | 827.5(5) |
| 18 | 15.072(2) | 7.9873(9) | 6.8410(9) | 90. c | 823.5(2) |
| SQDH | |||||
| 298 | 8.031(1) | 7.492(5) | 17.809(6) | 90.98(1) | 1071.3(7) |
| 293 | 8.025(1) | 7.485(1) | 17.797(3) | 90.98(2) | 1068.9(3) |
| 79 | 7.959(3) | 7.198(3) | 17.797(6) | 91.59(1) | 1019.2(7) |
| 25.5 | 7.955(5) | 7.155(4) | 17.802(4) | 91.65(1) | 1012.8(9) |
| 20 | 7.959(1) | 7.154(1) | 17.799(2) | 91.69(1) | 1013.0(2) |
| Compound | SQ | SQDH |
|---|---|---|
| Sample Information | ||
| Empirical formula | C8H12N2O2 | C8H12N2O2·2H2O |
| Formula wt/g mol−1 | 168.22 | 204.23 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pbcn | P21/c |
| Z | 4 | 4 |
| T/K | 18 (1) | 20 (1) |
| a/Å | 15.072 (2) | 7.959 (2) |
| b/Å | 7.9873 (9) | 7.154 (1) |
| c/Å | 6.8410 (9) | 17.799 (2) |
| β/ deg | 90.00 | 91.69 (1) |
| V/Å3 | 823.5 (2) | 1013.0 (2) |
| Dx/g cm−3 | 1.357 | 1.339 |
| F(000) | 360 | 440 |
| μ/mm−1 | 0.0895 | 0.101 |
| Data Collection | ||
| Cryostat | He/closed cycle | |
| λ/Å | 0.71073 | |
| (sinθ/λ)max | 1.15 | |
| Scan technique | ω/2θ | |
| Diffractometer | Syntex P1bar | Siemens P4 |
| Scan speed (2θ)/deg min−1 | 4 | 3 |
| No. collected reflections | 24625 | 13171 |
| No. unique reflns | 5163 | 5535 |
| No. observedd reflections (I > 0, Nobs) | 4761 | 5374 |
| Multipolar refinement (VALRAY) | ||
| Refinement Results on all data | ||
| GoF | 1.2462 | 0.7169 |
| R(F), R (F2) | 0.0233, 0.0217 | 0.0212, 0.0210 |
| Rw (F2) | 0.0326 | 0.0296 |
| No. variables (Nv) | 212 | 508 |
| Nobs/Nv | 22.5 | 10.6 |
| On data within the Cu sphere | ||
| No.obs reflns (I > 0) | 927 | 2225 |
| R (F), R (F2) | 0.0107, 0.0189 | 0.0146, 0.0147 |
| Rw (F2) | 0.0209 | 0.0203 |
| O···H-C | dO···H (Å) | Angle OHC (deg) | Position of CH Atom |
|---|---|---|---|
| Intramolecular | |||
| O5···H3C-C3 | 2.401 | 136.35 | x, y, z |
| O5···H4A-C4 | 2.456 | 133.86 | 1 − x, −y, −z |
| Intermolecular | |||
| O5···H4C-C4 | 2.487 | 130.56 | 1 − x, y, −½ − z |
| O5···H4B-C4 | 2.561 | 149.44 | ½ + x, ½ − y, −z |
| O5···H3B-C3 | 2.928 | 123.36 | 1 − x, y, −½ − z |
| O5···H3B-C3 | 3.017 | 95.70 | 1 − x, 1 − y, −z |
| O···H-C | dO···H (Å) | Angle OHC (deg) | Position of CH Atoms |
|---|---|---|---|
| Intramolecular | |||
| O1···H1-C3 | 2.370 | 135.08 | x, y, z |
| O1···H5-C4 | 2.536 | 131.30 | −x, −y, −z |
| O2···H8-C7 | 2.506 | 132.56 | x, y, z |
| O2···H11-C8 | 2.374 | 135.11 | −x, 1 − y, −z |
| Intermolecular | |||
| O1···H7-C7 | 2.465 | 141.66 | −x, −y, −z |
| O1···H9-C7 | 2.759 | 114.63 | 1 + x, y, z |
| O1···H10-C8 | 2.885 | 131.68 | −x, −y, −z |
| O2···H3-C3 | 2.741 | 122.29 | −x, 1 − y, −z |
| O2···H6-C4 | 2.920 | 113.53 | x, ½ − y, −½ + z |
| O2···H5-C4 | 3.033 | 100.31 | −x, −y, −z |
| O···H-O or O···H-C | dO···H (Å) | Angle OHO (deg) or Angle OHC | Position of CH (or OH) Atoms |
|---|---|---|---|
| Water···water | |||
| O3···H15-O4 | 1.772 | 169.32 | x, y, z |
| O4···H13-O3 | 1.759 | 178.42 | 1 − x, −½ + y, −½ − z |
| Water···Osquaraine | |||
| O1···H16-O4 | 1.830 | 169.44 | x, y, z |
| O2···H14-O3 | 1.803 | 179.02 | −1 + x, y, z |
| Water···Hmethyl | |||
| O3···H8-C7 | 2.542 | 156.87 | 1 + x, y, z |
| O3···H4-C4 | 2.660 | 146.27 | 1 − x, −y, −z |
| O4···H2-C3 | 2.630 | 167.08 | 1 − x, −y, −z |
| O4···H5-C4 | 2.466 | 156.15 | −x, −y, −z |
| O4···H3-C3 | 2.680 | 131.16 | x, ½ − y, −½ + z |
| SQ | SQDH | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sq1 | Sq2 | ||||||||||
| Bond | Length b. path | ρ(rb) | ∇2ρ(rb) | Bond | Length b. path | ρ(rb) | ∇2ρ(rb) | Bond | length b. path | ρ(rb) | ∇2ρ(rb) |
| O5-C1 | 1.237 1.237 | 2.85(1) | −36.2(6) | O1-C1 | 1.241 1.241 | 2.82(2) | −33(1) | O2−C5 | 1.239 1.239 | 2.87(2) | −34(1) |
| C1-C2 | 1.467 1.473 | 1.844(8) | −12.1(2) | C1-C2 | 1.467 1.472 | 1.81(1) | −10.2(3) | C5−C6 | 1.466 1.472 | 1.82(1) | −10.6(3) |
| C1-C2’ | 1.467 1.472 | 1.845(8) | −12.1(2) | C1-C2’ | 1.466 1.470 | 1.82(1) | −10.7(3) | C5−C6’ | 1.465 1.469 | 1.80(1) | −10.3(3) |
| C2-N6 | 1.318 1.318 | 2.48(1) | −29.8(5) | C2-N1 | 1.316 1.316 | 2.47(2) | −25.7(6) | C6−N2 | 1.319 1.319 | 2.48(2) | −25.2(6) |
| N6-C3 | 1.457 1.457 | 1.74(1) | −10.5(3) | N1-C3 | 1.462 1.462 | 1.70(1) | −8.2(5) | N2−C7 | 1.462 1.462 | 1.72(1) | −8.7(5) |
| N6-C4 | 1.458 1.458 | 1.77(1) | −10.9(3) | N1-C4 | 1.461 1.462 | 1.72(1) | −8.5(5) | N2−C8 | 1.463 1.463 | 1.70(1) | −7.9(5) |
| C3-H3A | 1.080 1.081 | 1.92(2) | −17.5(6) | C3-H1 | 1.072 1.072 | 1.92(2) | −17.4(8) | C7−H7 | 1.088 1.088 | 1.99(2) | −20.0(8) |
| C3-H3B | 1.087 1.088 | 1.90(2) | −17.9(6) | C3-H2 | 1.100 1.101 | 1.88(2) | −15.9(9) | C7−H8 | 1.078 1.079 | 1.90(2) | −16.3(9) |
| C3-H3C | 1.062 1.063 | 1.91(2) | −17.2(9) | C3-H3 | 1.092 1.092 | 1.94(2) | −18(1) | C7−H9 | 1.089 1.090 | 1.97(2) | −19.2(8) |
| C4-H4A | 1.063 1.063 | 1.96(2) | −19.0(6) | C4-H4 | 1.062 1.063 | 1.90(2) | −16(1) | C8−H10 | 1.106 1.107 | 1.92(2) | −18.0(8) |
| C4-H4B | 1.073 1.074 | 1.98(2) | −20.2(6) | C4-H5 | 1.089 1.089 | 1.85(2) | −14.8(8) | C8−H11 | 1.078 1.079 | 1.94(2) | −17.2(9) |
| C4-H4C | 1.115 1.115 | 1.91(1) | −17.8(5) | C4-H6 | 1.076 1.077 | 1.85(3) | −14(1) | C8−H12 | 1.091 1.092 | 1.91(2) | −16.4(8) |
| Intramolecular contacts | |||||||||||
| Contact | Distance b. path | ρ(rb) | ∇2ρ(rb) | Contact | distance b. path | ρ(rb) | ∇2ρ(rb) | Contact | distance b. path | ρ(rb) | ∇2ρ(rb) |
| O5···H3C | 2.401 2.413 | 0.070(2) | 1.03(1) | O1···H1 | 2.370 2.397 | 0.082(3) | 1.09(1) | O2···H11’ | 2.374 2.413 | 0.078(3) | 1.08(1) |
| O5···H4A | 2.456 2.464 | 0.061(1) | 0.91(1) | O1···H5’ | 2.536 | Not found | O2···H8 | 2.506 | Not found | ||
| Group | SQ | SQDH | |
|---|---|---|---|
| Squaraine #1 | Squaraine #2 | ||
 | 35.50(2) | 35.95(2) | 35.88(2) |
 | 70.32(2) | 70.29(2) | 69.94(2) |
 | 29.4(1) | 29.5(3) | 29.4(3) |
 | 70.6(1) | 70.4(4) | 70.4(4) |
| molecule | 211.5(2) | 211.1(5) | 210.7(5) |
| Group | SQ | SQDH | |
|---|---|---|---|
| Squaraine #1 | Squaraine #2 | ||
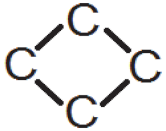 | 2.39(1) | 2.16(2) | 2.18(2) |
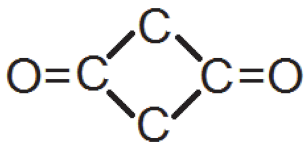 | 0.42(2) | 0.32(2) | 0.32(2) |
 | 0.37(3) | 0.35(2) | 0.37(2) |
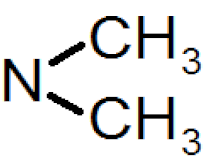 | −0.21(4) | −0.15(3) | −0.09(3) |
| molecule | 0.00(6) | +0.02(5) | +0.14(6) |
| Moment | SQ | SQDH | |
|---|---|---|---|
| Squaraine Sq1 | Squaraine Sq2 | ||
| Traceless Cartesian tensors | |||
| <xx> | 32 ± 4 | 22 ± 5 | 35 ± 5 |
| <xy> | −4 ± 3 | 0.6 ± 3.7 | −4 ± 4 |
| <yy> | −25 ± 2 | −20 ± 3 | −28 ± 3 |
| <xz> | 4 ± 2 | 3 ± 2 | 2 ± 2 |
| <yz> | 1.1 ± 0.7 | −0.4 ± 1.0 | 1 ± 1 |
| <zz> | −7 ± 2 | −2 ± 3 | −7 ± 3 |
| Anisotropy | 7.1 ± 0.6 | 6.0 ± 0.6 | 7.5 ± 0.5 |
| Spherical Harmonic Tensors | |||
| <2, 2> | 33 ± 5 | 24 ± 4 | 36 ± 5 |
| <2, −2> | −5 ± 5 | 0.7 ± 4.3 | −4 ± 4 |
| <2, 1> | 4 ± 3 | 3 ± 2 | 3 ± 2 |
| <2, −1> | 1 ± 1 | −0.4 ± 1.2 | 2 ± 1 |
| <2, 0> | −7 ± 3 | −2 ± 3 | −7 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Destro, R.; Roversi, P.; Soave, R.; Hovestad, A.; Lo Presti, L. Experimental Charge Density Analysis and Electrostatic Properties of Crystalline 1,3-Bis(Dimethylamino)Squaraine and Its Dihydrate from Low Temperature (T = 18 and 20 K) XRD Data. Crystals 2020, 10, 894. https://doi.org/10.3390/cryst10100894
Destro R, Roversi P, Soave R, Hovestad A, Lo Presti L. Experimental Charge Density Analysis and Electrostatic Properties of Crystalline 1,3-Bis(Dimethylamino)Squaraine and Its Dihydrate from Low Temperature (T = 18 and 20 K) XRD Data. Crystals. 2020; 10(10):894. https://doi.org/10.3390/cryst10100894
Chicago/Turabian StyleDestro, Riccardo, Pietro Roversi, Raffaella Soave, Arjan Hovestad, and Leonardo Lo Presti. 2020. "Experimental Charge Density Analysis and Electrostatic Properties of Crystalline 1,3-Bis(Dimethylamino)Squaraine and Its Dihydrate from Low Temperature (T = 18 and 20 K) XRD Data" Crystals 10, no. 10: 894. https://doi.org/10.3390/cryst10100894