Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity


Aix Marseille Univ, CNRS, Institut de Chimie Radicalaire ICR, UMR 7273, F-13397 Marseille, France
Catalysts 2019, 9(9), 736; https://doi.org/10.3390/catal9090736
Submission received: 12 August 2019
/
Revised: 27 August 2019
/
Accepted: 28 August 2019
/
Published: 30 August 2019
(This article belongs to the Section Photocatalysis)
Abstract
:In recent years, polymerization processes activated by light have attracted a great deal of interest due to the wide range of applications in which this polymerization technique is involved. Parallel to the traditional industrial applications ranging from inks, adhesives, and coatings, the development of high-tech applications such as nanotechnology and 3D-printing have given a revival of interest to this polymerization technique known for decades. To initiate a photochemical polymerization, the key element is the molecule capable to interact with light, i.e., the photoinitiator and more generally the photoinitiating system, as a combination of several components is often required to create the reactive species responsible for the polymerization process. With the aim of reducing the photoinitiator content while optimizing the polymerization yield and/or the polymerization speed, photocatalytic systems have been developed, enabling the photosensitizer to be regenerated during the polymerization process. In this review, an overview of the photocatalytic systems developed for polymerizations carried out under a low light intensity and visible light is provided. Over the years, a wide range of organometallic photocatalysts has been proposed, addressing both the polymerization efficiency and/or the toxicity, as well as environmental issues.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Photochemical reactions are chemical reactions occurring when a light-sensitive material present in a reaction mixture is excited by light. As the first manifestation of this photoexcitation, significant changes of its electronic states occur, enabling an electron from the ground state to be transferred in the excited state, inducing consecutive reactions. Depending on the energy of the absorbed photons, a cleavage of a chemical bond or the production of radicals can occur [1,2,3,4]. In an effort to drastically reduce the use or the generation of hazardous substances during chemical reactions, photo-assisted chemistry, and thus photochemistry, has been identified as an environmentally friendly synthetic method which is now commonly used in organic chemistry [5]. Indeed, numerous photochemical reactions can be carried out without taking recourse to acids, bases, transition metals, or catalysts, so that the formation of side products is drastically limited. Compared to the classical organic chemistry, reactivity of photoexcited compounds significantly differs from that of the ground state so that the formation of products that could not be obtained using conventional synthetic methods is possible. In fact, photochemistry does not compete with classical organic chemistry but offers additional synthetic routes to chemists. Since the beginning of the 21st century, the development of visible-light photochemical reactions, as well as the availability of cheap, compact, and low-consumption irradiation setups, has renewed the interest in photoinduced reactions [6,7,8,9,10] and discarded the classical UV-based photochemistry requiring bulky and expensive irradiation setups. Parallel to this, the use of UV light additionally raised safety concerns connected with the dangers of the UV radiations so that alternatives are actively researched [11,12,13]. Recently, a major breakthrough has been achieved with the emergence of visible light photoredox catalysis. It notably allowed to drastically reduce the content of the light-sensitive material by the possibility to regenerate it during the chemical reactions [14,15,16]. The concept of visible light photoredox catalysis has been separately developed by David MacMillan, Tehshik Yoon, and Corey Stephenson in late 2008 and early 2009. Using this approach, numerous synthetic problems in organic chemistry could be solved while addressing the energy consumption and safety issues [17]. Rapidly, these concepts of visible light photoredox catalysis have been transposed to the synthesis of macromolecules, historically based on UV processes [18,19]. Photopolymerization constitutes an effective and easy way to prepare polymers by transforming a liquid monomer or a liquid resin into a highly crosslinked solid polymer. From a theoretical point of view, photopolymerization is an appealing polymerization technique which exhibits numerous advantages compared to the solution-phase polymerization. Notably, photopolymerization can be carried out without any solvent and at room temperature so that no or lower amounts of volatile organic compounds (VOC) are released. A spatial [20] and temporal [21] control can also be obtained (see Figure 1). High polymerization rates and high final conversion values can also be reached. When photopolymerization is carried out under visible light, an improved curing depth resulting from a better light penetration inside the resin can be obtained, and safe irradiation conditions can be used [22].
In 2010, an important milestone was established for photopolymerization, with the first example of visible light photoredox catalysis applied to polymer synthesis [23]. Following this pioneering work, numerous visible light photoredox catalysts have been specifically designed for the purpose of photopolymerization. More precisely, a great deal of effort has been devoted to develop photoredox catalysts capable of initiating a polymerization under low light intensity and under air. These constitute difficult polymerization conditions, especially for the free radical polymerization (FRP) of acrylates where oxygen acts as an efficient inhibitor of polymerization [24]. Oxygen inhibition is a major drawback for polymerization, especially when liquid monomers are used due to the facile oxygen diffusion inside the resins. At present, examples of visible light photoredox catalysts for polymerization activatable under low light intensity still remain scarce, despite the possibility to initiate a polymerization with such compounds while significantly reducing the energy consumption [25]. Given that organic and organometallic photoredox catalysts of polymerization have been developed since 2010, availability of metal-based photocatalysts remains extremely low [11,25]. This is notably related to the potential toxicity of the metals which are used to design these photocatalysts. However, since 2010, a lot of effort has been made to reduce the toxicity by examining new metals. Additionally, it has to be mentioned that, contrary to organic photocatalysts which often exhibit a low molecular weight, metal complexes, due to the presence of the metal center and the peripheral ligands, possess generally a high molecular weight. This is an advantage for metal-based photocatalysts since in photopolymerization, the photocatalyst is classically introduced as a weight percent relative to the monomer so that, when converted into mole, the concentration of photocatalysts is extremely low.
With the aim to develop new photoinitiators, an interesting question remains concerning the exact role of the metal center in the catalytic activity of the photosensitizers. To examine this point, a wide range of metal complexes has been developed, aiming at establishing a structure‒performance relationship. The development of new photocatalysts exhibiting improved light absorption properties centered in the visible range and displaying improved reactivities in photoinitiation undoubtedly represents, at present, still a great challenge. In this review, an overview of the different organometallic visible light photocatalysts developed for polymerization processes carried out under low light intensity and in solvent-free conditions is presented. Especially, a great deal of effort is devoted to developing highly reactive photoinitiating systems enabling the avoidance of using inert atmosphere to perform the polymerization.
2. Organometallic Photocatalysts
Photocatalytic activity of photoinitiators is directly related to key parameters, which are the following:
(1) Their absorption properties, including both their absorption range as well as their molar extinction coefficients. This characteristic is of crucial importance to reduce the photoinitiator content, as a photoinitiator with a high molar extinction coefficient can be introduced in reduced quantity.
(2) Their excited state lifetimes. Notably, polymerization efficiency is improved by elongating the excited state lifetime of photoinitiators. By providing more time for the excited compound to react with the different additives, the production of reactive species is improved.
(3) Their electrochemical properties. Indeed, to react with the different additives, photoinitiators should exhibit adapted redox potentials. By adapting the redox potentials, the rate constant of interaction of the photocatalyst with the additives can be drastically increased, boosting the polymerization speed. To develop photocatalytic systems, metal complexes should also display reversible oxidation and reduction processes.
2.1. Ruthenium Complexes
As mentioned in the introduction section, numerous visible light organometallic photocatalysts have been developed to realize various chemical transformations in organic chemistry [26,27,28]. Concerning photopolymerization, the first report mentioning the use of a ruthenium complex as a photocatalyst was published in 2010 [23]. For the proof of concept, the benchmark Ru(II)(bpy)3Cl2 (where bpy stands for 2,2’-bipyridine) was used, and this complex is characterized by a strong absorption in the visible range, the absorption maximum being detected at 453 nm (14500 M−1·cm−1). In these conditions, Ru(II)(bpy)3Cl2 could efficiently promote the free radical promoted cationic polymerization (FRPCP) of epoxides when combined with an iodonium salt and a silane upon irradiation with laser diodes emitting at 405, 457, 473, and 532 nm (100 mW/cm²). Upon excitation of the three-component system Ru(II)(bpy)32+/TTMS3Si-H/Ph2I+ (0.2/3/2%, w/w/w) (where TTMS3Si-H and Ph2I+ stand for tris(trimethylsilyl)silane and diphenyliodonium salt, respectively) with light, the selected Ru(II) complex could react by electron transfer with the sacrificial quencher (i.e., the iodonium salt), generating a strong oxidant Ru(II)(bpy)33+. This strong oxidant is capable to oxidize the tris(trimethylsilyl)silane radical into the silylium cation TTMS3Si+ responsible of the cationic polymerization (CP) of (3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX), or biosourced limonene dioxide (LDO) (see Figure 2 and Scheme 1). In the present case, the CP of epoxides occurs according to a FRPCP mechanism. Indeed, the formation of Ph• radicals and subsequently TTMS3Si• radicals by hydrogen abstraction from TTMS3Si-H is required prior to the formation of TTMS3Si+ cations. In turn, TTMS3Si• (and Ph•) will initiate the CP of epoxides. Excellent polymerization profiles could be obtained, since final monomer conversions higher than 95% and 60% could be determined for the ring-opening polymerization of EPOX after 400 s of irradiation of the photocurable resins with a laser diode at 457 and 532 nm under air, respectively. Conversely, a lower final monomer conversion was obtained with LDO upon irradiation at 457 nm, the conversion value only peaking at 60% after 400 s of irradiation. Interestingly, the possibility to initiate the polymerization of EPOX with sunlight was examined, and a conversion of 60% could be obtained after 9 min of sun exposure, in contrast to 5 min for an irradiation with green light. The slower conversion under sunlight was ascribed to the higher intensity of the green bulb (18 mW/cm² vs. 5 mW/cm² for sunlight).
For future applications, the stability and possibility of storing the photocurable resins is an important parameter to consider. This point was also examined by the authors. In this study, after 21 days of storage, almost no modification of the photopolymerization profiles was observed, evidencing the suitability of Ru(II)(bpy)3Cl2 as photocatalyst (see Figure 3).
As another interesting feature, a slow bleaching of the film was observed, ascribed to the addition of silyl radicals onto the π-conjugated system of the bipyridine ligand of Ru(II)(bpy)3Cl2 [29]. Ability of photoinitiators to bleach during polymerization is actively researched by industrials who prefer the formation of colorless films. Indeed, the fabrication of colored polymers in industry should result from the incorporation of a dye that is carefully selected by the industrials and not from residual color imposed by the photoinitiators. As a consequence of the photobleaching, the polymerization process is not fully catalytic, since a slow consumption of the light-sensitive compound occurs during polymerization. However, this undesired consumption is sufficiently slow in the timescale of the polymerization process (a few hundred of seconds) not to impact the polymerization process.
As previously mentioned, Ru(II)(bpy)32+ can promote the formation of Ph• and TTMS3Si• radicals (see Scheme 1) [23]. These radicals can thus be exploited to initiate the FRP of acrylates. Especially, the rate constant of the addition of TTMS3Si• radicals on the double bond of methacrylates is relatively high (k > 106 M−1·s−1), so that the free radical polymerization (FRP) of trimethylolpropane triacrylate (TMPTA) can be obtained with these highly reactive radicals. A final monomer conversion of 35% within 120 s could be obtained with the three-component system Ru(II)(bpy)32+/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) [23]. In fact, these three-component systems are interesting formulations as they constitute dual radical/cation sources of polymerization.
In 2012, another Ru complex, i.e., Ru(II)(phen)3Cl2 (where phen stands for 1,10-phenanthroline), was tested under the same conditions as those used for Ru(II)(bpy)3Cl2 [30]. Despite similar absorption properties to those of Ru(II)(bpy)32+, a lower efficiency was found with this complex. Lower final monomer conversions were determined for the ring-opening polymerization of EPOX with a white light emitting diode LED (45% after 180 s) or a blue LED emitting at 450 nm (48% after 180 s) using the same composition, i.e., Ru(II)(phen)32+/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w). Counter performances of this complex were assigned to its higher oxidation potential, thus reacting less efficiently with the iodonium salt due to a lower rate constant of interaction. Examination of hydrogen donors other than the expensive TTMS3Si-H did not improve the polymerization efficiencies. The final monomer conversions decreased to 5% with ethyl dimethylaminobenzoate after 180 s of irradiation and 40% with 4-methoxybenzyl alcohol using the former blue LED emitting at 450 nm. The higher final monomer conversion obtained with TTMS3Si-H (48%) is attributable to its low ionization potential as well as its high oxidation rate constant by Ph2I+ [31,32].
Even if the performance of ruthenium complexes as photocatalysts was remarkable (see Figure 3), no other Ru(II) complexes were examined for photopolymerization. This is not so much attributable to the cost of the Ru precursor RuCl3.xH2O (~ 65 €/gram) used to prepare these complexes but to the potential toxicity of the ruthenium complexes. Similarly, platinum complexes were also only scarcely examined as photocatalysts of polymerization despite their good photoinitiating properties. However, in this last case, the toxicity was less concerning than the high cost of the Pt precursor PtCl2 (~ 165 €/gram).
2.2. Platinum Complexes
Platinum complexes are complexes of crucial interest for photopolymerization, as these complexes exhibit good absorption properties over the visible range. These complexes are also characterized by extremely long-living triplet excited states in the microsecond timescale, making these complexes ideal candidates for photopolymerization. The first examples of Pt-based photocatalysts for photopolymerization were reported in 2012 [33]. However, it has to be mentioned that Pt complexes were previously used in redox-initiating systems of polymerization [34,35]. Here again, the two Pt complexes Pt-1 and Pt-2 proved to be efficient photocatalysts for the CP of EPOX in three-component photoinitiating systems (see Figure 4). If the absorption properties of Pt-1 were limited in the visible range (the complex only absorbs until 440 nm), a broader absorption was found for Pt-2, extending from 400 to 540 nm. Due to the polyaromaticity of the ligand, a significant enhancement of the molar extinction coefficient was also found for Pt-2 (48,000 M−1 cm−1 for Pt-2 at 496 nm vs. 7400 M−1 cm−1 for Pt-1 at 385 nm). A triplet excited state lifetime of ~ 20 μs was determined by Laser Flash Photolysis (LFP) experiments for the two complexes. Upon excitation of the three-component system Pt complex/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) under air with a halogen lamp, final conversions of 59% and 70% were obtained after 1000 s of irradiation with Pt-1 and Pt-2, respectively, for the CP of EPOX. The higher photoinitiating ability of Pt-2 can be confidently assigned to its enhanced and broader absorption properties over the whole visible range. Regeneration of Pt-2 during polymerization was evidenced by flash laser photolysis (LFP) experiments. By comparing the LFP experiments done on the two-component system Pt-2/Ph2I+ and the three-component system Pt-2/TTMS3Si-H/Ph2I+, photodegradation of Pt-2 was determined as occurring more rapidly in the two-component system than in the three-component system. The regeneration of Pt-2 in the three-component system by the silyl radical TTMS3Si• was thus evidenced. Therefore, the chemical equation detailed below can be established as the reaction enabling the regeneration of Pt-2 by oxidation of TTMS3Si•.
TTMS3Si• + (Pt-2)•+ → TTMS3Si+ + Pt-2
The regeneration of Pt-2 was also confirmed by photoluminescence (PL) experiments, the PL spectrum of the final polymers superimposing that of the complex in solution. Finally, due to the simultaneous formation of cations and radicals during photoexcitation, the possibility to form an interpenetrated polymer network (IPN) was examined. While polymerizing a TMPTA/EPOX (50%/50%, w/w) blend in the previous conditions, final monomer conversions of 60% and 50% were respectively determined for EPOX and TMPTA after 1000 s of irradiation with the halogen lamp under air.
Once again, these excellent results with Pt complexes were not followed by subsequent works. This is notably attributable to the high cost of the Pt precursor (~165 €/gram), making this approach not applicable in industry, especially for polymers synthesis. However, other parameters also have to be considered to support the few examples of ruthenium and platinum complexes used as photocatalysts. Notably, the possibility to finely tune the absorption properties of photocatalysts is a fundamental criterion to adjust the absorption maximum of complexes with the emission wavelength of the irradiation sources. Concerning this point, absorption properties of Ru and Pt complexes are difficult to tune. The second point to consider is the reaction yields of synthesis of these different photocatalysts which can adversely affect the interest for such metal complexes. This is partially the case for Ru and Pt complexes which can only be obtained in moderate yields, around 40%–50% [36,37]. In fact, Ru and Pt complexes have rapidly been replaced by Ir complexes for which the tunability of their absorption properties was easier [38]. As the first consequence of this, numerous Ir complexes were also designed and synthesized, enabling to establish a structure–performance relationship.
2.3. Iridium Complexes
Iridium complexes have been the focus of numerous researches due to the facile tunability of their optical properties as well as their electrochemical properties [39,40,41,42,43,44,45,46]. Indeed, polypyridyl ligands can be easily modified by chemical engineering, so that the absorption of complexes can be shifted from the blue to the red region by replacing the well-known cyclometalated (2,4-difluorophenyl) pyridine (dfppy) ligand [47] with the 1-phenylisoquinoline (piq) ligand [48]. In contrast to ruthenium complexes, which are mostly prepared as homoleptic complexes, homoleptic, heteroleptic, but also tris(cyclometalated) complexes exhibiting three different ligands [49] can be prepared. In contrast to ruthenium complexes, which are only cationic complexes, cationic [50], anionic [51], soft salts [52,53], and neutral iridium complexes can be designed. This point is of importance, especially as the efficiency of the photoinitiating system is dependent on the ability for the metal complex to solubilize within the photocurable resin. Due to the different advantages listed above, the scope of applications of iridium complexes is extremely broad, ranging from organic electronics [54] to bioimaging [55] and photoredox catalysis. In fact, iridium complexes have revolutionized the photocatalysis applied to photopolymerization by the possibility to involve these complexes in oxidative and reductive processes. Indeed, when introduced in a three-component Ir complex/iodonium salt/silane system, Ir complexes are engaged in an oxidative mechanism [56,57]. Conversely, in the three-component Ir complex/amine/alkyl halide system, Ir complexes are involved in a reductive pathway [23,58,59,60,61]. As other advantages, which concern the efficiency of the initiating step, complexes with high triplet excited states and low oxidation potentials are actively researched. Iridium complexes clearly meet these requirements [62,63].
From a synthetic point of view, the design of iridium complexes remains a great challenge and, depending of the final charge of the complexes, major differences can be found concerning the reaction yields. Irrespective of the final charge of iridium complexes, the synthesis of all complexes starts with the same step, i.e., step 1 (see Scheme 2). It consists of the formation of the μ-dichloride-bridged iridium dimer formed by refluxing IrCl3·3H2O with 2.5 equivalent cyclometalating ligands in a mixture of 2-ethoxyethanol/water at a ratio of 3/1 by following the Nonoyama route [64]. In the second step, the third ligand is introduced by a bridge-splitting and substitution reaction of the cyclometalated iridium dimer (see Scheme 2). Depending of the nature of the third ligand, anionic, cationic, or neutral iridium complexes can be formed. Thus, when a cyclometalating ligand is introduced, tris(cyclometalated)iridium complexes can be synthesized with reaction yields higher than 70% [65,66] but require harsh synthetic conditions (reaction temperature around 200 °C). Difficult purifications are also encountered to isolate the complex in pure form. Conversely, heteroleptic complexes prepared with an acetylacetonate ligand can be obtained with similar reaction yields to the tris(cyclometalated) ones. However, the mild reaction conditions (heating at 65 °C) greatly simplify the purification by avoiding the formation of numerous side-products [67]. Finally, the best reaction yields for the second step are obtained for the synthesis of cationic and anionic complexes, the reaction yields ranging between 80% and 90% [68]. The first report mentioning the use of an iridium complex in photopolymerization was reported in 2011 (see Figure 5) [61]. The benchmark tris(2-phenylpyridine) iridium Ir(ppy)3 could promote the FRP of TMPTA and the CP of EPOX in an oxidative mechanism. The reaction of Ir(ppy)3 with Ph2I+ is extremely fast, since a rate constant of interaction of 5.4 × 109 M−1 s−1 could be determined by quenching of the luminescent triplet state of Ir(ppy)3. Based on the redox properties of the different additives composing the resin, a negative free energy change (ΔG < 0) could be calculated using the Rehm–Weller equation, favorable to a fast polymerization process [69]. Regeneration of Ir(ppy)3 in the three-component system Ir(ppy)3/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w), and thus the demonstration of the photocatalytic character of the polymerization process, was evidenced once again by flash laser photolysis. Thus, the absorption peak at approximately 580 nm characteristic of Ir(ppy)3+ could not be detected in the UV-visible spectrum during photolysis, demonstrating the fast reduction of the Ir(ppy)3+ cation by TTMS3Si•.
Excellent polymerization profiles were obtained for the CP of EPOX and the FRP of TMPTA. Final conversions of 80% after 400 s and 45% after 180 s were respectively obtained with the three-component Ir(ppy)3/TTMS3Si-H/Ph2I+ system (0.2%/3%/2%, w/w/w) upon irradiation under air with a xenon lamp (60 mW/cm²). While using soft irradiation conditions, i.e., LED bulbs (18 mW/cm²), a final conversion of 60% after 120 s of irradiation was still determined for EPOX using the same resin composition. A comparison with a modified photoinitiating system, comprising Ru(II)(bpy)32+ instead of Ir(ppy)3 as the photocatalyst, evidenced the superiority of Ir(ppy)3 over Ru(II)(bpy)3Cl2, and a final conversion of 60% was obtained after 180 s with Ru(II)(bpy)32+ as opposed to 120 s for Ir(ppy)3. The higher performance of Ir(ppy)3 relative to Ru(II)(bpy)32+ was assigned to its longer excited state lifetime (1.5 μs vs. 0.45 μs for Ru(II)(bpy)32+), its lower oxidation potential (0.77 V vs. 1.3 V for Ru(II)(bpy)32+), and its higher rate constant of interaction with Ph2I+. Following these pioneering works on photopolymerization, thirty iridium complexes (Ir-1-Ir-30) were examined as photocatalysts for the FRP of acrylates, the CP of epoxides, the FRPCP of N-vinylcarbazole (NVK), or the generation of IPN. Their chemical structures are presented in Figure 5 [56,57,58,70,71,72].
Among this series, the polymerization could only be carried out with a laser diode at 532 nm with Ir-10-Ir-12 and Ir-25, which exhibit the most red-shifted absorptions. Specifically, Ir-10-Ir-12 and Ir-25 possess phenylisoquinolines as the cyclometalated ligands, which is a ligand well-known to red-shift both the absorption and the emission of iridium complexes. For all others, the polymerization could be carried out with Xe lamps, LEDs emitting at 462 and 514 nm, laser diodes emitting at 457 nm, or even sunlight. At similar photophysical properties (molar extinction coefficient, redox potential, etc.), major differences and even irregularities were found between complexes during the FRP of acrylates or the FRPCP of epoxides. In fact, these differences of photoinitiating abilities were determined as originating from inequal solubilities of the different complexes in monomers (acrylates, epoxides). Here again, the solubility of photocatalysts proved to govern the reactivity of Ir complexes. For instance, the presence of fluorine atoms on iridium complexes is well known to drastically reduce the solubility in most of the common organic solvents.
However, a lower solubility can also originate from the use of polyaromatic cyclometalated ligands. This is the case when the phenylpyridine ligand was replaced by a benzo[h]quinoline or a 1-phenylisoquinoline. To illustrate this, comparisons between the photoinitiating ability of two three-component photoinitiating systems Ir complex/Ph2I+/TTMS3Si-H comprising Ir(btpy)3 Ir-4 or Ir(piq)3 Ir-3 as the photosensitizers during the FRPCP of an EPOX/NVK mixture (50%/50% w/w) upon irradiation with a LED at 462 nm were established. It revealed the final monomer conversion to reach 40% for Ir(btpy)3 Ir-4 instead of 85% for Ir(piq)3 Ir-3 after 400 s of irradiation simply due to the lower solubility of Ir(btpy)3 Ir-4 in the monomer mixture [72]. By chemical engineering, absorption spectra of iridium complexes could be adjusted to perfectly fit the emission of the light sources. A two-fold enhancement of the monomer conversion could be obtained for Ir-3 and Ir-5 relative to Ir-1, as a result of a better adequation of their absorption/emission spectra. To achieve this, polyaromatic ligands were used and the elaboration of a dinuclear complex Ir-5 was carried out. Thus, at 462 nm, corresponding to the emission of the laser diode, the molar extinction coefficients could be increased to 1100 M−1 cm−1 for Ir-1, to 7700 M−1 cm−1 for Ir(piq)3 Ir-3, and even 23,550 M−1 cm−1 for the dinuclear complex Ir-5. At identical weight percent in the photocurable resins, photopolymerization of an EPOX/NVK (50%/50% w/w) blend with the three-component system Ir complex/Ph2I+/TTMS3Si-H (0.2%/3%/2%, w/w/w) for 400 s with a LED at 462 nm (2 mW/cm²) resulted in vinylic conversion values of 50%, 80%, and 85% for Ir-1, Ir-3, and Ir-5, respectively (see Figure 6a). Once again, the lower final conversion for the dinuclear complex Ir-5 relative to that of Ir-3 resulted from its lower solubility in the monomer blend, adversely affecting its photoinitiating ability.
Another interesting finding is that the catalytic character of the polymerization process could be easily evidenced by comparing the photoluminescence (PL) of the polymer films with those of the corresponding photoinitiators. In all cases, a superimposition of both PL spectra could be obtained, revealing the regeneration of the different iridium complexes. A high reactivity of Ir-3 as a photocatalyst was also demonstrated by polymerizing NVK in aerated toluene (50%/50% w/w). A complete monomer conversion was obtained within 5 s with the three-component system Ir-3/Ph2I+/TTMS3Si-H (0.2%/3%/2%, w/w/w) upon irradiation with a halogen lamp (8 mW/cm²) (see Figure 6b).
Ir-3 and Ir-5 proved to be remarkable photocatalysts since the polymerization could be ended after approximately 100 s irrespective of the polymerization conditions. Conversely, for all the other complexes, reaction times ranging from 180 to 1000 s were required to get good final monomer conversions. For all these polymerizations, relatively short inhibition times could be found despite polymerizations being carried out under air, demonstrating the exceptional reactivity of TTMS3Si•. In fact, the high photoinitiating ability of Ir complexes is directly related to the use of TTMS3Si-H in the three-component systems, other additives resulting in lower final conversions. If the photoinitiating abilities of ruthenium, platinum, and iridium complexes were promising, the environmental concerns and toxicity issues raised by the use of transition metals have rapidly discarded these photocatalysts for industrial applications. Starting from 2013 (i.e., only three years after the pioneering work on Ru(II)(bpy)3Cl2 as a photocatalyst of polymerization), these issues were addressed with the development of less toxic and cheap photocatalysts based on abundant metals such as zinc, copper, and iron. In this field, the group of Jacques Lalevée is at the forefront of this research, developing successively zinc, copper, and iron photocatalysts for photopolymerization carried out under low light intensity and mostly under air in 2013, 2014, and 2015 respectively.
2.4. Zinc Complexes
In 2013, a new family of photocatalysts based on a cheap metal precursor, i.e. zinc, was developed as a photoinitiator of photopolymerization. To illustrate the difference of costs, the Pt precursor (PtCl2) is approximately 1100 times more expensive than Zn(OAc)2.2H2O (0.15 €/gram) which is the standard reagent for the synthesis of Schiff base Zn(II) chelates, and IrCl3.2H2O is 450 times more expensive than Zn(OAc)2.2H2O [73]. Considering that reaction yields for the synthesis of Schiff base Zn(II) chelates range between 60% and 90% [74], and that zinc is clearly less toxic than Ru, Ir, and Pt, Schiff base Zn(II) chelates have been thus examined for photopolymerization [75]. However, contrarily to the previous complexes that were phosphorescent materials, meaning that these complexes exhibit long-living excited states in the microsecond timescale, a different situation is found for Zn complexes. Indeed, the latter ones only exhibit short excited state lifetimes in the nanosecond timescale. When Zn-1-Zn-3 were examined as photocatalysts (see Figure 7), this severe reduction of the excited state lifetimes directly impacted their photoinitiating abilities during the FRP of TMPTA with the standard three-component system Zn complex/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w). Final monomer conversions around 30% were obtained after 200 s of irradiation with a halogen lamp (13 mW/cm²) in laminates (i.e., in oxygen-free conditions), far behind the conversion values determined with iridium photocatalysts, where the polymerization reactions were additionally performed under air (see Figure 8).
These results clearly show the crucial role of the excited state lifetimes in favoring the reaction between the photoinitiator and the different additives. Influence of this parameter on the photoinitiating ability is more clearly evidenced if a comparison of the molar extinction coefficients between Zn-1-Zn-3 is established. Indeed, Zn-1 and Zn-2 only exhibit small molar extinction coefficients (4700 and 5700 M−1 cm−1 at 350 nm, respectively) contrarily to Zn-3 which is characterized by a strong absorption (38,000 M−1 cm−1 at 550 nm). Despite the eight-fold enhancement of the absorption properties of Zn-3, similar polymerization profiles were obtained for Zn-1-Zn-3 (see Figure 8). It has to be noted that in the present case, Zn-1-Zn-3 exhibited similar solubility properties in TMPTA and that this parameter has not to be considered to explain the difference of reactivities. Low efficiencies only result from unfavorable rate constants of interaction with Ph2I+.
Finally, by replacing TTMS3Si-H with tri (ethylene glycol) divinyl ether (DVE-3) as the additive in the following three-component system Zn-3/DVE-3/Ph2I+ (0.2%/6%/2%, w/w/w), the monomer conversion could be increased up to 45% after 400 s of irradiation with a laser diode at 532 nm. Noticeably, a colorless polymer was obtained after polymerization. In fact, the formation of a colorless coating after polymerization is indicative of a fast degradation of Zn-3 during the polymerization process. This result also supports the low final conversions obtained with the three Zn complexes while using TTMS3Si-H as the reductant, the photostability of Schiff base Zn(II) chelates being limited. Facing these considerations (low stability, moderate ability to initiate the FRP of acrylates), more stable complexes based on copper were then examined.
2.5. Copper Complexes
Copper complexes are metal complexes that have been widely used for thermal atom transfer radical polymerizations (ATRP) [76,77,78]. More recently, these complexes have also been used for a new mechanism of polymerization named MABLI (metal acetylacetonate–bidentate ligand interaction) involving the generation of acetylacetonate (acac•) radicals by a redox process enabling the initiation of the FRP of acrylates [79,80,81,82,83]. This point will be discussed later in this paragraph. In the context of photoactivated polymerization processes, the first report was published in 2014 with a series of complexes bearing bis(2-(diphenylphosphino) phenyl) ether (DPEPhos) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos) as the phosphorylated ligands (see Figure 9) [84,85,86,87]. From a synthetic point of view, these complexes are easy to synthesize and can be obtained in two steps, the first one consisting of the complexation of the Cu(I) cation with the phosphorylated ligand, followed by the second step introducing the ancillary ligand. Overall, copper complexes can be prepared with reaction yields ranging between 75% and 90% (see Scheme 3).
These complexes proved to be efficient photoinitiators. Good final conversions could be obtained for the polymerization of EPOX or TMPTA even with the use of two-component photoinitiating systems Cu complex/iod (0.5%/1%, w/w) (where iod stands for (methyl-4-phenyl (methyl-1-ethyl)-4-phenyl) iodonium tetrakis (pentafluorophenyl) borate). However, it has to be noted that these results are obtained in laminates and not under air. While using three-component systems, an enhancement of approximately 10% of the final conversions could be obtained with all complexes. Thus, if a final conversion of 48% was obtained for the polymerization of TMPTA after 500 s of irradiation with a halogen lamp in laminate with the Cu-2/Ph2I+ (0.5%/1%, w/w) system. This conversion could be improved until 56% with the Cu-2/Ph2I+ (0.2%/2%, w/w) system where the copper concentration has been divided by a factor of 2.5 to reduce the inner filter effects [84].
Comparison of the final conversions obtained for the photopolymerization of TMPTA with Cu-2 and bisacylphosphine oxide (BAPO, also named Irgacure 819) used as a reference in the three-component system photocatalyst/iod/NVK (0.2%/2%/3%, w/w/w) (where iod stands for (methyl-4-phenyl (methyl-1-ethyl)-4-phenyl) iodonium) revealed the exceptional photoinitiating ability of Cu-2. For polymerization carried out at identical weight percent and under irradiation at 405 nm, Cu-2 could outperform BAPO (see Figure 10). While carefully examining the results obtained with the Cu-1-Cu22 series, two main groups of complexes could be identified. As the first group, those comprising bulky groups at the inner positions of the bipyridine ligand, such as Cu-2, Cu-3, Cu-5, Cu-6, Cu-11, and Cu-13, could give higher photoinitiating performances than those of the second group only composed of complexes that are not sterically hindered. It has to be noted that all complexes from the first group showed longer excited state lifetimes than those of the second one. But these results have to be considered in light of the recent development concerning copper complexes.
Indeed, the differences in photoinitiating abilities observed between the two groups directly arise from the possibility or not for copper complexes to give rise to a major structural rearrangement upon excitation. Indeed, when excited, hindered complexes cannot promote a structural rearrangement. However, for the non-hindered complexes, a modification of their geometrical structures from a tetrahedral geometry to a square planar geometry can occur. It has to be noted that, in the literature, numerous copper complexes have been reported as being phosphorescent materials due to their long excited state lifetimes. In fact, most of these complexes were thermally activated delayed fluorescence (TADF) materials [88]. In 2012, Chihaya Adachi evidenced with a series of organic and organometallic compounds the property of thermally activated delayed fluorescence (TADF) resulting from a small energy splitting existing between the singlet and the triplet excited states, so that the singlet state can be thermally repopulated from the triplet state by reverse intersystem crossing (RISC) [89,90]. Even though this property has been known since 1961 [91], it had not been exploited before 2012 and was only valorized by the design of the third generation of emitters for organic light-emitting diodes OLEDs, enabling devices to reach 100% internal quantum efficiency [92]. As the first manifestation of this property, an elongation of the excited state lifetime combined with an enhancement of the photoluminescence quantum yield are observed. Therefore, if numerous copper complexes have been reported as phosphorescent materials prior to the demonstration of the TADF property, numerous complexes were in fact TADF materials. The difference of photoinitiating ability between TADF/non-TADF photoinitiators was rationalized in 2018, with the comparison of two copper complexes, Cu-23 and Cu-24 (see Figure 11) [93]. For this demonstration, two complexes were selected for the similarity of their molar extinction coefficients as well as their absorption spectra. In these conditions, evidence of the contribution of the TADF properties on photoinitiation could be demonstrated. Notably, a significant enhancement of the excited state lifetime was found for the TADF complex, i.e., 2.5 μs for Cu-24 vs. 0.02 μs for Cu-23. Benefits of this 125-fold enhancement of the excited state lifetime was evidenced during the FRPCP of EPOX using the three-component system Cu complex/(tert-Bu)Ph2I+/NVK (0.5%/1%/1% w/w/w) (30% of conversion for Cu-24 vs. 12% for Cu-23 upon irradiation with a LED at 405 nm for 400 s under air) or during the FRP of the acrylate-based dental resin 2,2-bis[4-(methacryloxy-2-hydroxypropoxy) phenyl]propane (BisGMA), triethylene glycol dimethacrylate (TEGDMA) 50%/50% (65% of conversion for Cu-24 vs. 35% for Cu-23 upon irradiation with a LED at 405 nm for 400 s under air) using the three-component system Cu complex/(tert-Bu)Ph2I+/EDB (0.5%/1%/1% w/w/w) (where (tert-Bu)Ph2I+ and EDB stand for bis(4-tert-butylphenyl)iodonium and ethyl 4-(dimethylamino)benzoate, respectively).
It has to be noted that among additives widely used in three-component systems, NVK is one of those [94]. Indeed, after sensitization of the iodonium salt, phenyl radicals Ph• are produced, which are species efficiently initiating the FRP of acrylates. However, Ph• can also be added to NVK and produce a radical Ph-NVK•, stabilized by the mesomeric effect and being of lower reactivity than Ph•. This radical can also be converted to a cation by reduction of the oxidized form of the photocatalyst. Consequently, a Ph-NVK+ cation of high reactivity is obtained, efficiently promoting the ring opening of epoxides by FRPCP (see Scheme 4).
Among TADF complexes, Cu-2 (also known as the “G1” complex) has been the focus of numerous publications due to its unique photoinitiating properties. This complex, which has been patented in 2015, is even now commercialized by Lambson Ltd. in different photocurable resins [95]. To establish its remarkable photocatalytic properties, a comparison was established with the benchmark photoinitiator Irgacure 819 (BAPO) which also absorbs at 405 nm. The comparison was done under industrial conditions, namely with a model resin (MR 1) and under air (see Figure 12) [96]. More recently, a comparison of Cu-2 with the non-TADF Cu-1 complex has also been established [97]. However, the trends determined during this second series of comparisons were the same as those obtained with BAPO; Cu-2 was outperforming all photoinitiators with which a comparison was established. While using the three-component photoinitiator/(tert-Bu)Ph2I+/NVK (1.7%/5%/1%, w/w/w) system, a final conversion close to 100% was obtained upon photopolymerizing a model resin (MR1) at 405 nm (230 mW/cm²) for 800 s under air with Cu-2. Conversely, a conversion of only 60% was obtained with BAPO (see Figure 13a). Considering the higher molecular weight of Cu-2 relative to BAPO, the superiority of Cu-2 at comparable weight percent was established. Further experiments consisting of decreasing the photocatalyst content revealed that photoinitiating performance with Cu-2 remained higher than that of BAPO, even at 0.12 w%, constituting a 30-fold decrease of the photocatalyst content (see Figure 13b).
Another finding of crucial industrial interest is the possibility for the photoinitiating system to give rise to a dark polymerization, occurring when the light is switched off. Indeed, during light irradiation, radicals and cations are photogenerated and these latter can continue to polymerize the resin after the end of the irradiation. Evidence of a living polymerization process was clearly demonstrated by irradiating the resin for a short time and by following the final conversion increase when the light was switched off. A 22% increase of the monomer conversion was determined with Cu-2, in contrast to 12% for the three-component system comprising BAPO, 400 s after the end of irradiation (see Figure 13c).
Finally, green synthesis of photoinitiators is another topic attracting both academic and industrial interest. The possibility to prepare Cu-2 by mechanosynthesis, by grinding the different reagents in an agate mortar, was examined, and the performance of the mechanosynthesized complex was remarkable [98]. First, the complex obtained by grinding was of high purity (higher than 95%), which was demonstrated by nuclear magnetic resonance (NMR). Parallel to this, a 170-fold enhancement of the reaction time was obtained by mortar grinding. Indeed, if the synthesis in solution phase can be done in two hours, the reaction time could be reduced to 310 s by mortar grinding. Considering the high purity of the mechanosynthesized complex, a similar performance of the photoinitiator formed by grinding or in solution could be obtained for the FRPCP of epoxides or the FRP of acrylates. Another aspect of interest of mechanosynthesis is the possibility to realize the synthesis of compounds that could not be obtained in the traditional solution phase (see Scheme 5).
In this work, the synthesis of Cu-2 bearing I− as the counteranion could be obtained upon grinding for 30 min. However, a lower purity was obtained (only 72% yield) (see Figure 14). In fact, the counteranion used to generate cationic photocatalysts is not without influence on the photoinitiating ability of the latter [99]. Indeed, when a stronger ion pair is formed between the cation and the anion, a lower reactivity is observed for the photocatalyst. This difference of reactivity was notably demonstrated by examining the photoinitiating ability of Cu-2 under the form of PF6−, BF4−, and I− salts for the FRP of a model methacrylate resin (MR2) of low viscosity (0.053 Pa·s) composed of 1,4-butanedioldimethacrylate (33.3 wt%), hydroxypropylmethacrylate (33.3 wt%), and urethane-dimethacrylate (33.3 wt%) using the two-component Cu-2/(tert-Bu)Ph2I+ (0.3%/0.8%, w/w) system (see Figure 15).
Indeed, as previously mentioned, one drawback of the FRP of acrylates is the inhibition of the polymerization process by oxygen. This phenomenon is amplified when a resin of low viscosity is used due to the facile diffusion of oxygen and when the polymerization is carried out under air. Consequently, use of such liquid resins is often selected to evidence the high reactivity of the photoinitiating system, the three-component system thus operating in difficult conditions. In the present case, upon irradiation with a LED at 405 nm under air, a short inhibition time was found for Cu-2.PF6 and Cu-2.BF4, around 10 s, which increased to 40 s for Cu-2.I. Despite these significant differences, and a slower polymerization process with Cu-2.I, identical final monomer conversions could be found with the three photoinitiating systems after only 160 s of irradiation.
Control of the production of radicals is a great challenge especially for photopolymerization. Indeed, for 3D-printing applications, a spatial and temporal control is researched meaning that the polymerization should only occur in the irradiated zone and for the time that the surface is exposed to light. The polymerization process should stop immediately after the light is switched off. In these conditions, precise figures can be photopolymerized. As exemplified in Figure 16, a precise figure could be obtained using Cu-2 for the FRP and the FRPCP of a blend of TMPTA/MR1 30%/70% upon exposure to a LED projector at 405 nm. Well-defined 3D written patterns were obtained with the three-component photoinitiating system Cu-2/(tert-Bu)Ph2I+/NVK (0.2%/5%/1%, w/w/w), notably due to the copolymerization of epoxides and acrylates avoiding the shrinkage effects.
However, for numerous industrial applications and especially for the photopolymerization of coatings on complex structures for which the surface cannot be fully exposed to light, the possibility to initiate a polymerization in shadowed area is actively researched [100]. Concerning this point, a remarkable example was reported with Cu-2 [101].
Once again, Cu-2 was tested in difficult conditions since the low viscosity resin MR2 was used for this study. To design a propagating three-component photocurable resin, NVK or EDB were not selected as the reducing agents but tin(II) 2-ethylhexanoate or bismuth(III) 2-ethylhexanoate. A comparison of the two photoinitiating systems Cu-2/(tert-Bu)Ph2I+/tin(II) 2-ethylhexanoate (0.2%/1%/5%, w/w/w) and Cu-2/(tert-Bu)Ph2I+/ bismuth(III) 2-ethylhexanoate (0.2%/1%/5%, w/w/w) revealed the FRP of MR2 upon irradiation with a LED at 405 nm under air to be extremely fast, since a conversion of 83% was obtained within 30 s for the first photoinitiating system and 70% in 150 s for the second one. Considering the better performance of tin(II) 2-ethylhexanoate as the reducing agent, tests of radical photopolymerization in shadowed areas were only carried out with this reductant. From an experimental point of view, the setup is extremely simple since a Pasteur pipette is filled with the photocurable resin and only the extremity of the pipette is exposed to light, the rest of the pipette being protected from light. After polymerization, the pipette is broken, and the length of the polymer is measured. Using this procedure and by irradiating a three-component photoinitiating system in which the Cu-2 concentration has been reduced to 0.15 wt% to get a better light penetration, the polymerization of the MR2 resin was determined as propagating until 9 cm from the irradiation point after irradiating for 4 minutes (see Figure 17). A remarkable final monomer conversion could be determined even at the extremity of the polymerized resin (77%). Interestingly, the polymerization of a MR2 resin comprising 45 wt% of barium glass fillers could also be performed in the same conditions and a polymer extending over 8.5 cm was obtained, with a monomer conversion of 55% over the whole polymer after only 4 minutes of irradiation. This result is exceptional considering that the light penetration inside the filled sample is considerably reduced due to light diffusion by the fillers. To monitor the progress of the polymerization process, an infrared thermal imaging camera was used, as the polymerization of acrylate is exothermic. Using the camera, a polymerization temperature increasing to 70°C could be determined, and the progress of the polymerization front inside the resin could followed “live” (see Figure 18).
In 2016, an interesting concept concerning the development of a new redox initiating system was proposed. Precisely, a particular reaction between copper (II) acetate and 2-dppba was identified as being capable to generate acac• as a radical source to initiate the FRP of acrylates [102,103]. This unprecedented route to produce carbon-centered acac• radicals was named by the authors “metal acetylacetonate–bidentate ligand interaction” (MABLI) and this initial concept was largely exploited so that its photoactivation was rendered possible in 2018 [79,82,83,104]. As shown in the Figure 19a, by mixing copper (II) acetate and 2-dppba, a ligand exchange, a modification of the oxidation degree of copper accompanied with the release of an acac• was demonstrated with this two-component system. Ligand exchange in a solution is extremely fast, occurring within 5 seconds in THF (see Figure 19b).
This unexpected reaction is of crucial interest to promote the redox FRP of acrylates without taking recourse to the standard amine/peroxide combination raising numerous toxicity and safety concerns. Notably, depending on the substitution pattern, amines can be extremely toxic, and peroxides are also known to be potentially explosive. For these reasons, long-term storage of these resins is a major issue for industry, and alternatives to amines and peroxides are thus actively researched by both the industrial and the academic communities [105]. By introducing Ph2I+ in the two-component Cu(acac)2/2-dppba system, re-oxydation of Cu(I) to Cu(II) was possible. A photoactivation of the polymerization of the methacrylate (1,4-BDMA/ HPMA/UDMA) mixture with this three-component system (Cu(acac)2/Ph2I+/2-dppba) could also be realized by irradiating at 405 nm, as Cu(acac)2 absorbs at this wavelength. Proof of the exceptional efficiency of the photoactivation was also evidenced by adding 0.024 wt% of a strong polymerization inhibitor (tempol) and polymerizing under air. In fact, tempol was introduced to reduce the polymerization efficiency and thus evidence the contribution of light irradiation. Besides these difficult polymerization conditions, an enhancement of 40% of the monomer conversion was obtained upon irradiation, increasing the monomer conversion from 40% to 80% (see Figure 20). However, this redox photoactivated system was not catalytic. This is notably due to the release of an acac• from the coordination sphere of the metal center, which happens irreversibility.
This issue was addressed in 2018 by totally modifying the formulation and by replacing the three partners in the former three-component system (Cu(acac)2/Ph2I+/2-dppba), namely 2-dppba by vitamin-C (Vit-C), Ph2I+ by dibenzoyl peroxide, and Cu(acac)2 by copper (II) 2-ethylhexanoate (Cu(Oct)2 or copper (II) acetate (Cu(OAc)2) [79]. Using this new three-component system, no acac• radicals are formed but Ph(C=O)O• resulting from the decomposition of BPO. Conversely, regeneration of the metal center is facilitated in this last case, as 2-ethylhexanoate and acetate ligands are labile ligands.
If the mechanism of polymerization was now catalytic, FRP efficiency of the three-component (Cu(OAc)2/BPO/Vit-C) remained lower than that of the reference two-component 4-N,N-trimethylaniline/BPO system. A lower exothermicity was measured by pyrometry (58 °C vs. 100 °C). Photoactivation of the polymerization of IPNs was also examined with the simultaneous polymerization of EPOX with the methacrylate mixture (1,4-BDMA/HPMA/UDMA) using the four-component system (Cu(Oct)2/BPO/Vit-C/Ph2I+). Due to the limited absorption of Cu(Oct)2 in the visible range, photoactivation of the resin could only be carried out at 405 nm. As shown in Figure 21, the accelerating effect of light was obvious, even if no real impact on the exothermicity of the polymerization was found. However, a higher final conversion for the acrylate functions was nevertheless obtained (70% of final monomer conversion upon irradiation vs. 50% without light). Considering that light does not contribute to a higher exothermicity of the reaction, the origin of this unexpected behavior was researched.
While getting a deeper insight into the chemical mechanism, the cationic system Cu(Oct)2/Ar2I+/reductant proved to be an effective combination to initiate the CP of epoxides, even in the absence of light. Electron spin resonance (ESR) analyses revealed that the Cu(Oct)2/Ph2I+/reductant combination does not produce radicals, as no electron paramagnetic resonance (EPR) signal was detected. Conversely, presence of a pH-indicator (ethyl red) in a solution of Cu(Oct)2 and Ph2I+ revealed the reaction media to become acidic upon addition of the reductant as the result of the formation of H+. From the different experiments, it could be concluded that without light, Cu(II)(Oct)2 could be reduced to Cu(I) by Vit-C and that the reduced form of Cu(Oct)2 could interact with Ph2I+ to produce protons (see Scheme 6). Parallel to this, upon light excitation, Cu(Oct)2 can directly interact in its excited state with Ph2I+, producing Ph•, and thus improving the final monomer conversion of acrylates. In light of these results, light activation only marginally contributes to the formation of radicals. The main source of radicals results from the redox process between Cu(II) and BPO.
The combination of a redox and a photoinitiated polymerization within a single three-component system is quite unique and relies on the dual role of copper complexes in these systems, acting both as a redox agent and a photosensitizer. However, efficiency of these double-mode polymerization systems is directly related to the generation of radicals out of the coordination sphere of the metal center (Ph(C=O)O• instead of acac•) and the use of weakly coordinating ligands (acetate ligands). This last parameter is of crucial importance, especially for the redox system where a modification of the oxidation degree of copper is observed. Parallel to this, efficiency of these systems is also based on the absorption ability of copper complexes in the visible range.
These results are consistent with those previously reported for another copper (I) complex, namely Cu-25 (see Figure 22). In the four-component Cu-25/Vit-C/BPO/(t-Bu)Ph2I+ system, the redox reaction was again found to be more important for the FRP of the methacrylate mixture (1,4-BDMA/ HPMA/UDMA) than the interaction in the excited state of the Cu(I)/(t-Bu)Ph2I+ combination (producing Ph•) [106].
As shown in Figure 22, the contribution of the photoactivated process is marginal to the overall polymerization process. Indeed, almost no enhancement of the reaction temperature as well as no modification of the reaction time was found, demonstrating the dark polymerization to be mainly responsible of the FRP of acrylates.
2.6. Iron Complexes
Following the different works done on ruthenium, iridium, zinc, and copper complexes, iron complexes have been identified as potential candidates for polymerization and tested for the first time in 2015 in the context of photoredox catalysis [107,108,109,110,111,112,113]. Indeed, prior to this work, photopolymerization of acrylates or styrene with iron complexes have already been reported in the literature [114,115,116]. Similarly, numerous combinations of ferrocenium salts have also been used for the FRP of acrylates under visible light in the past [117,118,119,120]. As a main advantage, iron is one of the cheapest metals. This element is also extremely abundant on Earth. Contrarily to ruthenium, iridium, and copper complexes, iron complexes exhibit a major drawback, namely, their relatively short excited-state lifetimes in the nanosecond timescale or even below, what could result in inefficient interactions with the iodonium salt during light irradiation and a poor reactivity [121]. However, as previously detailed, the excited state lifetime is not the only parameter governing the reactivity of photocatalysts. The molar extinction coefficient, the rate constant of interaction with the iodonium salt, or the solubility also have to be considered. As observed for iridium complexes, iron-based photocatalysts could be used both in oxidative and reductive cycles for the FRP of acrylates. Thus, parallel to the classical oxidative cycle Fe complex/Ph2I+/NVK, reactive radicals could also be produced with the following three-component Fe complex/EDB/R-Cl system (where R-Cl is a halogenated aromatic ring). In this last case, R• and EDB(-H)• are both formed as initiating species (see Scheme 7). Since the regeneration of Fe complexes likely occurs, an extremely low photocatalyst content could be used, as low as 0.02 wt%. Considering that all iron complexes absorb around 400 nm, these complexes are thus good candidates for photopolymerization carried out at 405 nm (see Figure 23).
As often observed, the two-component systems Fe complex/Ph2I+ (0.2%/2%, w/w) were less efficient than the three-component photoinitiating system Fe complex/Ph2I+/NVK (0.2%/2%/3%, w/w/w) for the FRP of TMPTA. An improvement of the final monomer conversion of approximately 15% was observed for all complexes. The highest conversion could be obtained with Fe-4, peaking at 59% upon irradiation at 405 nm for 400 s in laminate. After storage of the photocurable resin for a month at room temperature, a final conversion of 57% could be still obtained, demonstrating the stability of the formulation. A comparison with BAPO as a reference revealed only the nitro-substituted iron photocatalysts, such as Fe-4 and Fe-8, to rival with BAPO (53% of conversion at identical percentage in weight) for the FRP of TMPTA.
The superiority of the nitro-substituted complexes Fe-4 and Fe-8 over the other photoinitiators was confirmed during the FRPCP of EPOX. Final conversions ranged between 42% (for Fe-8) and 54% (for Fe-4), in contrast to 17% (for Fe-2) or 19% (for Fe-1) for the non-substituted ones. By decreasing the photocatalyst content to 0.02 wt%, the remarkable photoinitiating ability of Fe-4 could be revealed. A final conversion of 42% could still be obtained, in contrast to the dramatic decrease observed for BAPO (18%). Interestingly, photocatalysts bearing nitro-substituents (Fe-4, Fe-8, Fe-19) could outperform all the other iron photocatalysts. This trend is not only observed with iron complexes but also with purely organic photoinitiators [122,123]. If this structure‒performance relationship could be established numerous times by comparing the results obtained for photoinitiators bearing nitro groups or not, the origin of this improvement has not been clearly established by the authors. Notably, the photophysical properties of photocatalysts are only marginally impacted by the introduction of the nitro groups.
In the past, photopolymerization has been almost exclusively used to prepare coatings and in this aim, multifunctional monomers were typically employed so that a robust crosslinked polymer could be obtained. However, based on the recent environmental concerns and the desire to develop green syntheses of polymers, numerous attempts have been carried out to develop a controlled polymerization of monofunctional monomers using light as the activation source. Contrary to polymers obtained with multifunctional monomers that form an insoluble crosslinked polymer network, soluble polymers with linear chains can be obtained using a monofunctional monomer. Analyses of the chain length as well as the polydispersity of polymers can be thus performed.
At present, only a low or moderate control of the polymer size is obtained by photopolymerization, high polydispersity indices being determined by size exclusion chromatography (SEC) [124,125]. This result is counterintuitive since, from a theoretical point of view, a higher control of the polymerization process should be obtained by using light, when the polymerization reaction is carried out at room temperature. In these conditions, the competition between polymerization and side-reactions should be avoided [126,127,128,129,130,131,132,133,134,135,136]. However, the production of highly reactive radicals still limits the control of the polymerization process, even if some attempts have been made to introduce control agents, as exemplified with photoassisted nitroxide-mediated polymerization (NMP2) [124,125]. Another interesting feature is that dormant species which can only be activated by light can also be created, enabling a subsequent chemical modification of the initial polymer. The first report mentioning the use of iron complexes for the controlled radical photopolymerization (CRP2) of different acrylates was reported in 2016 [110]. In this series, Fe-4 proved again to be an interesting candidate. A polymerization “on-demand” of methyl methacrylate (MMA) using the three-component system Fe-4/EBr/EDB (where EBr stands for ethyl α-bromo-phenylacetate) could be obtained (see Figure 24a). A good linearity of the MMA conversion as a function of time was also demonstrated (see Figure 24b). Due to the living character of the photopolymerization, a re-initiation of the polymerization process was possible with butyl acrylate (BA). Block copolymers poly(MMA)-b-poly(BA) could be prepared and the Mn increased from 55,000 g/mol for poly(MMA) to 250,000 g/mol for the copolymer (see Figure 24c). However, the polydispersity for these polymers remained high.
Finally, surface modification was also examined with Fe-4. To evidence this, the hydrophilic surface of a polyacrylate was converted to a hydrophobic one by re-initiating the polymerization with a fluorinated monomer. The contact angle of the polymer surface changed from 60° to 115°, demonstrating the hydrophobicity of the surface after photochemical modification. To further evidence the polymerization of a second layer on top of the first one, a mask was used so that the first layer was only partially exposed to light. A spatial control of the polymerization could be obtained. Analysis by X-ray photoelectron spectroscopic (XPS) confirmed only the surface exposed to light to be chemically modified and fluorine to be only found in this area (see Figure 25). Finally, the iron (III)-based metal-organic framework (MOF), which is a porous structure, was also used as a photoinitiator of polymerization in combination with an iodonium salt and NVK to promote the FRPCP of epoxides or the FRP of acrylates under visible light [137]. The photocatalytic activity of MOFs is well-known in the literature [138,139,140], as these catalysts have been used for the alkoxylation of propylene glycol [141] or acrylic acid [142], or the degradation of Rhodanine 6G [143]. The use of these coordination polymers is of crucial interest for the design of composite materials, these organic/inorganic hybrid materials acting as fillers capable of modifying the mechanical properties of the final polymers [144,145,146,147,148,149]
However, as a drawback, MOFs were not soluble within the resin, resulting in a heterogenous catalysis. Among the MOF series, MIL53 and MIL88A were the most efficient photoinitiators, with a final monomer conversion of 58% and 53% being respectively obtained for the FRPCP of epoxides with the three-component MIL/Ph2I+/NVK (0.2%/2%/3%, w/w/w) system upon irradiation for 400 s under air with a LED at 405 nm (see Figure 26).
Specifically, MIL53 and MIL88A are characterized by flexible structures capable to reversibly modify their pore size. While getting a deeper insight into the photocatalytic activity of the different MOFs, the iron content, the size of the pores, or the particles size were not determined as strongly influencing the catalytic activity. The main factor influencing the photoinitiating abilities of MOFs was determined as being the organic part (higher final conversions are obtained with flexible diacids than aromatic diacids) and the presence of hydroxy-bridging groups. Concerning this last factor, this trend is consistent with previous results reported in the literature [150].
As shown in this review, numerous photocatalysts have been designed for polymerization processes carried out at 405 nm. Interest for this specific wavelength is supported by the fact that this is presently the wavelength under use in 3D printers [151,152]. However, as far as the light penetration is concerned, polymerization at a long wavelength is of higher interest than 405 nm simply due to the problem of light penetration inside the resin. Indeed, if the light penetration is typically 1 mm at 405 nm, it can reach 5 cm at 800 nm (see Figure 27) [22]. Therefore, from an industrial point of view, a thicker layer can be polymerized in once, possibly addressing the low polymerization peed of 3D-printers. Additionally, resins with high filler contents can also be polymerized, which is not possible with photocurable resins activatable at 405 nm. Therefore, polymerization in the near-infrared region (NIR) region paves the way towards the fabrication of composites by photopolymerization.
Concerning iron complexes, ferrocene which is one of the simplest metallocene comprising an iron (II) cation sandwiched between two cyclopentadienyl moieties was notably used in 2019 for the design of a series of push‒pull dyes capable of initiating a polymerization in the near-infrared region (NIR) [153]. A series of nine dyes, namely Fe-23‒Fe-31, was examined for photopolymerization carried out at 405 and 785 nm (see Figure 28). NIR dyes based on ferrocene have been extensively studied in the literature due to the reversibility of the oxidation process centered on the ferrocene moiety [154].
From a synthetic point of view, push‒pull chromophores are interesting compounds as these structures can be easily synthesized in one ste, by means of a Knoevenagel reaction. By opposing ferrocene-carboxaldehyde with one equivalent of the appropriate electron acceptor in the presence of a catalytic amount of piperidine, the dyes can be synthesized. Using this procedure, Fe-26‒Fe31 could be obtained in high yields, ranging from 89% to 98% (see Scheme 8). The lower reaction yield for Fe-31 (89% yield) can be confidently assigned to the steric hindrance generated by the pentamethylcyclopentadienyl group of the ferrocene donor.
In this series, the most red-shifted absorption was found for Fe-30 so that its photoinitiating ability at 785 nm was examined. While using the three-component photoinitiating systems Fe-30/(tert-Bu)Ph2I+/2-dppba (0.2%/2%/1.5%, w/w/w) or Fe-30/(tert-Bu)Ph2I+/DMABA (0.2%/2%/1.5%, w/w/w), where 2-dppba stands for 2-diphenylphosphinobenzoic acid and DMABA for 3-(dimethylamino) benzyl alcohol, final monomer conversions of 52% could be obtained for the FRP of BisGMA/TEGDMA (30/70 wt%) blend with the two systems after irradiation with a LED at 785 nm for 400 s under air. By increasing the lamp intensity from 400 mW/cm² to 2.55 W/cm², the final conversion could be increased up to 85% after only 200 s with the three-component system Fe-30/(tert-Bu)Ph2I+/DMABA (0.2%/2%/1.5%, w/w/w). Dependence of the monomer conversion on the light intensity is clearly evidenced. Parallel to photopolymerization, Fe-30 was demonstrated to be capable of initiating a redox polymerization (i.e., without light) using the three-component system Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w) where APS stands for ammonium persulfate. In this case, a second resin was used, composed of a methacrylate mixture containing 33.3 wt% of 1,4-butanediol-dimethacrylate (1,4-BDMA), 33.3 wt% of hydroxypropylmethacrylate (HPMA), and 33.3 wt% of urethane-dimethacrylate (UDMA) (see Figure 29).
To monitor the progress of the polymerization reaction, optical pyrometric measurements were done, as the reaction is exothermic (see Figure 30a). Depending on the ferrocene derivatives, a reaction temperature ranging between 35 and 40 °C was found. However, the reaction was slow, requiring 600 s with Fe-26 and 1200 s for Fe-30. The ability of catalysts to be used in both redox and photoredox polymerization is unusual. The two polymerization modes possess their own advantages. As shown in Figure 30b, in the case of a photopolymerization reaction carried out under air, the surface is more polymerized than the bottom part of the sample due to the problem of light penetration inside the resin. Only the surface, which is exposed to light, can polymerize efficiently. A spatial control is obtained, where only the surface exposed to light polymerizes. However, for redox polymerization, the surface is less polymerized than the bottom part due to oxygen inhibition. No spatial control was obtained. The whole sample was polymerized due to the diffusion of radicals. By combining the two polymerization modes, the global monomer conversion can be significantly improved and photoacceleration of the polymerization process is also possible “on demand”.
By applying this concept of photoactivated redox polymerization and by irradiation of a four-component system Fe-30/2-dppba/APS/(tert-Bu)Ph2I+ (0.1%/1.5%/2%/1%, w/w/w/w), a two-fold reduction of the reaction time was obtained, associated with a major increase of the reaction temperature (+ 20 °C), demonstrating the efficiency of the photoactivation and the production of numerous radicals (see Figure 30c). Notably, the remarkable efficiency of the photoactivated redox polymerization can confidently assigned to the regeneration of Fe-30 during the oxidative cycle but also to the faster photoinitiated process relative to the redox process (see Scheme 9).
In fact, this work on ferrocene photocatalysts is described in the continuation of a previous work concerning the photoactivation of redox polymerizations with light using copper complexes (see previous section on copper complexes).
3. Conclusions
In this review, approximately 70 metal-based photocatalysts of polymerization have been reviewed. While the photoinitiating performances have governed the initial works on platinum, ruthenium, or iridium complexes, rapidly, environmental and toxicity concerns have taken precedence over performance. At present, copper or iron complexes are clearly less efficient than iridium complexes, high monomer conversions being obtained in laminate, whereas a similar performance was previously obtained with iridium complexes under air. Besides, by chemical engineering, TADF photocatalysts based on copper or iron photocatalysts specifically designed for photoassisted redox polymerization have greatly improved the final monomer conversions. At present, some challenges remain concerning photopolymerization. Notably, the polymerization at long wavelength is only scarcely studied, even if it paves the way towards photocomposites. Parallel to this, controlled photopolymerization is still far behind that of the thermally controlled polymerization. These two challenges will certainly be addressed in the forthcoming years.
Author Contributions
Conceptualization, F.D.; writing—original draft preparation, F.D.; writing—review and editing, F.D.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Hoffmann, N. Electron and hydrogen transfer in organic photochemical reactions. J. Phys. Org. Chem. 2015, 28, 121–136. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Hoffmann, N. Studies in organic and physical photochemistry—an interdisciplinary approach. Org. Biomol. Chem. 2016, 14, 7392–7442. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lu, W.; Devalankar, D.; Ding, Z. Photochemical formation and cleavage of C–N bond. Org. Lett. 2015, 17, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, N.; Mizuno, K.; Otsuji, Y.; Caldwell, R.A.; Helms, A.M. Photochemical C−C bond cleavage of 1,2-diarylcyclopropanes bearing an acetylphenyl group. generation and observation of triplet 1,3-biradicals. J. Org. Chem. 1998, 63, 3176–3184. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical reactions as key steps in organic synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5323. [Google Scholar] [CrossRef]
- Narayanama, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef]
- Bogdos, M.K.; Pinard, E.; Murphy, J.A. Applications of organocatalysed visible-light photoredox reactions for medicinal chemistry. Beilstein J. Org. Chem. 2018, 14, 2035–2064. [Google Scholar] [CrossRef]
- Parasram, M.; Gevorgyan, V. Visible light-induced transition metal-catalyzed transformations: Beyond conventional photosensitizers. Chem. Soc. Rev. 2017, 46, 6227–6240. [Google Scholar] [CrossRef] [PubMed]
- Islamova, R.M.; Dobrynin, M.V.; Vlasov, A.V.; Eremina, A.A.; Kinzhalov, M.A.; Kolesnikov, I.E.; Zolotarev, A.A.; Masloborodova, E.A.; Luzyanin, K.V. Iridium (III)-catalysed cross-linking of polysiloxanes leading to the thermally resistant luminescent silicone rubbers. Catal. Sci. Technol. 2017, 7, 5843–5846. [Google Scholar] [CrossRef]
- Gee, J.C.; Fuller, B.A.; Lockett, H.M.; Sedghi, G.; Robertson, C.M.; Luzyanin, K.V. Visible light accelerated hydrosilylation of alkynes using platinum-[acyclic diaminocarbene] photocatalysts. Chem. Commun. 2018, 54, 9450–9453. [Google Scholar] [CrossRef] [PubMed]
- Ischay, M.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient visible light photocatalysis of [2 + 2] enone cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef] [PubMed]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging photoredox catalysis with organocatalysis: The direct asymmetric alkylation of aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef]
- Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R.J. Electron-transfer photoredox catalysis: Development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- Oelgemöller, M. Solar Photochemical synthesis: From the beginnings of organic photochemistry to the solar manufacturing of commodity chemicals. Chem. Rev. 2016, 116, 9664–9682. [Google Scholar] [CrossRef]
- Dadashi-Silab, S.; Doran, S.; Yagci, Y. Photoinduced electron transfer reactions for macromolecular syntheses. Chem. Rev. 2016, 116, 10212–10275. [Google Scholar] [CrossRef]
- Chen, M.; Zhong, M.; Johnson, J.A. Light-controlled radical polymerization: Mechanisms, methods, and applications. Chem. Rev. 2016, 116, 10167–10211. [Google Scholar] [CrossRef]
- Poelma, J.E.; Fors, B.P.; Meyers, G.F.; Kramer, J.W.; Hawker, C.J. Fabrication of complex three-dimensional polymer brush nanostructures through light-mediated living radical polymerization. Angew. Chem. Int. Ed. 2013, 52, 6844–6848. [Google Scholar] [CrossRef]
- Anastasaki, A.; Nikolaou, V.; Brandford-Adams, F.; Nurumbetov, G.; Zhang, Q.; Clarkson, G.J.; Fox, D.J.; Wilson, P.; Kempe, K.; Haddleton, D.M. Photo-induced living radical polymerization ofacrylates utilizing a discrete copper(II)-formate complex. Chem. Commun. 2015, 51, 5626–5629. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, A.H.; Dumur, F.; Grant, T.M.; Noirbent, G.; Gigmes, D.; Lessard, B.H.; Fouassier, J.P.; Lalevée, J. High Performance Near-infrared (NIR) photoinitiating systems operating under low light intensity and in the presence of oxygen. Macromolecules 2018, 51, 1314–1324. [Google Scholar] [CrossRef]
- Lalevée, J.; Blanchard, N.; Tehfe, M.A.; Morlet-Savary, F.; Fouassier, J.P. Green bulb light source induced epoxy cationic polymerization under air using tris(2,2′-bipyridine)ruthenium(II) and silyl radicals. Macromolecules 2010, 43, 10191–10195. [Google Scholar] [CrossRef]
- Ligon, S.C.; Husár, B.; Wutzel, H.; Holman, R.; Liska, R. Strategies to reduce oxygen inhibition in photoinduced polymerization. Chem. Rev. 2014, 114, 557–589. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, A.H.; Dumur, F.; Noirbent, G.; Lalevée, J.; Gigmes, D. Organometallic vs organic photoredox catalysts for photocuring reactions in the visible region. Beilstein J. Org. Chem. 2018, 14, 3025–3046. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nicewicz, D.A.; Nguyen, T.M. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. [Google Scholar] [CrossRef]
- Tucker, J.W.; Stephenson, C.R.J. Shining light on photoredox catalysis: Theory and synthetic applications. J. Org. Chem. 2012, 77, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C. Organosilanes in Radical Chemistry; John Wiley & Sons: Chicheste, UK, 2004. [Google Scholar]
- Lalevée, J.; Blanchard, N.; Tehfe, M.A.; Peter, M.; Morlet-Savary, F.; Fouassier, J.P. Household LED irradiation under air: Cationic polymerization using iridium or ruthenium complex photocatalysts. Polym. Bull. 2012, 68, 341–347. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Lalevée, J.; Gigmes, D.; Fouassier, J.P. Green chemistry: Sunlight-induced cationic polymerization of renewable epoxy monomers under air. Macromolecules 2010, 43, 1364–1370. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Lalevée, J.; Gigmes, D.; Fouassier, J.P. Combination of transition metal carbonyls and silanes: New photoinitiating systems. J. Polym. Sci. A Polym. Chem. 2010, 48, 1830–1837. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Ma, L.; Graff, B.; Morlet-Savary, F.; Fouassier, J.P.; Zhao, J.; Lalevée, J. Cyclometallated Pt(II) complexes in visible-light photoredox catalysis: New polymerization initiating systems. Macromol. Chem. Phys. 2012, 213, 2282–2286. [Google Scholar] [CrossRef]
- Crivello, J.V. Silane reduction of onium salts. Appl. Organomet. Chem. 2010, 24, 169–178. [Google Scholar] [CrossRef]
- Bamford, C.H.; Mullik, S.U.; Puddephatt, R.J. Photo-initiation of free-radical polymerization by dimethyl-(2,2′-bipyridyl)platinum(II)+ tetrafluoroethylene. J. Chem. Soc. Faraday Trans. 1975, 71, 2213–2225. [Google Scholar] [CrossRef]
- Ziessel, R.; Diring, S.; Retailleau, P. Terpyridine-platinum(II) acetylide complexes bearing pendent coordination units. Dalton Trans. 2006, 3285–3329. [Google Scholar] [CrossRef]
- Li, K.; Ming Tong, G.S.; Wan, Q.; Cheng, G.; Tong, W.Y.; Ang, W.H.; Kwong, W.L.; Che, C.M. Highly phosphorescent platinum (II) emitters: photophysics, materials and biological applications. Chem. Sci. 2016, 7, 1653–1673. [Google Scholar] [CrossRef]
- Kappaun, S.; Slugovc, C.; List, E.J.W. Phosphorescent organic light-emitting devices: Working principle and iridium-based emitter materials. Int. J. Mol. Sci. 2008, 9, 1527–1547. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Lepeltier, M.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Structural effects in the iridium complex series: Photoredox catalysis and photoinitiation of polymerization reactions under visible lights. Macromol. Chem. Phys. 2017, 218, 1700192. [Google Scholar] [CrossRef]
- Aspley, C.J.; Williams, J.A.G. Palladium-catalysed cross-coupling reactions of ruthenium bis-terpyridyl complexes: Strategies for the incorporation and exploitation of boronic acid functionality. New J. Chem. 2001, 25, 1136–1147. [Google Scholar] [CrossRef]
- Arm, K.J.; Williams, J.A.G. Boronic acid-substituted metal complexes: Versatile building blocks for the synthesis of multimetallic assemblies. Chem. Commun. 2005, 2, 230–232. [Google Scholar] [CrossRef]
- Arm, K.J.; Williams, J.A.G. A cross-coupling strategy for the synthesis of dimetallic assemblies containing mixed bipyridine-terpyridine bridging ligands: Luminescence and energy transfer properties. Dalton Trans. 2006, 18, 2172–2174. [Google Scholar] [CrossRef] [PubMed]
- Leslie, W.; Batsanov, A.S.; Howard, J.A.K.; Williams, J.A.G. Cross-couplings in the elaboration of luminescent bis-terpyridyl iridium complexes: The effect of extended or inhibited conjugation on emission. Dalton Trans. 2004, 4, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Whittle, V.L.; Williams, J.A.G. A new class of iridium complexes suitable for stepwise incorporation into linear assemblies: Synthesis, electrochemistry, and luminescence. Inorg. Chem. 2008, 47, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.G.; Wilkinson, A.J.; Whittle, V.L. Light-emitting iridium complexes with tridentate ligands. Dalton Trans. 2008, 16, 2081–2099. [Google Scholar] [CrossRef] [PubMed]
- Whittle, V.L.; Williams, J.A.G. Cyclometallated, bis-terdentate iridium complexes as linearly expandable cores for the construction of multimetallic assemblies. Dalton Trans. 2009, 20, 3929–3940. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wei, L.; Wang, J.; Li, Y.; Wu, H.; Yang, W.; Cao, Y. Novel heteroleptic iridium(III) complexes with a 2-(1H-pyrazol-5-yl)pyridine derivative containing a carbazole group as ancillary ligand: Synthesis and application for polymer light-emitting diodes. Synth. Met. 2014, 187, 209–216. [Google Scholar] [CrossRef]
- Lepeltier, M.; Dumur, F.; Marrot, J.; Contal, E.; Bertin, D.; Gigmes, D.; Mayer, C.R. Unprecedented combination of regioselective hydrodefluorination and ligand exchange reaction during the syntheses of tris-cyclometalated iridium(III) complexes. Dalton Trans. 2013, 42, 4479–4486. [Google Scholar] [CrossRef] [PubMed]
- Lepeltier, M.; Dumur, F.; Graff, B.; Xiao, P.; Gigmes, D.; Lalevée, J.; Mayer, C.R. Tris-cyclometalated iridium(III) complexes with three different ligands: A new example with 2-(2,4-difluorophenyl)pyridine-based complex. Helv. Chim. Acta 2014, 97, 939–956. [Google Scholar] [CrossRef]
- Dumur, F.; Bertin, D.; Gigmes, D. Iridium (III) complexes as promising emitters for solid-state Light-Emitting Electrochemical Cells (LECs). Int. J. Nanotechnol. 2012, 9, 377–395. [Google Scholar] [CrossRef]
- Dumur, F.; Yuskevitch, Y.; Wantz, G.; Mayer, C.R.; Bertin, D.; Gigmes, D. Light-emitting electrochemical cells based on a solution-processed multilayered device and an anionic iridium (III) complex. Synth. Met. 2013, 177, 100–104. [Google Scholar] [CrossRef]
- Dumur, F.; Nasr, G.; Wantz, G.; Mayer, C.R.; Dumas, E.; Guerlin, A.; Miomandre, F.; Clavier, G.; Bertin, D.; Gigmes, D. Cationic iridium complex for the design of soft salt-based phosphorescent OLEDs and color-tunable light-emitting electrochemical cells. Org. Electron. 2011, 12, 1683–1694. [Google Scholar] [CrossRef]
- Nasr, G.; Guerlin, A.; Dumur, F.; Beouch, L.; Dumas, E.; Clavier, G.; Miomandre, F.; Goubard, F.; Gigmes, D.; Bertin, D.; et al. Iridium(III) soft salts from dinuclear cationic and mononuclear anionic complexes for OLED devices. Chem. Commun. 2011, 47, 10698–10700. [Google Scholar] [CrossRef] [PubMed]
- Lepeltier, M.; Graff, B.; Lalevée, J.; Wantz, G.; Ibrahim-Ouali, M.; Gigmes, D.; Dumur, F. Heteroleptic iridium (III) complexes with three different ligands: Unusual triplet emitters for light-emitting electrochemical cells. Org. Electron. 2016, 37, 24–34. [Google Scholar] [CrossRef]
- Lepeltier, M.; Appaix, F.; Liao, Y.Y.; Dumur, F.; Marrot, J.; Le Bahers, T.; Andraud, C.; Monnereau, C. Carbazole-substituted iridium complex as a solid state emitter for two-photon intravital imaging. Inorg. Chem. 2016, 55, 9586–9595. [Google Scholar] [CrossRef] [PubMed]
- Lalevée, J.; Peter, M.; Dumur, F.; Gigmes, D.; Blanchard, N.; Tehfe, M.A.; Morlet-Savary, F.; Fouassier, J.P. Subtle ligand effects in oxidative photocatalysis with iridium complexes: Application to photopolymerization. Chem. Eur. J. 2011, 17, 15027–15031. [Google Scholar] [CrossRef] [PubMed]
- Telitel, S.; Dumur, F.; Lepeltier, M.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Photoredox process induced polymerization reactions: Iridium complexes for panchromatic photoinitiating systems. Comptes Rendus Chim. 2016, 19, 71–78. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Z.; Xu, D.; Yu, Y.; Tang, X.; Guo, H. Studies of free radical polymerization initiated by visible light photoredox catalysis. Macromol. Chem. Phys. 2015, 216, 1055–1060. [Google Scholar] [CrossRef]
- Zhang, G.; Song, I.Y.; Ahn, K.H.; Park, T.; Choi, W. Free radical polymerization initiated and controlled by visible light photocatalysis at ambient temperature. Macromolecules 2011, 44, 7594–7599. [Google Scholar] [CrossRef]
- Lalevée, J.; Tehfe, M.A.; Dumur, F.; Gigmes, D.; Blanchard, N.; Morlet-Savary, F.; Fouassier, J.P. Iridium photocatalysts in free radical photopolymerization under visible lights. ACS Macro Lett. 2012, 1, 286–290. [Google Scholar] [CrossRef]
- Lalevée, J.; Blanchard, N.; Tehfe, M.A.; Peter, M.; Morlet-Savary, F.; Fouassier, J.P. A novel photopolymerization initiating system based on an iridium complex photocatalyst. Macromol. Rapid Commun. 2011, 32, 917–920. [Google Scholar] [CrossRef]
- Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Photochemistry and photophysics of coordination compounds: Iridium. Top. Curr. Chem. 2007, 281, 143–203. [Google Scholar] [CrossRef]
- Okada, S.; Okinaka, K.; Iwawaki, H.; Furugori, M.; Hashimoto, M.; Mukaide, T.; Kamatani, J.; Igawa, S.; Tsuboyama, A.; Takiguchi, T.; et al. Substituent effects of iridium complexes for highly efficient red OLEDs. Dalton Trans. 2005, 9, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Nonoyama, M. Benzo[h]quinolin-10-yl-N Iridium(III) Complexes. Bull. Chem. Soc. Jpn. 1974, 47, 767–768. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, X.; Cao, Y.; Shi, C.; Tao, P.; Zhao, Q.; Yuan, A. An oxygen-bridged triarylamine polycyclic unit based tris-cyclometalated heteroleptic iridium(III) complex: Correlation between the structure and photophysical properties. Dalton Trans. 2019, 48, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, A.B.; Alleyne, B.D.; Djurovich, P.I.; Lamansky, S.; Tsyba, I.; Ho, N.N.; Bau, R.; Thompson, M.E. Synthesis and characterization of facial and meridional tris-cyclometalated iridium(III) complexes. J. Am. Chem. Soc. 2003, 125, 7377–7387. [Google Scholar] [CrossRef] [PubMed]
- Edkins, R.M.; Wriglesworth, A.; Fucke, K.; Bettington, S.L.; Beeby, A. The synthesis and photophysics of tris-heteroleptic cyclometalated iridium complexes. Dalton Trans. 2011, 40, 9672–9678. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Meng, G.; Zhou, Y.; Tang, H.; Zhao, J.; Wang, Z. The photoluminescent properties of new cationic iridium (III) complexes using different anions and their applications in white light-emitting diodes. Materials 2015, 8, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- Rehm, D.; Weller, A. Kinetics of fluorescence quenching by electron and H.-atom transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- Lalevée, J.; Tehfe, M.A.; Morlet-Savary, F.; Graff, B.; Dumur, F.; Gigmes, D.; Blanchard, N.; Fouassier, J.P. Photoredox catalysis for polymerization reactions. Chimia 2012, 66, 439–441. [Google Scholar] [CrossRef]
- Lalevée, J.; Telitel, S.; Xiao, P.; Lepeltier, M.; Dumur, F.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P. Metal and metal-free photocatalysts: Mechanistic approach and application as photoinitiators of photopolymerization. Beilstein J. Org. Chem. 2014, 10, 863–876. [Google Scholar] [CrossRef]
- Lalevée, J.; Dumur, F.; Mayer, C.R.; Gigmes, D.; Nasr, G.; Tehfe, M.A.; Telitel, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P. Photopolymerization of N-Vinylcarbazole Using Visible-Light Harvesting Iridium Complexes as Photoinitiators. Macromolecules 2012, 45, 4134–4141. [Google Scholar] [CrossRef]
- Dumur, F.; Beouch, L.; Tehfe, M.A.; Contal, E.; Lepeltier, M.; Wantz, G.; Graff, B.; Goubard, F.; Mayer, C.R.; Lalevée, J.; et al. Low-cost zinc complexes for white organic light-emitting devices. Thin Solid Films 2014, 564, 351–360. [Google Scholar] [CrossRef]
- Dumur, F.; Contal, E.; Wantz, G.; Gigmes, D. Photoluminescence of zinc complexes: Easily tunable optical properties by variation of the bridge between the imido groups of schiff base ligands. Eur. J. Inorg. Chem. 2014, 25, 4186–4198. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Telitel, S.; Gigmes, D.; Contal, E.; Bertin, D.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; Lalevée, J. Zinc-based metal complexes as new photocatalysts in polymerization initiating systems. Eur. Polym. J. 2013, 49, 1040–1049. [Google Scholar] [CrossRef]
- Poli, R.; Allan, L.E.N.; Shaver, M.P. Iron-mediated reversible deactivation controlled radical polymerization. Prog. Polym. Sci. 2014, 39, 1827–1845. [Google Scholar] [CrossRef]
- Wang, J.S.; Matyjaszewski, K. Controlled/”living” radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3746. [Google Scholar] [CrossRef] [PubMed]
- Garra, P.; Dumur, F.; Nechab, M.; Morlet-Savary, F.; Dietlin, C.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Stable copper acetylacetonate-based oxidizing agents in redox (NIR photoactivated) polymerization: An opportunity for the one pot grafting from approach and an example on a 3D printed object. Polym. Chem. 2018, 9, 2173–2182. [Google Scholar] [CrossRef]
- Garra, P.; Carré, M.; Dumur, F.; Morlet-Savary, F.; Dietlin, C.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Copper-based (photo)redox initiating systems as highly efficient systems for interpenetrating polymer network preparation. Macromolecules 2018, 51, 679–688. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Morlet-Savary, F.; Dietlin, C.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Mechanosynthesis of a Copper complex for redox initiating systems with a unique near infrared light activation. J. Polym. Sci. A Polym. Chem. 2017, 55, 3646–3655. [Google Scholar] [CrossRef]
- Garra, P.; Morlet-Savary, F.; Graff, B.; Dumur, F.; Monnier, V.; Dietlin, C.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Metal Acetylacetonate–Bidentate Ligand Interaction (MABLI) as highly efficient free radical generating systems for polymer synthesis. Polym. Chem. 2018, 9, 1371–1378. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Gigmes, D.; Nechab, M.; Morlet-Savary, F.; Dietlin, C.; Gree, S.; Fouassier, J.P.; Lalevée, J. Metal acetylacetonate–bidentate ligand interaction (mabli) (photo)activated polymerization: Toward high performance amine-free, peroxide-free redox radical (photo)initiating systems. Macromolecules 2018, 51, 2706–2715. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Copper Complexes in Radical Photoinitiating Systems: Applications to Free Radical and Cationic Polymerization upon Visible LEDs. Macromolecules 2014, 47, 3837–3844. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Kermagoret, A.; Versace, D.L.; Toufaily, J.; Hamieh, T.; Graff, B.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Copper photoredox catalysts for polymerization upon near UV or visible light: Structure/reactivity/efficiency relationships and use in LED projector 3D printing resins. Polym. Chem. 2017, 8, 568–580. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Copper complexes: The effect of ligands on their photoinitiation efficiencies in radical polymerization reactions under visible light. Polym. Chem. 2014, 5, 6350–6357. [Google Scholar] [CrossRef]
- Yang, Q.; Balverde, S.; Dumur, F.; Lalevée, J.; Poly, J. Synergetic effect of the epoxide functional groups in the photocatalyzed atom transfer radical copolymerization of glycidyl methacrylate. Polym. Chem. 2016, 7, 6084–6093. [Google Scholar] [CrossRef]
- Dumur, F. Recent advances in organic light-emitting devices comprising copper complexes: A realistic approach for low-cost and highly emissive devices? Org. Electron. 2015, 21, 27–39. [Google Scholar] [CrossRef]
- Zhang, Q.; Komino, T.; Huang, S.; Matsunami, S.; Goushi, K.; Adachi, C. Triplet exciton confinement in green organic light-emitting diodes containing luminescent charge-transfer cu (i) complexes. Adv. Funct. Mater. 2012, 22, 2327–2336. [Google Scholar] [CrossRef]
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. [Google Scholar] [CrossRef]
- Parker, C.A.; Hatchard, C.G. Triplet-singlet emission in fluid solutions. Phosphorescence of eosin. Trans. Faraday Soc. 1961, 57, 1894–1904. [Google Scholar] [CrossRef]
- Bui, T.T.; Goubard, F.; Ibrahim-Ouali, M.; Gigmes, D.; Dumur, F. Recent advances on organic blue thermally activated delayed fluorescence (TADF) emitters for organic light-emitting diodes (OLEDs). Beilstein J. Org. Chem. 2018, 14, 282–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzrati-Zerelli, M.; Noirbent, G.; Goubard, G.; Bui, T.T.; Villotte, S.; Dietlin, C.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P.; Dumur, F.; et al. A novel class of photoinitiators with a thermally activated delayed fluorescence (TADF) property. New J. Chem. 2018, 42, 8261–8270. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Lalevée, J.; Zhao, J.; Stenzel, M.H. N-Vinylcarbazole as Versatile Photoinaddimer of Photopolymerization under Household UV LED Bulb (392 nm). Macromol. Rapid Commun. 2015, 36, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Lalevée, J.; Xiao, P.; Dumur, F.; Gigmes, D. Light induced free radical and/or cationic photopolymerization method. Patent WO2015132295 A1, 2015. [Google Scholar]
- Mokbel, H.; Anderson, D.; Plenderleith, R.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Copper photoredox catalyst “G1”: A new high performance photoinitiator for near-UV and visible LEDs. J. Polym. Chem. 2017, 8, 5580–5592. [Google Scholar] [CrossRef]
- Mokbel, H.; Anderson, D.; Plenderleith, R.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Simultaneous initiation of radical and cationic polymerization reactions using the “G1” copper complex as photoredox catalyst: Applications of free radical/cationic hybrid photopolymerization in the composites and 3D printing fields. Prog. Org. Coat. 2019, 132, 50–61. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Mokbel, H.; Monnier, V.; Morlet-Savary, F.; Dietlin, C.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. New synthetic route to an highly efficient photoredox catalyst by mechanosynthesis. ACS Omega 2018, 3, 10938–10944. [Google Scholar] [CrossRef]
- Mokbel, H.; Toufaily, J.; Hamieh, T.; Dumur, F.; Campolo, D.; Gigmes, D.; Fouassier, J.P.; Ortyl, J.; Lalevée, J. Specific cationic photoinitiators for near UV and visible LEDs: Iodonium versus ferrocenium structures. J. Appl. Polym. Sci. 2015, 42759. [Google Scholar] [CrossRef]
- Garra, P.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Photopolymerization processes of thick films and in shadow areas: A review for the access to composites. Polym. Chem. 2017, 8, 7088–7101. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Gigmes, D.; Al Mousawi, A.; Morlet-Savary, F.; Dietlin, C.; Fouassier, J.P.; Lalevée, J. Copper (photo)redox catalyst for radical photopolymerization in shadowed areas and access to thick and filled samples. Macromolecules 2017, 50, 3761–3771. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Morlet-Savary, F.; Dietlin, C.; Fouassier, J.P.; Laleveée, J. A new highly efficient amine-free and peroxide-free redox system for free radical polymerization under air with possible light activation. Macromolecules 2016, 49, 6296–6309. [Google Scholar] [CrossRef]
- Garra, P.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Redox two-component initiated free radical and cationic polymerizations: Concepts, reactions and applications. Prog. Polym. Sci. 2019, 94, 33–56. [Google Scholar] [CrossRef]
- Garra, P.; Dumur, F.; Nechab, M.; Morlet-Savary, F.; Dietlin, C.; Graff, B.; Doronina, E.P.; Sidorkin, V.F.; Gigmes, D.; Fouassier, J.P.; et al. Peroxide-free and amine-free redox free radical polymerization: Metal acetylacetonates/stable carbonyl compounds for highly efficient synthesis of composites. Macromolecules 2018, 51, 6395–6404. [Google Scholar] [CrossRef]
- Achilias, D.S.; Sideridou, I. Study of the effect of two BPO/amine initiation systems on the free-radical polymerization of MMA used in dental resins and bone cements. J. Macromol. Sci. Part A Pure Appl. Chem. 2002, 39, 1435–1450. [Google Scholar] [CrossRef]
- Garra, P.; Kermagoret, A.; Al Mousawi, A.; Dumur, F.; Gigmes, D.; Morlet-Savary, F.; Dietlin, C.; Fouassier, J.P.; Lalevée, J. New copper(I) complex based initiating systems in redox polymerization and comparison with the amine/benzoyl peroxide reference. Polym. Chem. 2017, 8, 4088–4097. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Iron complexes as photoinitiators for radical and cationic polymerization through photoredox catalysis processes. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 42–49. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Visible-light-sensitive photoredox catalysis by iron complexes: Applications in cationic and radical polymerization reactions. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2247–2253. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Iron complexes in visible-light-sensitive photoredox catalysis: Effect of ligands on their photoinitiation efficiencies. ChemCatChem 2016, 8, 2227–2233. [Google Scholar] [CrossRef]
- Telitel, S.; Dumur, F.; Campolo, D.; Poly, J.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Iron complexes as potential photocatalysts for controlled radical photopolymerizations: A tool for modifications and patterning of surfaces. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 702–713. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, J.; Campolo, D.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Copper and iron complexes as visible-light-sensitive photoinitiators of polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2673–2684. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. The carbazole-bound ferrocenium salt as a specific cationic photoinitiator upon near-UV and visible LEDs (365–405 nm). Polym. Bull. 2016, 73, 493–507. [Google Scholar] [CrossRef]
- Lalevée, J.; Dumur, F.; Nechab, M.; Gigmes, D.; Fouassier, J.P. Metal complex based photocatalyst systems: A quest for new possibilities and application to photopolymerization reactions. Trends Photochem. Photobiol. 2012, 14, 27–38. Available online: http://www.researchtrends.net/tia/article_pdf.asp?in=0&vn=14&tid=15&aid=3797 (accessed on 5 August 2019).
- Cunningham, A.F.; Desobry, V. Radiation Curing in Polymer Science and Technology; Fouassier, J.P., Rabek, J.F., Eds.; Elsevier: Barking, UK, 1993; Volume 2, pp. 323–374. [Google Scholar]
- Anderson, J.; Hardgrove, E.; Cavitt, T.B.; Reeves, P. Photoinitiation of multifunctional acrylates via ferrocene-alkyl chloride charge transfer complexes. J. Coat. Technol. Res. 2007, 4, 43–49. [Google Scholar] [CrossRef]
- Neumann, M.G.; Schmitt, C.C.; Rigoli, I.C. The photoinitiation of MMA polymerization in the presence of iron complexes. J. Photochem. Photobiol. A 2003, 159, 145–150. [Google Scholar] [CrossRef]
- Burget, D.; Fouassier, J.P. Laser flash photolysis studies of the interaction of Rose Bengal with an iron arene complex. J. Chem. Soc. Faraday Trans. 1998, 94, 1849–1854. [Google Scholar] [CrossRef]
- Grotzinger, C.; Burget, D.; Jacques, P.; Fouassier, J.P. A novel and efficient xanthenic dye–organometallic ion-pair complex for photoinitiating polymerization. J. Appl. Polym. Sci. 2001, 81, 2368–2376. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Morlet-Savary, F.; Yamashita, K.; Imahashi, S. Visible light-induced polymerization reactions: The seven-role of the electron transfer process in the dye/iron arene complex/amine system. J. Appl. Polym. Sci. 1996, 62, 1877–1885. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Morlet-Savary, F.; Yamashita, K.; Imahashi, S. The role of the dye/iron arene complex/amine system as a photoinitiator for photopolymerization reactions. Polymer 1997, 38, 1415–1421. [Google Scholar] [CrossRef]
- Fredin, L.A.; Papai, M.; Rozsalyi, E.; Vanko, G.; Warnmark, K.; Sundstrçm, V.; Persson, P. Exceptional excited-state lifetime of an iron (II)–N-heterocyclic carbene complex explained. J. Phys. Chem. Lett. 2014, 5, 2066–2071. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Dumur, F.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Carbazole scaffold based photoinitiators/photoredox catalysts for new LED projector 3D printing resins. Macromolecules 2017, 50, 2747–2758. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Zhang, J.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Amino and nitro substituted 2-amino-1H-benzo[de]isoquinoline-1,3(2H)-diones: As versatile photo-initiators of polymerization: From violet-blue LEDs absorption to a panchromatic behavior. J. Polym. Chem. 2015, 6, 1171–1179. [Google Scholar] [CrossRef]
- Morris, J.; Telitel, S.; Fairfull-Smith, K.E.; Bottle, S.E.; Lalevée, J.; Clément, J.L.; Guillaneuf, Y.; Gigmes, D. Novel polymer synthesis methodologies using combinations of thermally- and photochemically-induced nitroxide mediated polymerization. Polym. Chem. 2015, 6, 754–763. [Google Scholar] [CrossRef]
- Telitel, S.; Telitel, S.; Bosson, J.; Lalevée, J.; Clément, J.L.; Godfroy, M.; Fillaut, J.L.; Akdas-Kilig, H.; Guillaneuf, Y.; Gigmes, D.; et al. UV-induced micropatterning of complex functional surfaces by photopolymerization controlled by alkoxyamines. Langmuir 2015, 31, 10026–10036. [Google Scholar] [CrossRef] [PubMed]
- Konkolewicz, D.; Schroeder, K.; Buback, J.; Bernhard, S.; Matyjaszewski, K. Visible light and sunlight photoinduced ATRP with ppm of Cu catalyst. ACS Macro Lett. 2012, 1, 1219–1223. [Google Scholar] [CrossRef]
- Fors, B.P.; Hawker, C.J. Control of a living radical polymerization of methacrylates by light. Angew. Chem. Int. Ed. 2012, 51, 8850–8853. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Kumar, P.; Sharma, C.D.; Ray, S.S. Light-induced controlled free radical polymerization of methacrylates using iron-based photocatalyst in visible light. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Tasdelen, M.A.; Uygun, M.; Yagci, Y. Photoinduced controlled radical polymerization. Macromol. Rapid Commun. 2011, 32, 58–62. [Google Scholar] [CrossRef]
- Ishizu, K.; Kakinuma, H. Synthesis of nanocylinders consisting of graft block copolymers by the photo-induced ATRP technique. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 63–70. [Google Scholar] [CrossRef]
- Kwak, Y.; Matyjaszewski, K. Photoirradiated atom transfer radical polymerization with an alkyl dithiocarbamate at ambient temperature. Macromolecules 2010, 43, 5180–5183. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Uygun, M.; Yagci, Y. Photoinduced controlled radical polymerization in methanol. Macromol. Chem. Phys. 2010, 211, 2271–2275. [Google Scholar] [CrossRef]
- Dadashi-Silab, S.; Tasdelen, M.A.; Kiskan, B.; Wang, X.; Antonietti, M.; Yagci, Y. Photochemically mediated atom transfer radical polymerization using polymeric semiconductor mesoporous graphitic carbon nitride. Macromol. Chem. Phys. 2014, 215, 675–681. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Konkolewicz, D.; Pan, X.; Matyjaszewski, K. Contribution of photochemistry to activator regeneration in ATRP. Macromolecules 2014, 47, 6316–6321. [Google Scholar] [CrossRef]
- Chantasirichot, S.; Inoue, Y.; Ishihara, K. Photoinduced atom transfer radical polymerization in a polar solvent to synthesize a water-soluble poly (2-methacryloyloxyethyl phosphorylcholine) and its block-type copolymers. Polymer 2015, 61, 55–60. [Google Scholar] [CrossRef]
- Doran, S.; Taskin, O.S.; Tasdelen, M.A.; Yagci, Y. Controlled photopolymerization and novel architectures. In Dyes and Chromophores in Polymer Science; Lalevee, J., Fouassier, J.P., Eds.; ISTE Wiley: London, UK, 2015. [Google Scholar]
- Zhang, J.; Dumur, F.; Horcajada, P.; Livage, C.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Iron-based metal-organic frameworks (MOF) as photocatalysts for radical and cationic polymerizations under near UV and visible LEDs (385-405 nm). Macromol. Chem. Phys. 2016, 217, 2534–2540. [Google Scholar] [CrossRef]
- Wang, C.C.; Li, J.R.; Lv, X.L.; Zhang, Y.Q.; Guo, G. Photocatalytic organic pollutants degradation in metal-organic frameworks. Energy Environ. Sci. 2014, 7, 2831–2867. [Google Scholar] [CrossRef]
- Garcia, H.; Ferrer, B. Metal Organic Frameworks as Heterogeneous Catalysts; The Royal Society of Chemistry: London, UK, 2013; p. 365. [Google Scholar]
- Wang, J.L.; Wang, C.; Lin, W. Metal-organic frameworks for light harvesting and photocatalysis. ACS Catal. 2012, 2, 2630–2640. [Google Scholar] [CrossRef]
- Farrusseng, D.; Aguado, S.; Pinel, C. Metal-organic frameworks: Opportunities for catalysis. Angew. Chem. Int. Ed. 2009, 48, 7502–7513. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Stoesser, M.; Ruppel, R.; Baum, E.; Bohres, E.; Sigl, M.; Lobree, L.; Yaghi, O.M.; Eddaoudi, M. Process for the Alkoxylation of Organic Compounds in the Presence of Novel Framework Materials. U.S. Patent US7279517 B2, 10 October 2001. [Google Scholar]
- Laurier, K.G.M.; Vermoortele, F.; Ameloot, R.; De Vos, D.E.; Hofkens, J.; Roeffaers, M.B.J. Iron (III)-based metal-organic frameworks as visible light photocatalysts. J. Am. Chem. Soc. 2013, 135, 14488–14491. [Google Scholar] [CrossRef]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef]
- Czaja, A.U.; Trukhan, N.; Muller, U. Industrial applications of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1284–1293. [Google Scholar] [CrossRef]
- Gu, Z.Y.; Park, J.; Raiff, A.; Wei, Z.; Zhou, H.C. Metal-organic frameworks as biomimetic catalysts. ChemCatChem 2014, 6, 67–75. [Google Scholar] [CrossRef]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Cirujano, F.G.; Leyva-Pérez, A.; Corma, A.; Llabrés i Xamena, F.X. MOFs as multifunctional catalysts: Synthesis of secondary arylamines, quinolines, pyrroles, and arylpyrrolidines over bifunctional MIL-101. ChemCatChem 2013, 5, 538–549. [Google Scholar] [CrossRef]
- Zhao, Z.; Qian, L.; Lv, H.; Wang, Y.; Zhao, G. Introduction of a Fe3O4 core enhances the photocatalytic activity of MIL-100(Fe) with tunable shell thickness in the presence of H2O2. ChemCatChem 2015, 7, 4148–4155. [Google Scholar] [CrossRef]
- Ai, L.; Zhang, C.; Li, L.; Jiang, J. Iron terephthalate metal–organic framework: Revealing the effective activation of hydrogen peroxide for the degradation of organic dye under visible light irradiation. J. Appl. Catal. B 2014, 148–149, 191–200. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, P. 3D printing of photopolymers. Polym. Chem. 2018, 9, 1530–1540. [Google Scholar] [CrossRef]
- Bagheri, A.; Jin, J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 2019, 1, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Garra, P.; Brunel, D.; Noirbent, G.; Graff, B.; Morlet-Savary, F.; Dietlin, C.; Sidorkin, V.F.; Dumur, F.; Duché, D.; Gigmes, D.; et al. Ferrocene-based (photo)redox polymerization under long wavelengths. Polym. Chem. 2019, 10, 1431–1441. [Google Scholar] [CrossRef]
- Brunel, D.; Noirbent, G.; Dumur, F. Ferrocene: An unrivaled electroactive building block for the design of push-pull dyes with near-infrared and infrared absorptions. Dyes Pigment. 2019, 170, 107611. [Google Scholar] [CrossRef]
Figure 1.
Advantages of photopolymerization. (A) Fast monomer conversion vs. time. (B) Spatial control of the polymerization by use of a mask. Reprinted with permission of Poelma et al. [20]. Copyright © 2013 John Wiley & Sons, Inc. (C) Monomer conversion vs. time by switching on/off the light. No monomer conversion occurs when the light is off. Reproduced with permission from [21] Copyright The Royal Society of Chemistry, 2015.
Figure 1.
Advantages of photopolymerization. (A) Fast monomer conversion vs. time. (B) Spatial control of the polymerization by use of a mask. Reprinted with permission of Poelma et al. [20]. Copyright © 2013 John Wiley & Sons, Inc. (C) Monomer conversion vs. time by switching on/off the light. No monomer conversion occurs when the light is off. Reproduced with permission from [21] Copyright The Royal Society of Chemistry, 2015.

Figure 2.
Chemical structures of Ru(II)(bpy)3Cl2 and the different monomers.

Scheme 1.
Catalytic cycle involving Ru(II)(bpy)3Cl2 as the photocatalyst in an oxidative process with the three-component system Ru(II)(bpy)32+/TTMS3Si-H/Ph2I+.
Scheme 1.
Catalytic cycle involving Ru(II)(bpy)3Cl2 as the photocatalyst in an oxidative process with the three-component system Ru(II)(bpy)32+/TTMS3Si-H/Ph2I+.
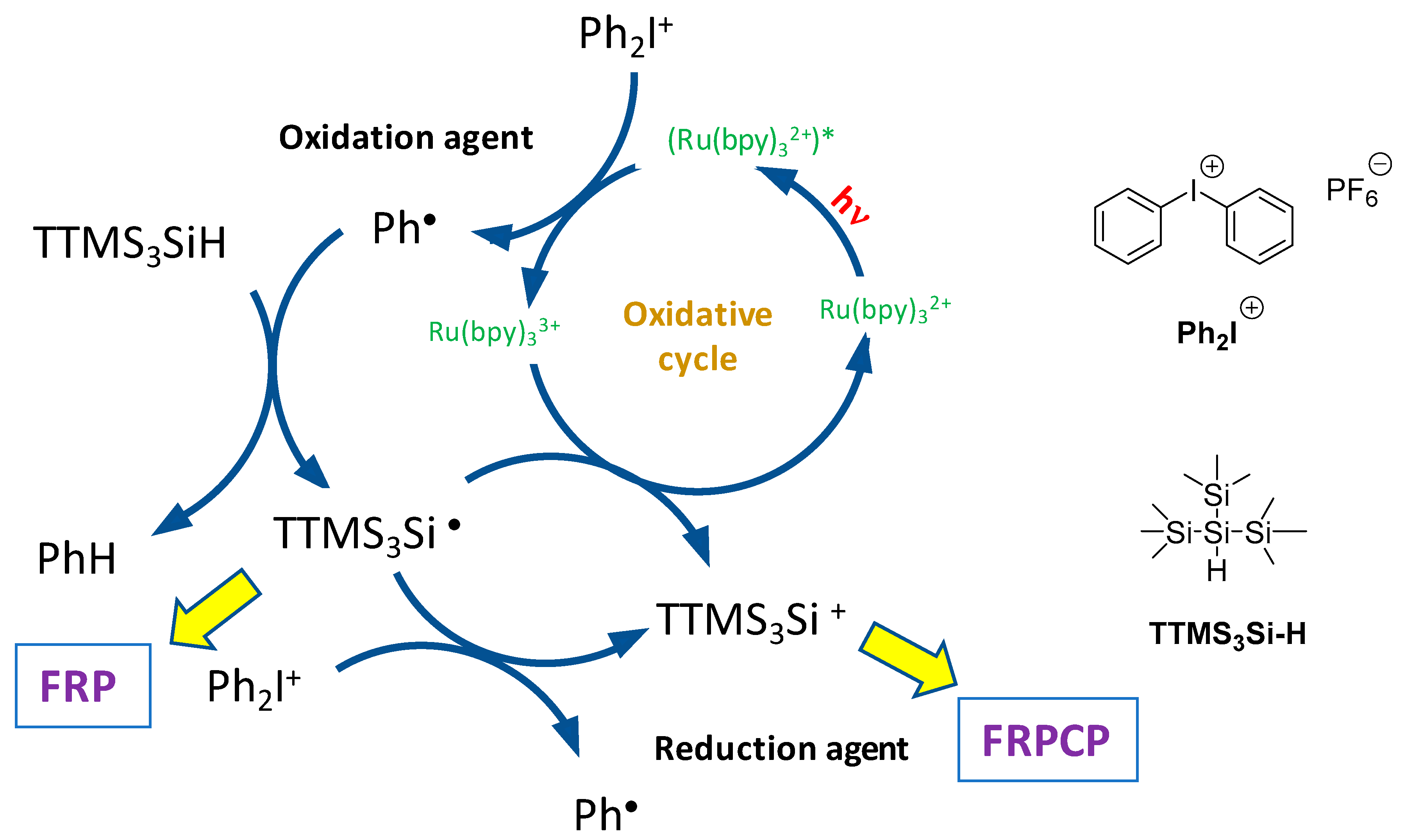
Figure 3.
Monomer conversions vs. time for the polymerization of 3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX) upon irradiation with a xenon lamp under air using the three-component system Ru(II)(bpy)3Cl2/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) immediately after preparation (1) and after 21 days of storage (2). Adapted with permission from Lalevée et al. [23]. Copyright 2010 American Chemical Society.
Figure 3.
Monomer conversions vs. time for the polymerization of 3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX) upon irradiation with a xenon lamp under air using the three-component system Ru(II)(bpy)3Cl2/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) immediately after preparation (1) and after 21 days of storage (2). Adapted with permission from Lalevée et al. [23]. Copyright 2010 American Chemical Society.

Figure 4.
Chemical structures of Pt-1 and Pt-2.
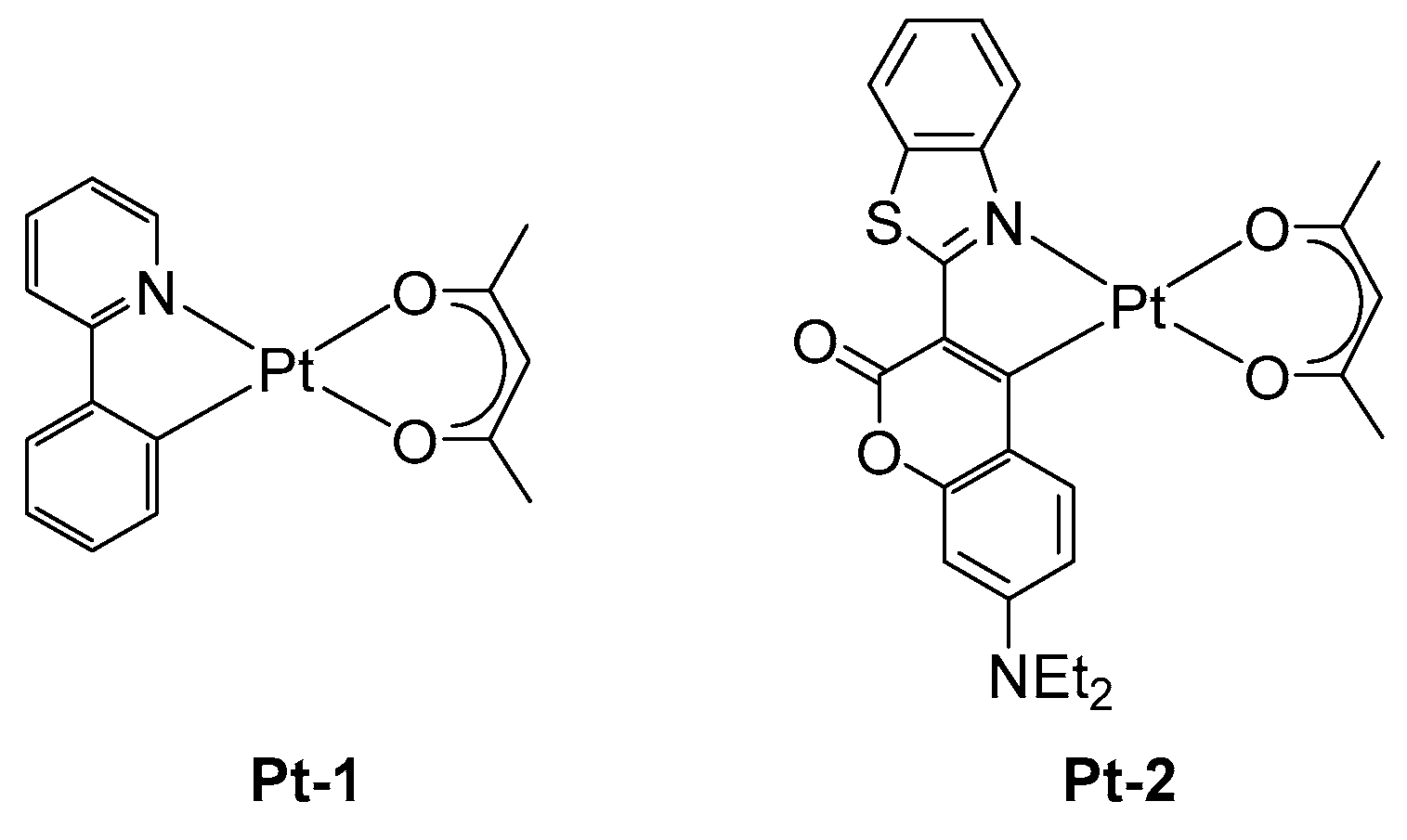
Scheme 2.
Synthetic route to cationic, anionic, and neutral iridium (III) complexes.

Figure 5.
Chemical structures of iridium-based photocatalysts Ir-1-Ir-30.
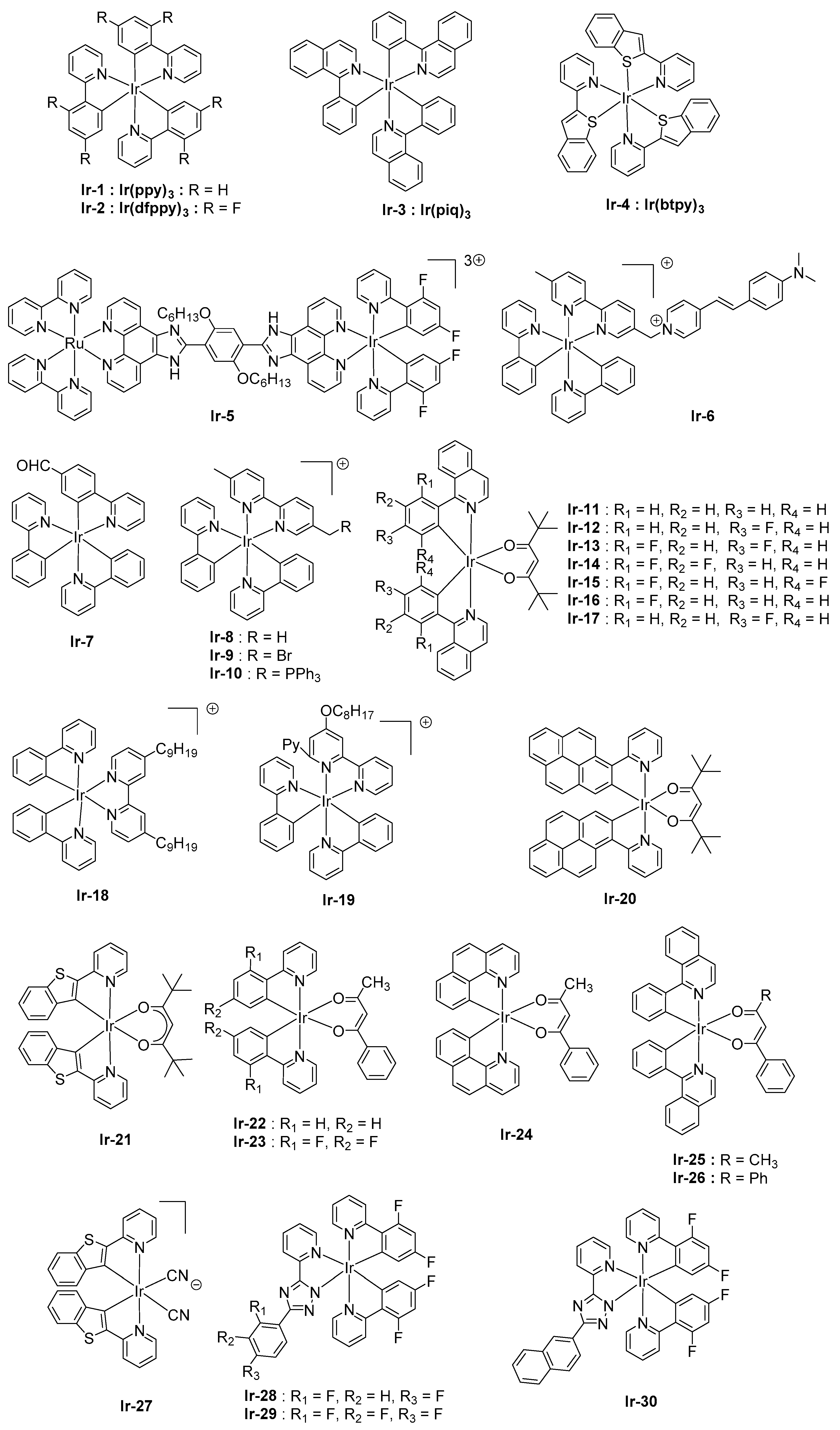
Figure 6.
(A) Conversion of the vinylic function vs. time for an EPOX/NVK (N-vinylcarbazole) (50%/50% w/w) blend using the three-component photoinitiating system Ir complex/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with (1) Ir-1, (2) Ir-5, and (3) Ir-3. Photopolymerization under air, upon irradiation with a LED at 462 nm; (B) photopolymerization of NVK in toluene (50%/50% w/w) under air using the three-component photoinitiating system Ir-3/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with a halogen lamp. Monomer conversion vs. time. Adapted with permission from Lalevée et al. [72]. Copyright 2012 American Chemical Society.
Figure 6.
(A) Conversion of the vinylic function vs. time for an EPOX/NVK (N-vinylcarbazole) (50%/50% w/w) blend using the three-component photoinitiating system Ir complex/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with (1) Ir-1, (2) Ir-5, and (3) Ir-3. Photopolymerization under air, upon irradiation with a LED at 462 nm; (B) photopolymerization of NVK in toluene (50%/50% w/w) under air using the three-component photoinitiating system Ir-3/TTMS3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with a halogen lamp. Monomer conversion vs. time. Adapted with permission from Lalevée et al. [72]. Copyright 2012 American Chemical Society.
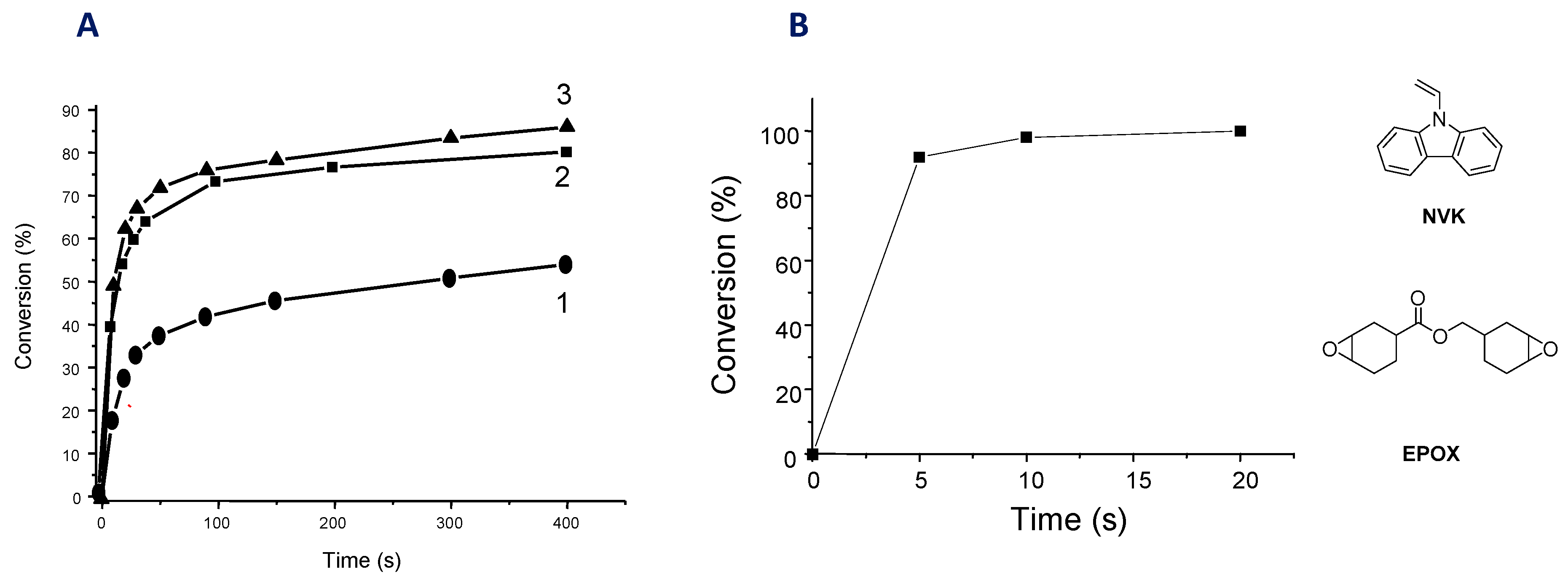
Figure 7.
Chemical structures of zinc-based photocatalysts Zn-1-Zn-3 and tri (ethylene glycol) divinyl ether (DVE-3) additive.
Figure 7.
Chemical structures of zinc-based photocatalysts Zn-1-Zn-3 and tri (ethylene glycol) divinyl ether (DVE-3) additive.
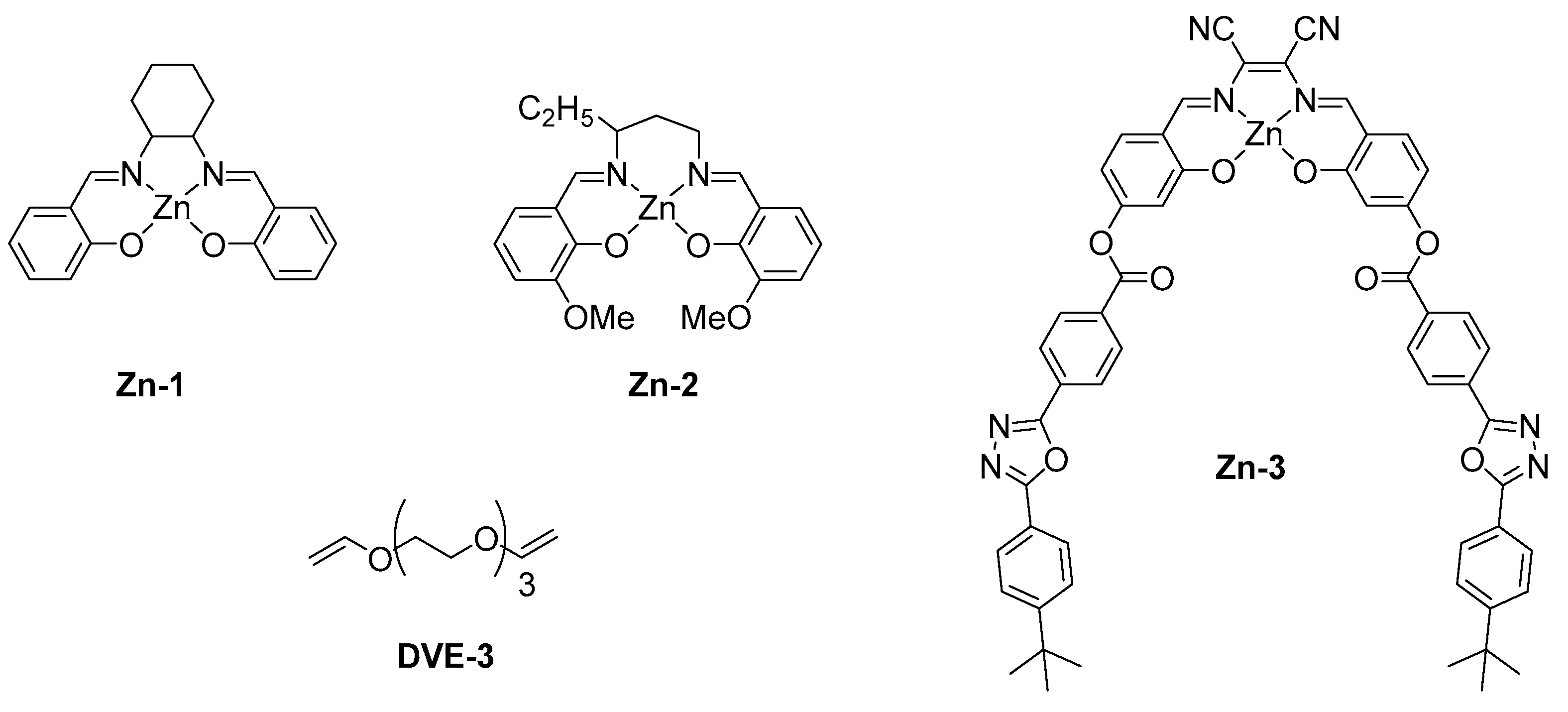
Figure 8.
(A) Absorption spectra of Zn-1-Zn-3 in acetonitrile with (1) Zn-3, (2) Zn-2, and (3) Zn-1. (B) Polymerization profiles of trimethylolpropane triacrylate (TMPTA) in laminate (A) upon irradiation with a halogen lamp using the three-component system Zn complex/(TMS)3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with (2) Zn-1, (3) Zn-2, and (4) Zn-3. Reprinted from [75]. Copyright (2013) with permission from Elsevier.
Figure 8.
(A) Absorption spectra of Zn-1-Zn-3 in acetonitrile with (1) Zn-3, (2) Zn-2, and (3) Zn-1. (B) Polymerization profiles of trimethylolpropane triacrylate (TMPTA) in laminate (A) upon irradiation with a halogen lamp using the three-component system Zn complex/(TMS)3Si-H/Ph2I+ (0.2%/3%/2%, w/w/w) with (2) Zn-1, (3) Zn-2, and (4) Zn-3. Reprinted from [75]. Copyright (2013) with permission from Elsevier.
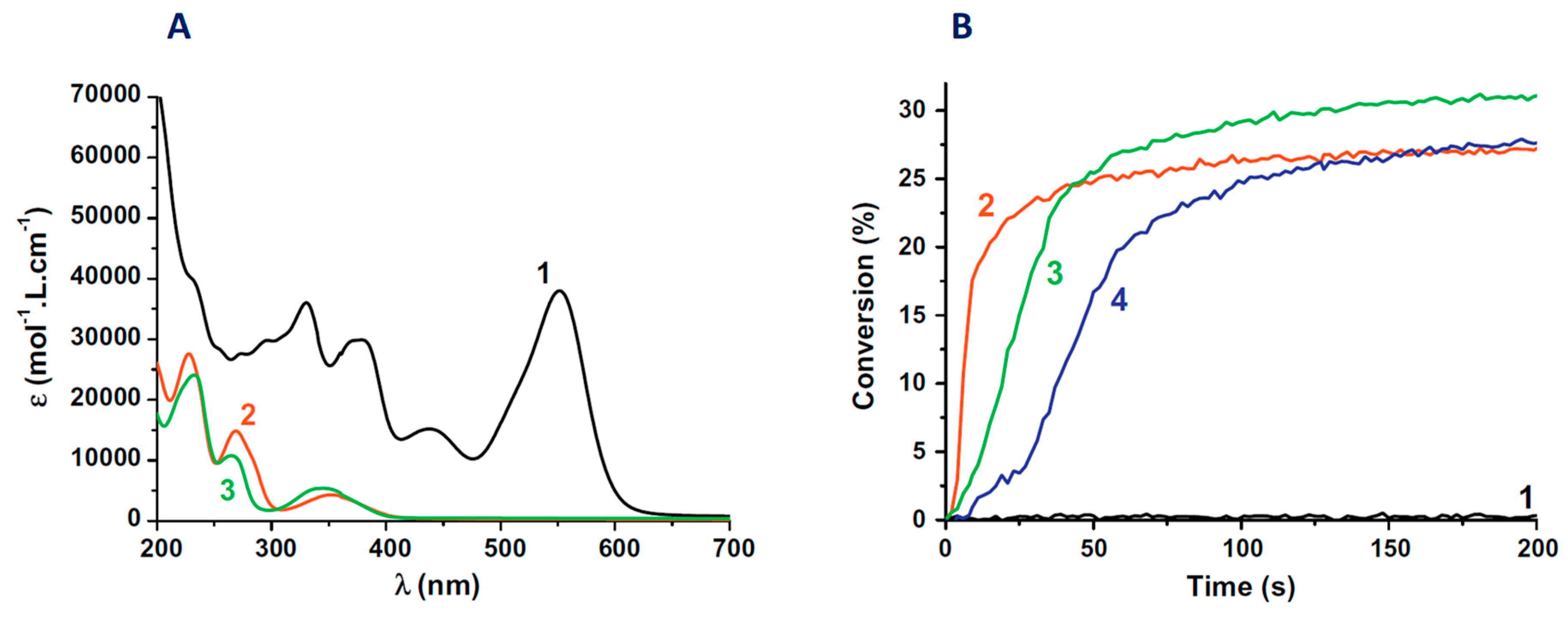
Figure 9.
Chemical structures of copper-based photocatalysts Cu-1-Cu22.
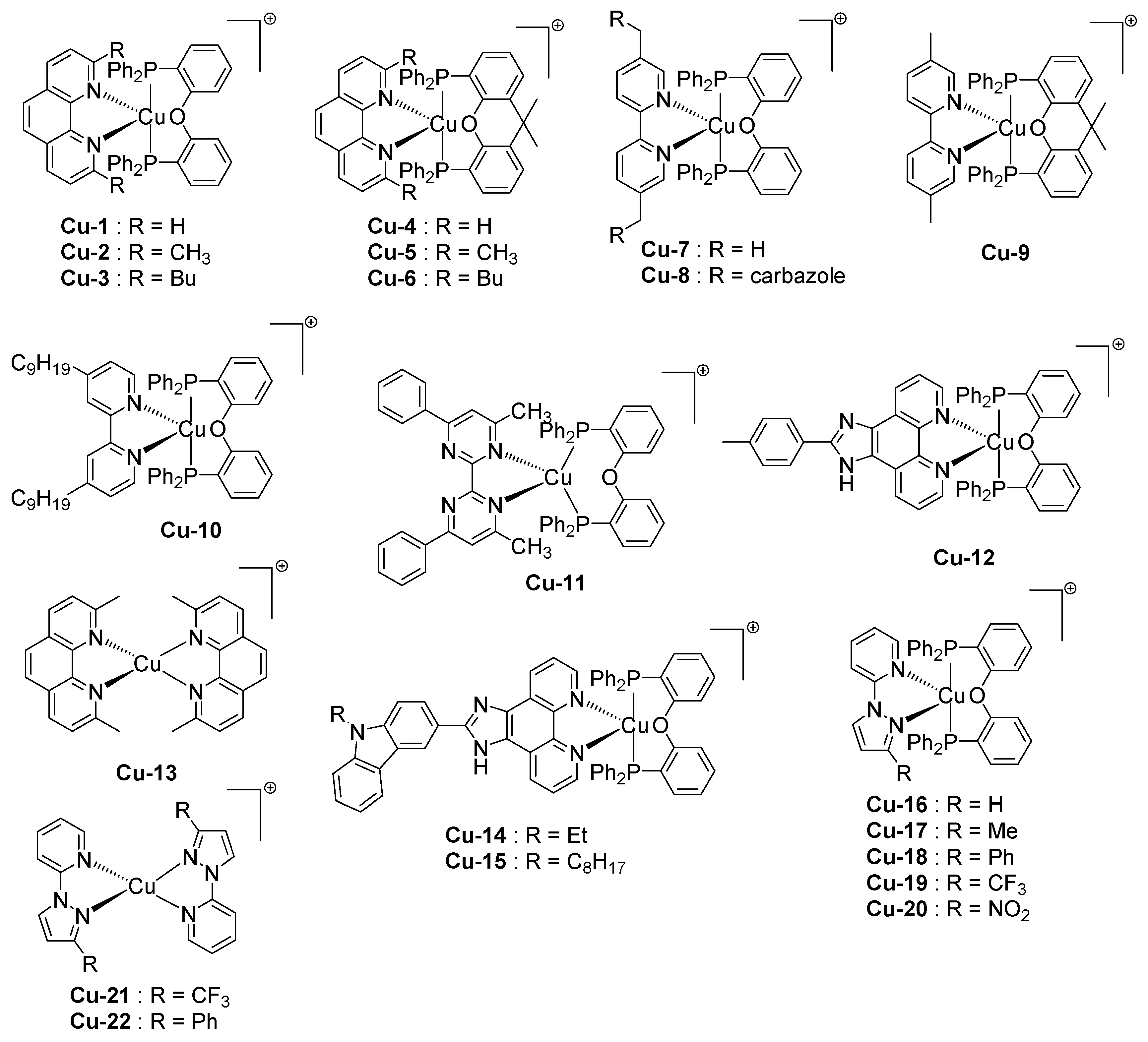
Scheme 3.
Synthetic route to copper (I) complexes.
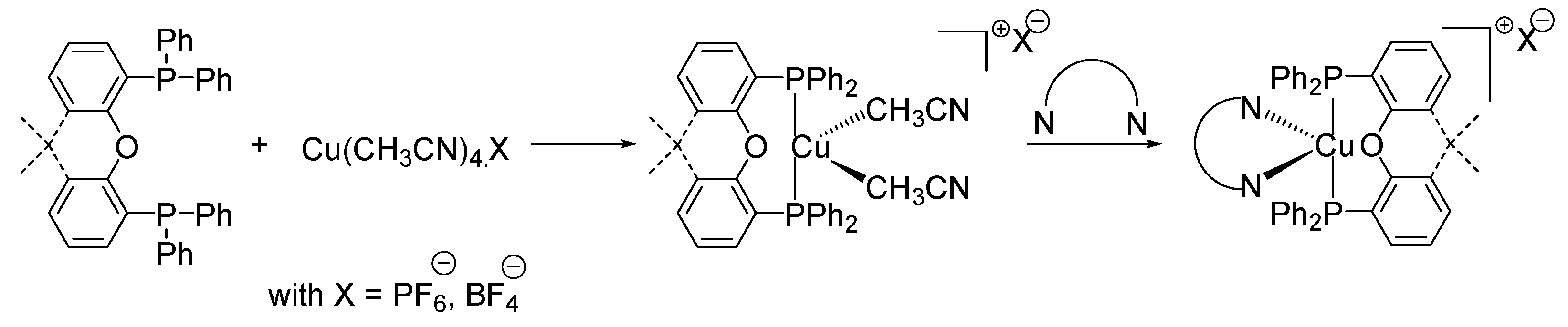
Figure 10.
Photopolymerization profiles for the polymerization of TMPTA in laminate using the three-component system Cu-2/Iod/NVK (0.2%/2%/3%, w/w/w) upon irradiation with a LED at 405 nm (1), a LED at 455 nm (2). Polymerization profile for the polymerization of TMPTA with the three-component system bisacylphosphine oxide (BAPO)/Iod/NVK (0.2%/2%/3%, w/w/w) upon irradiation with a LED at 405 nm. Adapted with permission from Xiao et al. [84]. Copyright 2014 American Chemical Society.
Figure 10.
Photopolymerization profiles for the polymerization of TMPTA in laminate using the three-component system Cu-2/Iod/NVK (0.2%/2%/3%, w/w/w) upon irradiation with a LED at 405 nm (1), a LED at 455 nm (2). Polymerization profile for the polymerization of TMPTA with the three-component system bisacylphosphine oxide (BAPO)/Iod/NVK (0.2%/2%/3%, w/w/w) upon irradiation with a LED at 405 nm. Adapted with permission from Xiao et al. [84]. Copyright 2014 American Chemical Society.
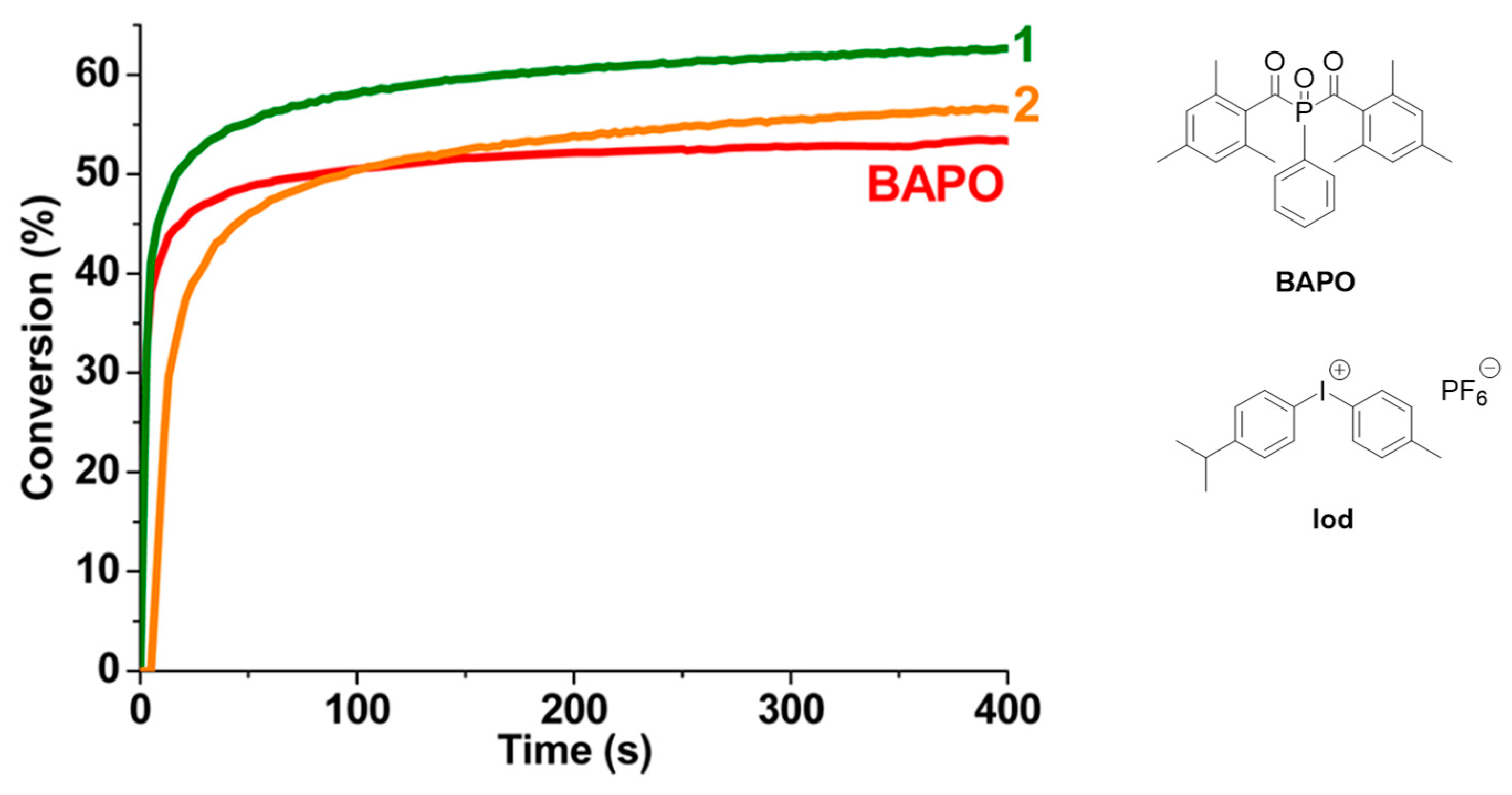
Figure 11.
Chemical structures of copper-based photocatalysts Cu-23 and Cu24.
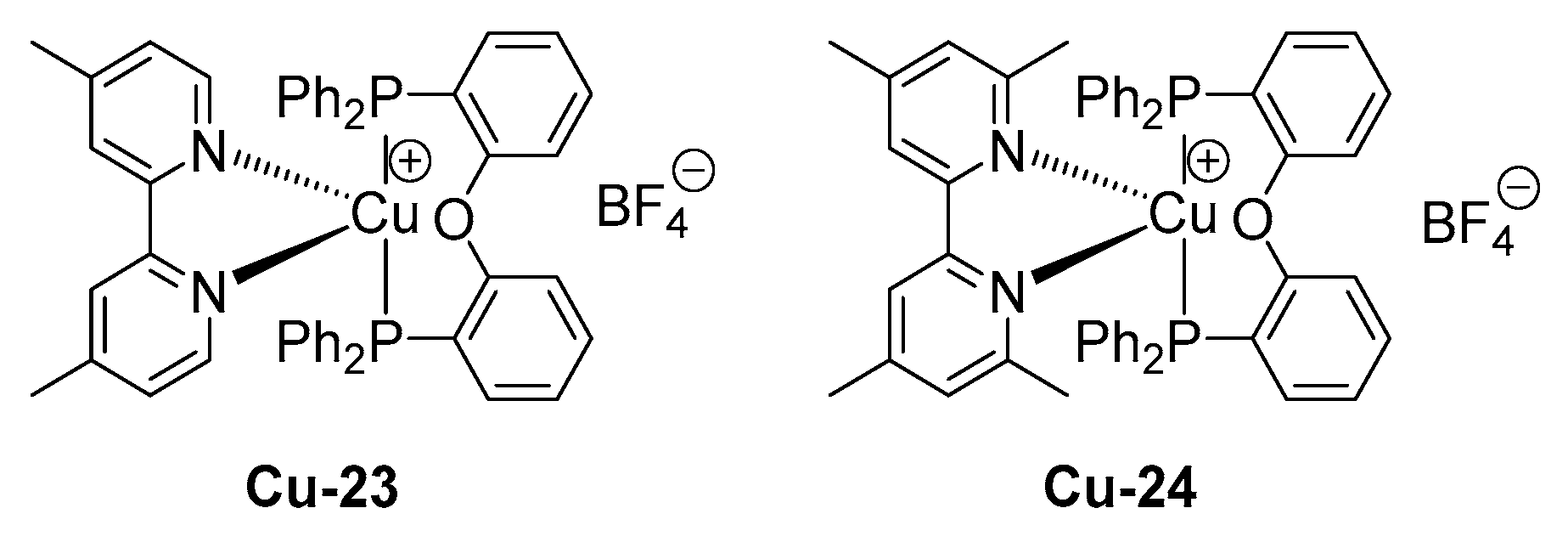
Scheme 4.
Catalytic cycle involving copper complexes as photocatalysts in an oxidative process.

Figure 12.
Chemical structures of copper-based photocatalysts Cu-2, BAPO, and the different monomers in the model resin (MR1).
Figure 12.
Chemical structures of copper-based photocatalysts Cu-2, BAPO, and the different monomers in the model resin (MR1).
Figure 13.
(A) Epoxide conversion vs. time for the model resin (MR1) under air using the three-component systems Cu-2/Iod/NVK (1.7%/4.6%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w) upon irradiation with a LED at 405 nm. (B) Comparison of the final monomer conversion vs. time between the three-component systems Cu-2/Iod/NVK (0.05%‒0.22%/5%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w). (C) Epoxide conversion vs. time for the model resin (MR1) under air using the three-component systems Cu-2/Iod/NVK (1.7%/4.6%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w) upon irradiation with a LED at 405 nm. The light was turned off after 60 seconds. Adapted from [96] with permission from The Royal Society of Chemistry.
Figure 13.
(A) Epoxide conversion vs. time for the model resin (MR1) under air using the three-component systems Cu-2/Iod/NVK (1.7%/4.6%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w) upon irradiation with a LED at 405 nm. (B) Comparison of the final monomer conversion vs. time between the three-component systems Cu-2/Iod/NVK (0.05%‒0.22%/5%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w). (C) Epoxide conversion vs. time for the model resin (MR1) under air using the three-component systems Cu-2/Iod/NVK (1.7%/4.6%/1%, w/w/w) and BAPO/Iod/NVK (1.8%/5%/1%, w/w/w) upon irradiation with a LED at 405 nm. The light was turned off after 60 seconds. Adapted from [96] with permission from The Royal Society of Chemistry.
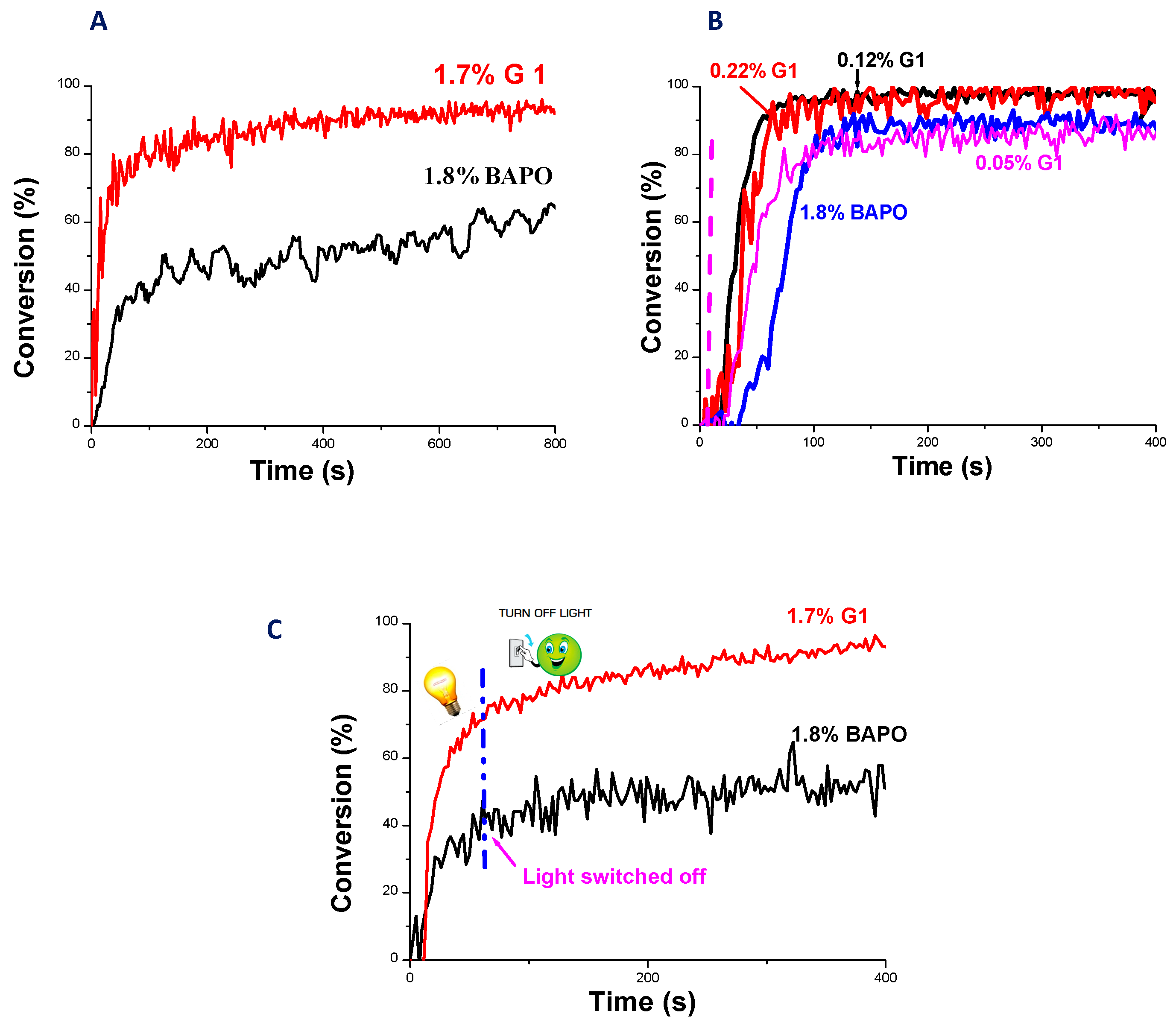
Scheme 5.
Synthetic route to Cu-2.
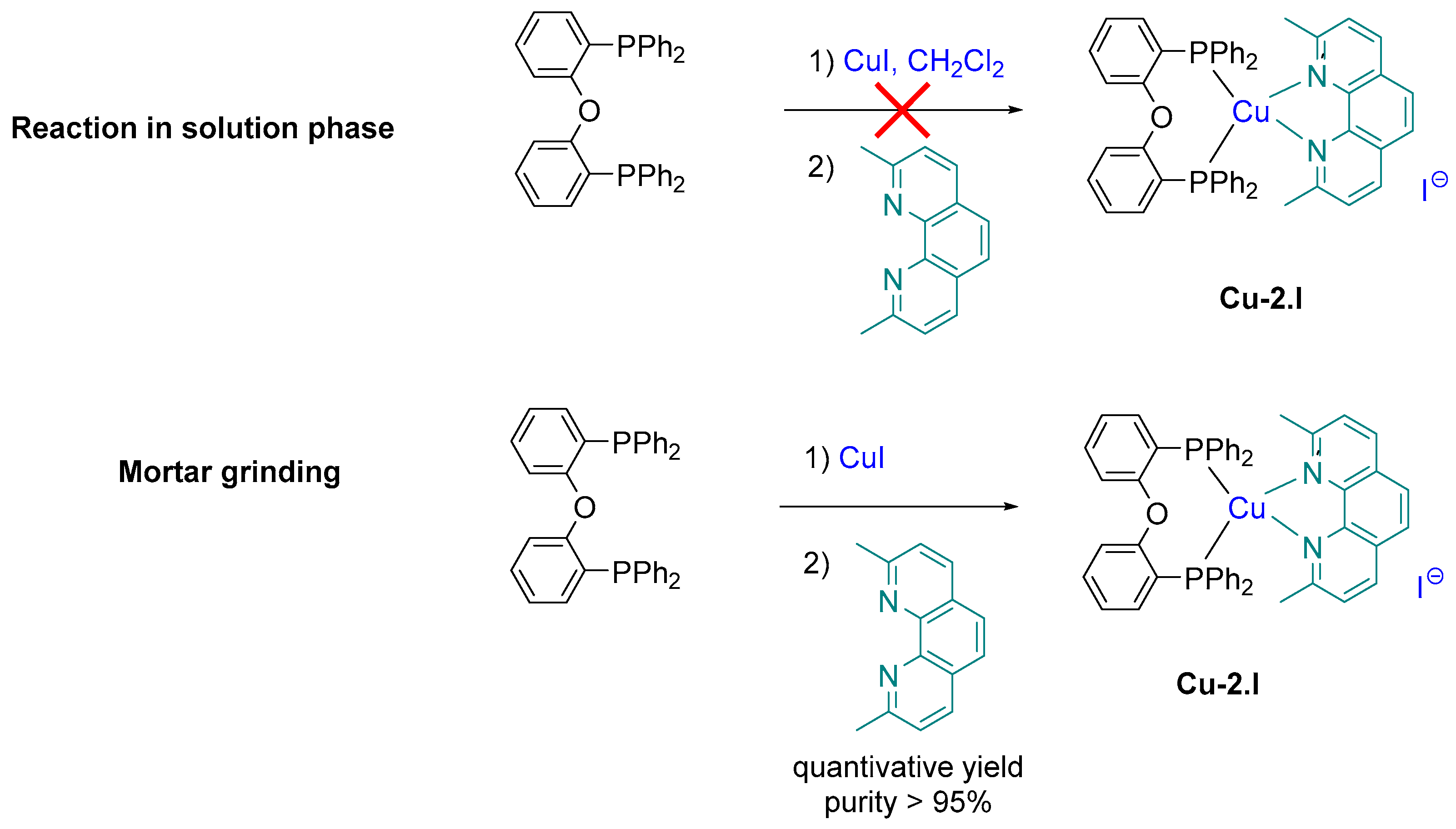
Figure 14.
Mechanosynthesis of Cu-2.BF4 (A) and Cu-2.I (B). Adapted with permission from Garra et al. [98]. Copyright 2018 American Chemical Society.
Figure 14.
Mechanosynthesis of Cu-2.BF4 (A) and Cu-2.I (B). Adapted with permission from Garra et al. [98]. Copyright 2018 American Chemical Society.

Figure 15.
Chemical structures of the monomers used in the model resin (MR2).
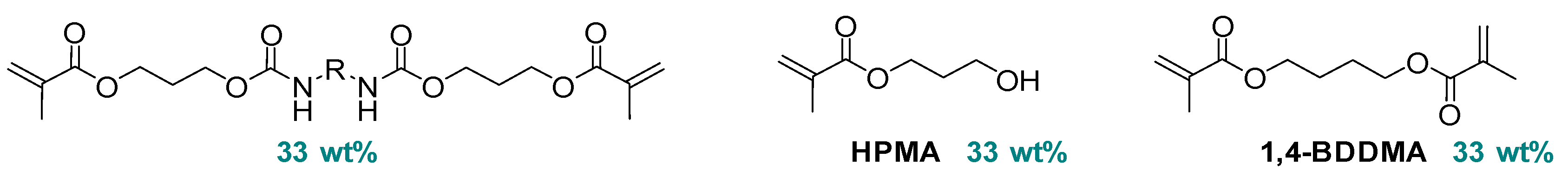
Figure 16.
3D-polymerization experiments with a LED projector at 405 nm (I = 110 mW/cm²). (A) different 3D figures imaged by numerical microscopy. (B) A 3D figure characterized by profilometry. Reprinted from [97]. Copyright (2019) with permission from Elsevier.
Figure 16.
3D-polymerization experiments with a LED projector at 405 nm (I = 110 mW/cm²). (A) different 3D figures imaged by numerical microscopy. (B) A 3D figure characterized by profilometry. Reprinted from [97]. Copyright (2019) with permission from Elsevier.
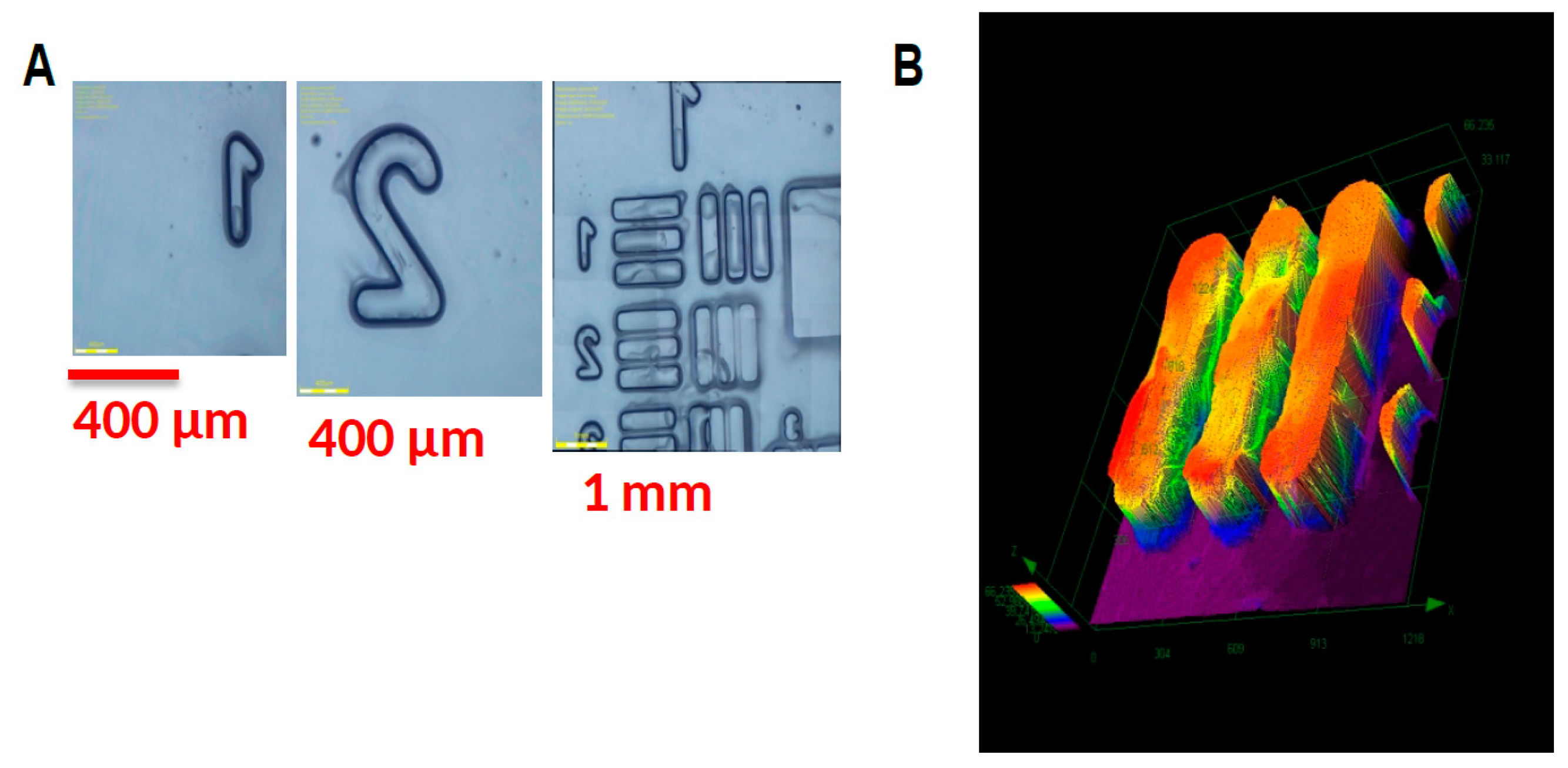
Figure 17.
(A) Experimental setup used for the polymerization in shadowed areas; (B) the Pasteur pipette containing the resin before irradiation; (C) the polymer obtained after breaking the Pasteur pipette for measuring the length; (D) monomer conversion as a function of the distance. Adapted with permission from Garra et al. [101]. Copyright 2017 American Chemical Society.
Figure 17.
(A) Experimental setup used for the polymerization in shadowed areas; (B) the Pasteur pipette containing the resin before irradiation; (C) the polymer obtained after breaking the Pasteur pipette for measuring the length; (D) monomer conversion as a function of the distance. Adapted with permission from Garra et al. [101]. Copyright 2017 American Chemical Society.

Figure 18.
Pasteur pipette containing the filled MR2 resin before (A) and after (B) breaking the pipette; (C) monomer conversion as a function of the distance; (D) evolution of the temperature as a function of the reaction time followed by infrared thermal imaging camera. Adapted with permission from Garra et al. [101]. Copyright 2017 American Chemical Society.
Figure 18.
Pasteur pipette containing the filled MR2 resin before (A) and after (B) breaking the pipette; (C) monomer conversion as a function of the distance; (D) evolution of the temperature as a function of the reaction time followed by infrared thermal imaging camera. Adapted with permission from Garra et al. [101]. Copyright 2017 American Chemical Society.

Figure 19.
(A) Redox reaction occurring between Cu(acac)2 and 2-dppba. (B) Ligand exchange reaction in tetrahydrofuran (THF) solution. (a) Solution color after addition of 2-dppba to a Cu(acac)2 solution. (b) Solution color before addition of 2-dppba. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society.
Figure 19.
(A) Redox reaction occurring between Cu(acac)2 and 2-dppba. (B) Ligand exchange reaction in tetrahydrofuran (THF) solution. (a) Solution color after addition of 2-dppba to a Cu(acac)2 solution. (b) Solution color before addition of 2-dppba. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society.
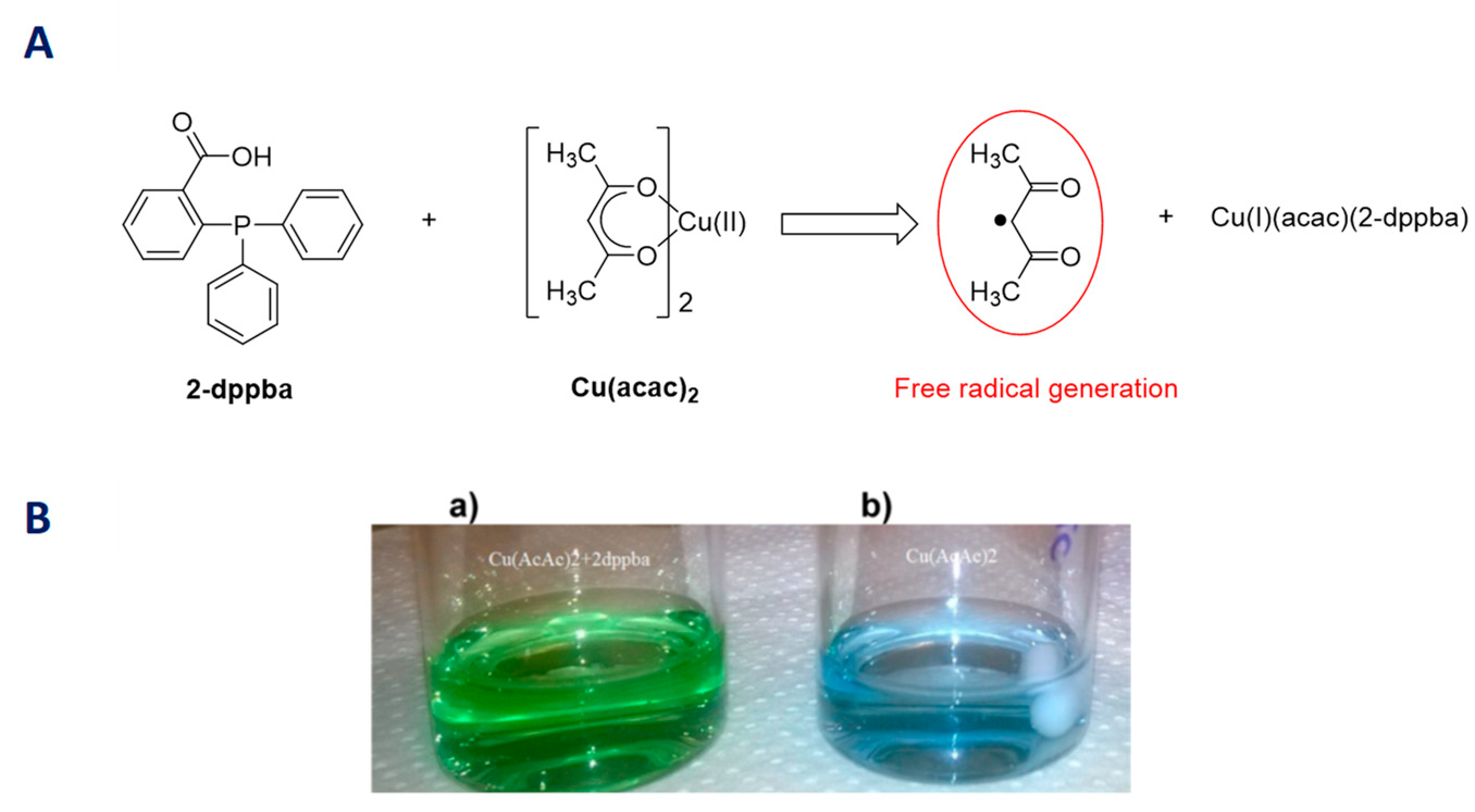
Figure 20.
(A) Redox polymerization and photoactivated redox polymerization of the methacrylate (1,4-BDMA/HPMA/UDMA) mixture with the three-component system Cu(acac)2/Ph2I+/2-dppba (0.75%/1.1%/1.5%, w/w/w) with and without irradiation with a LED at 405 nm. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society.
Figure 20.
(A) Redox polymerization and photoactivated redox polymerization of the methacrylate (1,4-BDMA/HPMA/UDMA) mixture with the three-component system Cu(acac)2/Ph2I+/2-dppba (0.75%/1.1%/1.5%, w/w/w) with and without irradiation with a LED at 405 nm. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society.
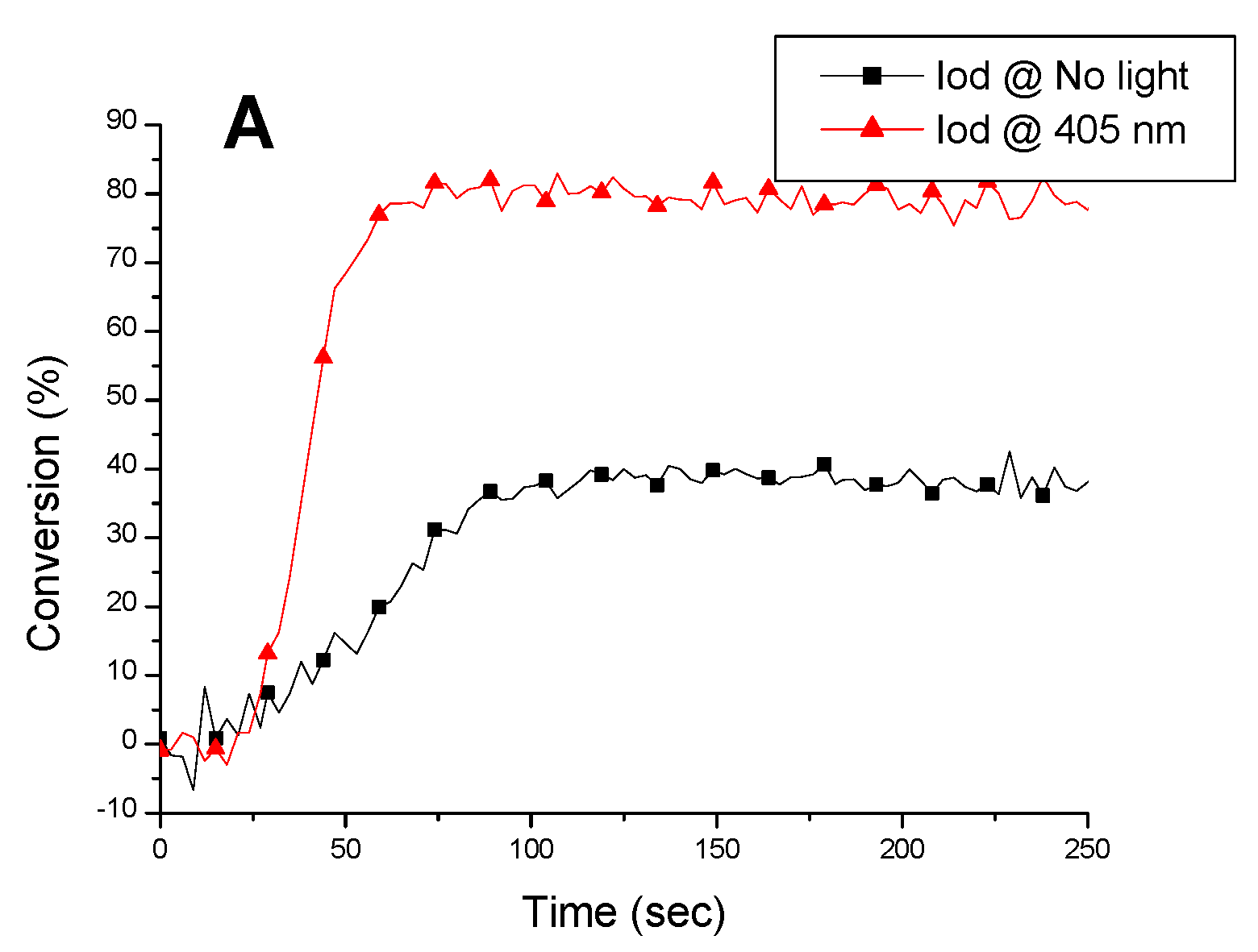
Figure 21.
(A) Optical pyrometric measurements for the polymerization of EPOX/methacrylate mixture (1,4-BDMA/HPMA/UDMA) blend under air using a four component system (Cu(Oct)2/BPO/Vit-C/Ph2I+, 0.75%/1.1%/1.5%/1%, w/w/w/w) with and without light irradiation at 405 nm; (B) Raman surface analysis of the samples. Adapted with permission from Garra et al. [80]. Copyright 2018 American Chemical Society.
Figure 21.
(A) Optical pyrometric measurements for the polymerization of EPOX/methacrylate mixture (1,4-BDMA/HPMA/UDMA) blend under air using a four component system (Cu(Oct)2/BPO/Vit-C/Ph2I+, 0.75%/1.1%/1.5%/1%, w/w/w/w) with and without light irradiation at 405 nm; (B) Raman surface analysis of the samples. Adapted with permission from Garra et al. [80]. Copyright 2018 American Chemical Society.
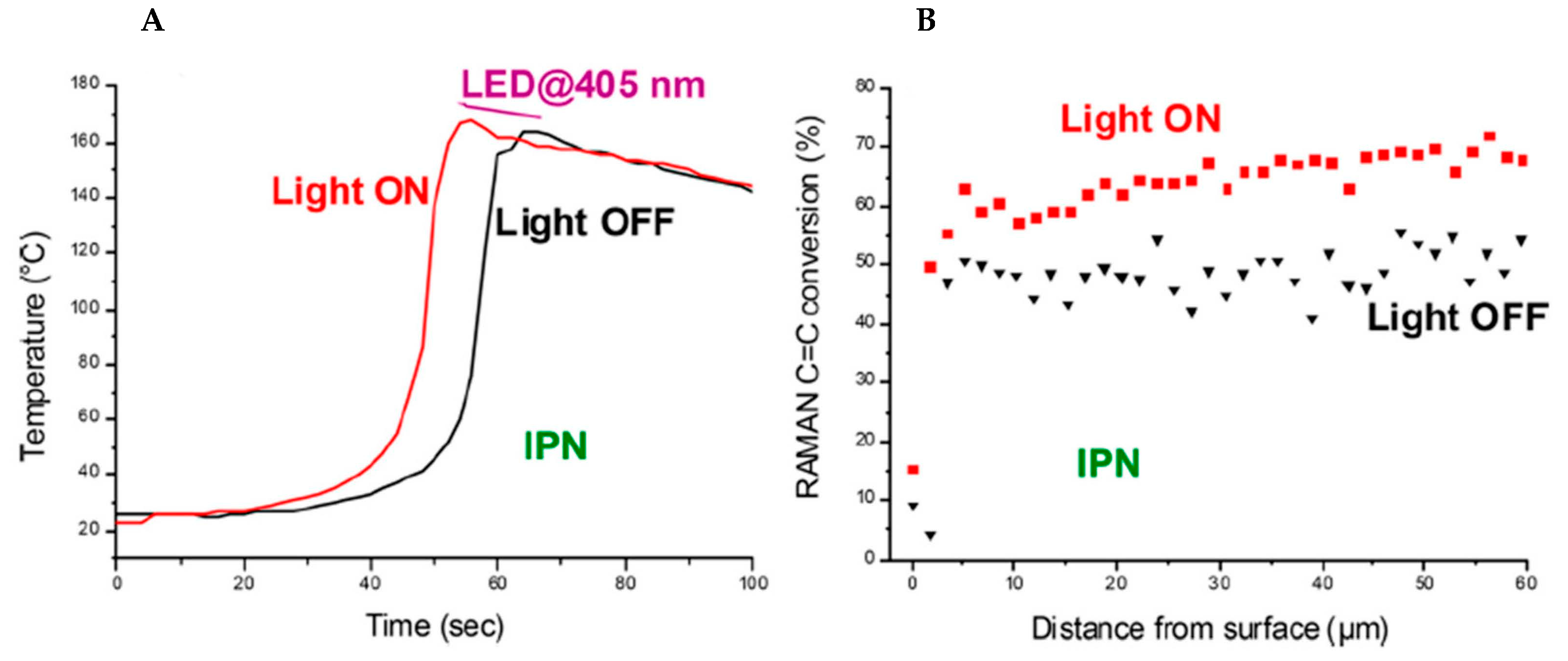
Scheme 6.
The different catalytic cycles involved in the photoactivated redox polymerization of the methacrylate (1,4-BDMAHPMA/UDMA)/EPOX blend using Cu(Oct)2 as the catalyst in the four-component system Cu(Oct)2/BPO/Vit-C/Ph2I+.
Scheme 6.
The different catalytic cycles involved in the photoactivated redox polymerization of the methacrylate (1,4-BDMAHPMA/UDMA)/EPOX blend using Cu(Oct)2 as the catalyst in the four-component system Cu(Oct)2/BPO/Vit-C/Ph2I+.
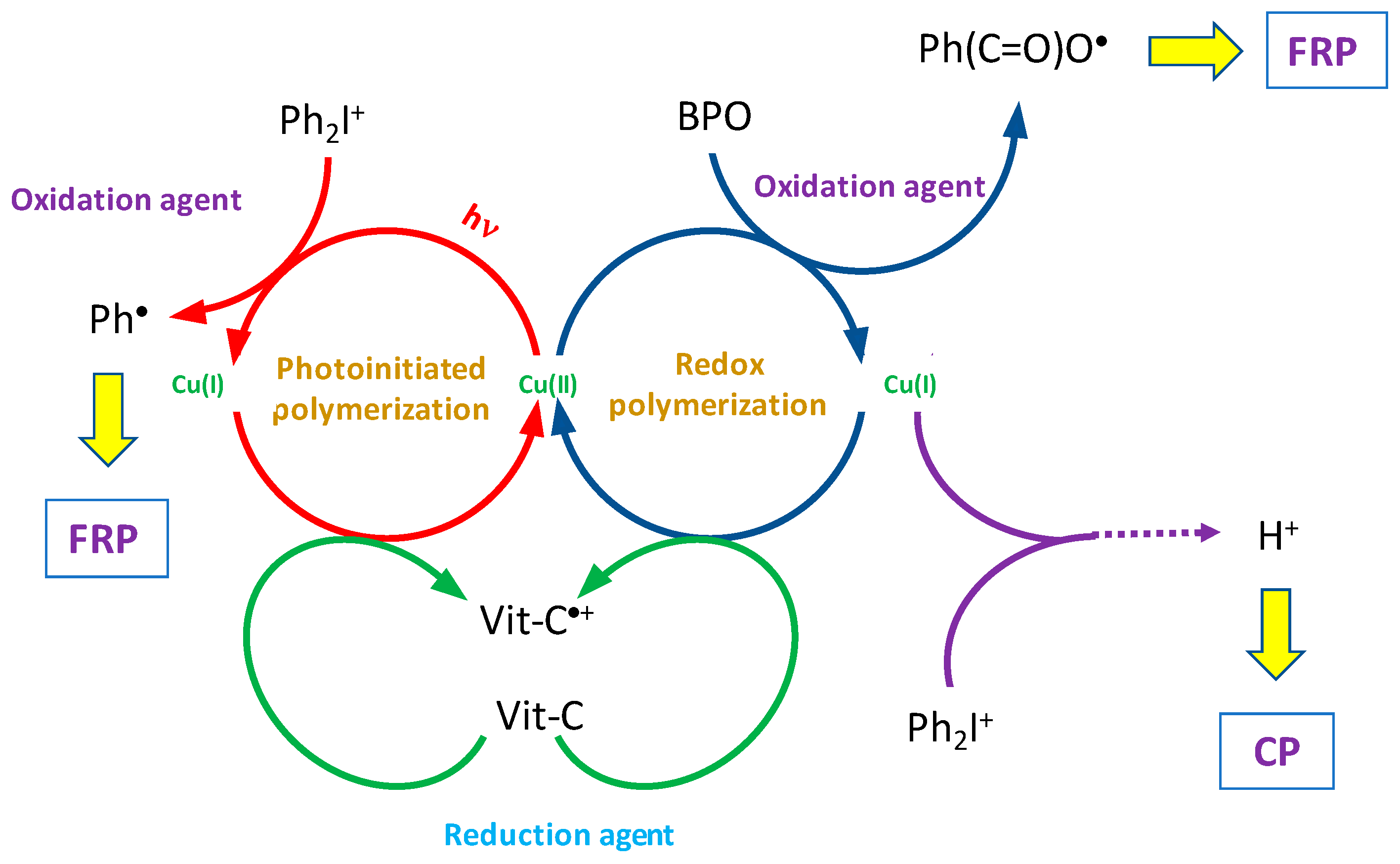
Figure 22.
Optical pyrometric measurements for the polymerization of the methacrylate mixture (1,4-BDMA/HPMA/UDMA) under air using the four-component system Cu-25/Vit-C/BPO/(t-Bu)Ph2I+ (0.5%/1.3%/1.4%/2%, w/w/w/w) in the presence of tempol (0.13–0.15 w%) upon irradiation with a LED at 405 nm (A), LED at 470 nm (B), and LED at 530 nm (C). Adapted from [106] with permission from The Royal Society of Chemistry.
Figure 22.
Optical pyrometric measurements for the polymerization of the methacrylate mixture (1,4-BDMA/HPMA/UDMA) under air using the four-component system Cu-25/Vit-C/BPO/(t-Bu)Ph2I+ (0.5%/1.3%/1.4%/2%, w/w/w/w) in the presence of tempol (0.13–0.15 w%) upon irradiation with a LED at 405 nm (A), LED at 470 nm (B), and LED at 530 nm (C). Adapted from [106] with permission from The Royal Society of Chemistry.

Scheme 7.
Catalytic cycle involving a Fe complex as the photocatalyst in a reductive process.
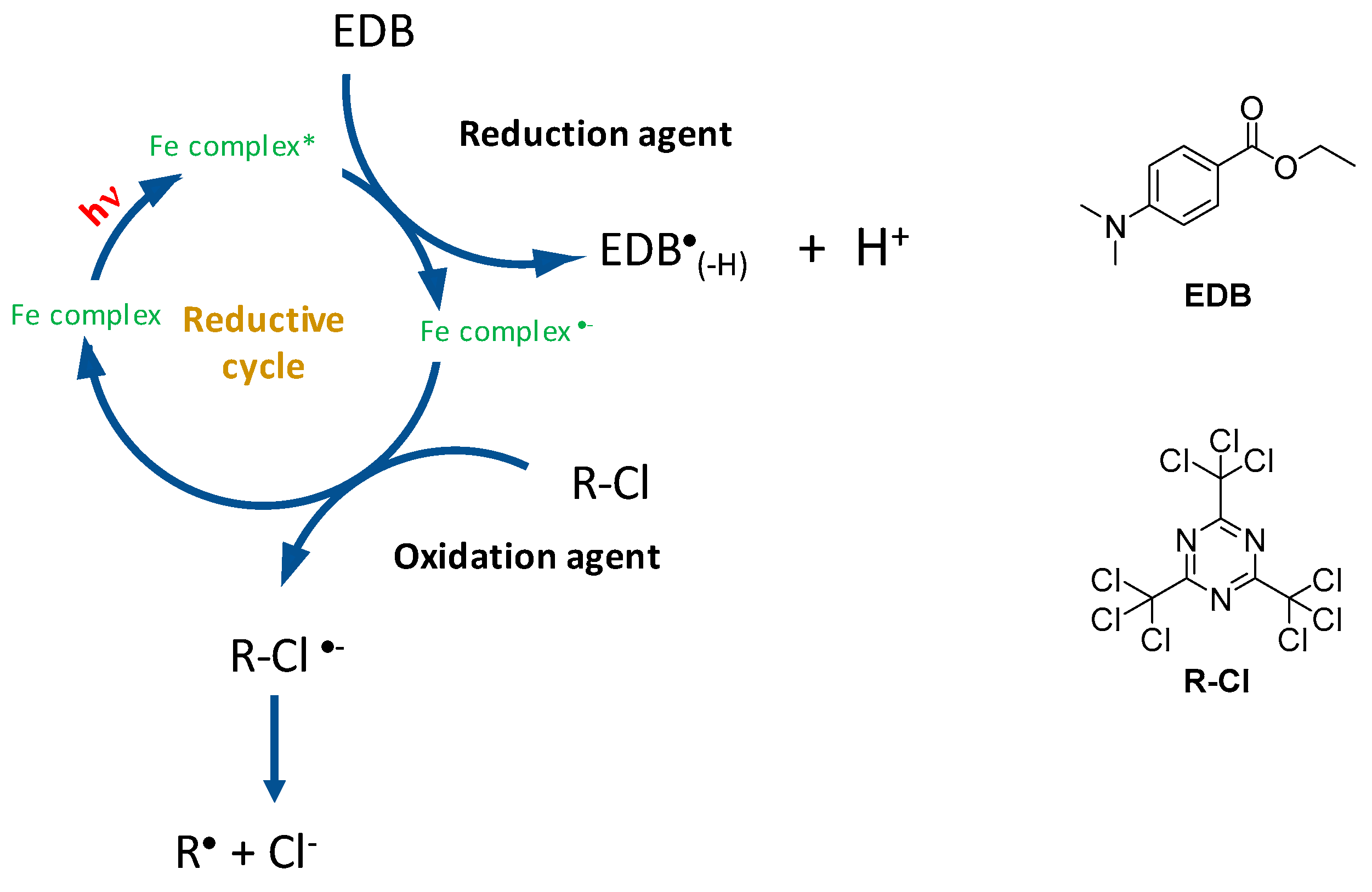
Figure 23.
Chemical structures of iron-based photocatalysts Fe-1-Fe22.
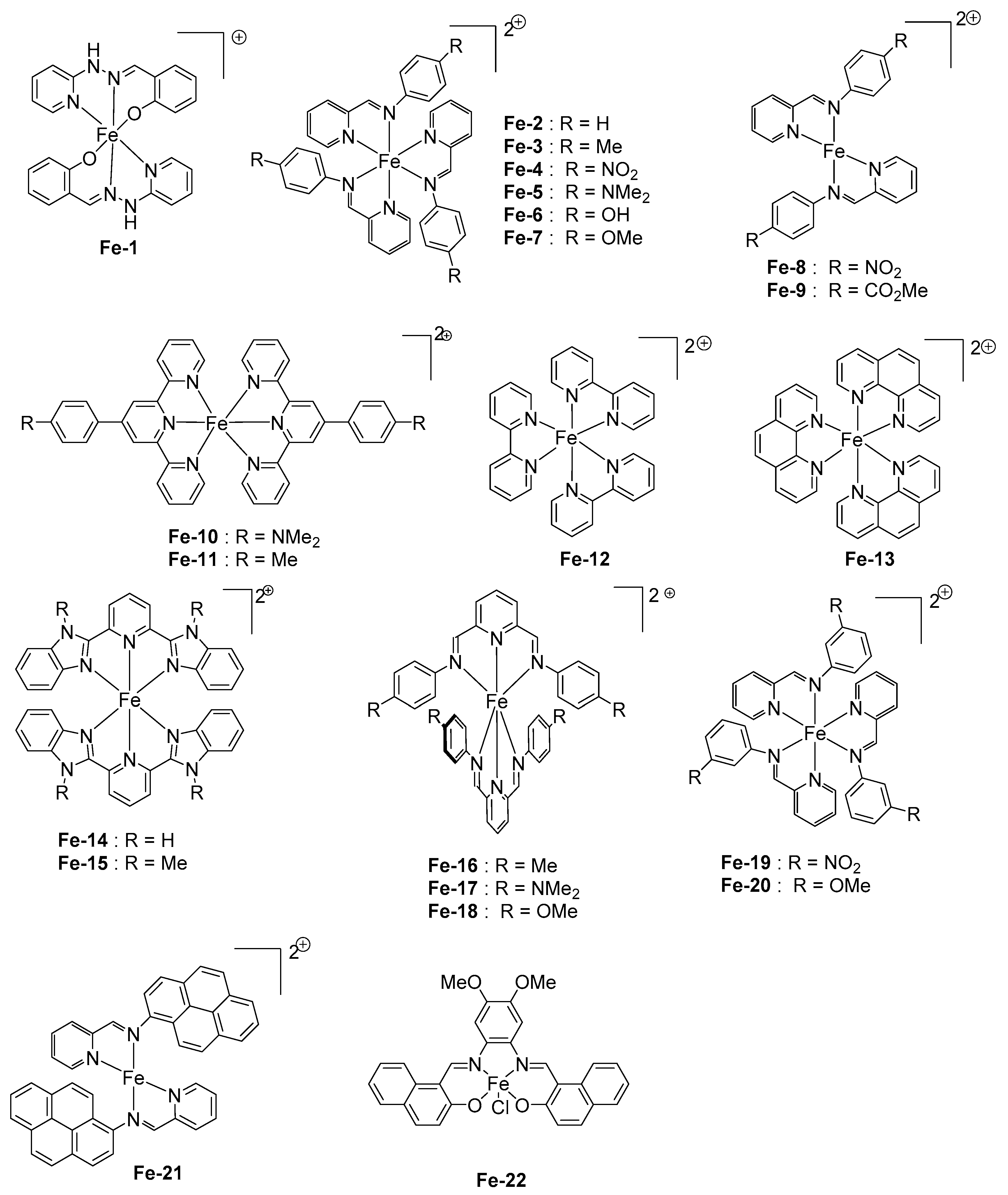
Figure 24.
Polymerization of methyl methacrylate (MMA) in toluene using the three-component photoinitiating system Fe-4/EBr/EDB upon irradiation at 405 nm with a LED. (A) Evolution of the monomer conversion vs. time; (B) evolution of Mn with the monomer conversion; (C) re-initiation experiments of polymerization of a poly(MMA) using butyl acrylate. Insert: chemical structure of the poly(MMA)-b-poly(BA) copolymer. Reprinted with permission from Telitel et al. [110]. Copyright © 2016 John Wiley & Sons, Inc.
Figure 24.
Polymerization of methyl methacrylate (MMA) in toluene using the three-component photoinitiating system Fe-4/EBr/EDB upon irradiation at 405 nm with a LED. (A) Evolution of the monomer conversion vs. time; (B) evolution of Mn with the monomer conversion; (C) re-initiation experiments of polymerization of a poly(MMA) using butyl acrylate. Insert: chemical structure of the poly(MMA)-b-poly(BA) copolymer. Reprinted with permission from Telitel et al. [110]. Copyright © 2016 John Wiley & Sons, Inc.
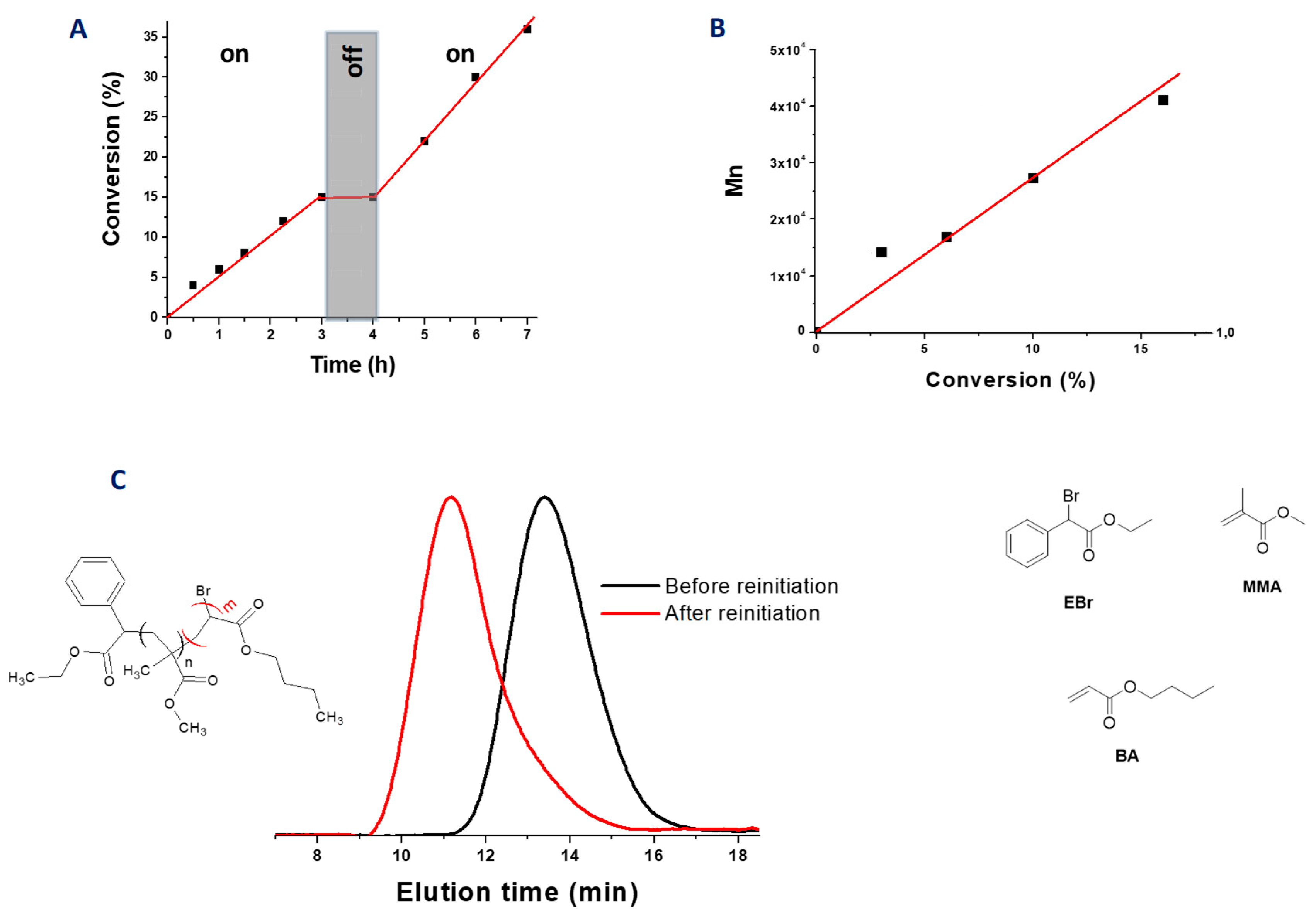
Figure 25.
(A) Contact angles for the polymer surface before and after re-initiation of the polymerization with a fluorinated monomer. (B) Profilometric analysis of the second layer formed by irradiation through a mask. Reprinted with permission from Telitel et al. [110]. Copyright © 2016 John Wiley & Sons, Inc.
Figure 25.
(A) Contact angles for the polymer surface before and after re-initiation of the polymerization with a fluorinated monomer. (B) Profilometric analysis of the second layer formed by irradiation through a mask. Reprinted with permission from Telitel et al. [110]. Copyright © 2016 John Wiley & Sons, Inc.

Figure 26.
Chemical structures of the different metal-organic frameworks (MOFs) used as photocatalysts of polymerization under visible light, and the organic linkers used in these structures.
Figure 26.
Chemical structures of the different metal-organic frameworks (MOFs) used as photocatalysts of polymerization under visible light, and the organic linkers used in these structures.

Figure 27.
Light penetration inside a photocurable resin. Adapted with permission from Bonardi et al. [22]. Copyright 2018 American Chemical Society.
Figure 27.
Light penetration inside a photocurable resin. Adapted with permission from Bonardi et al. [22]. Copyright 2018 American Chemical Society.

Figure 28.
Chemical structures of the different ferrocene-based dyes used as photocatalysts of polymerization under near-infrared region (NIR) light.
Figure 28.
Chemical structures of the different ferrocene-based dyes used as photocatalysts of polymerization under near-infrared region (NIR) light.
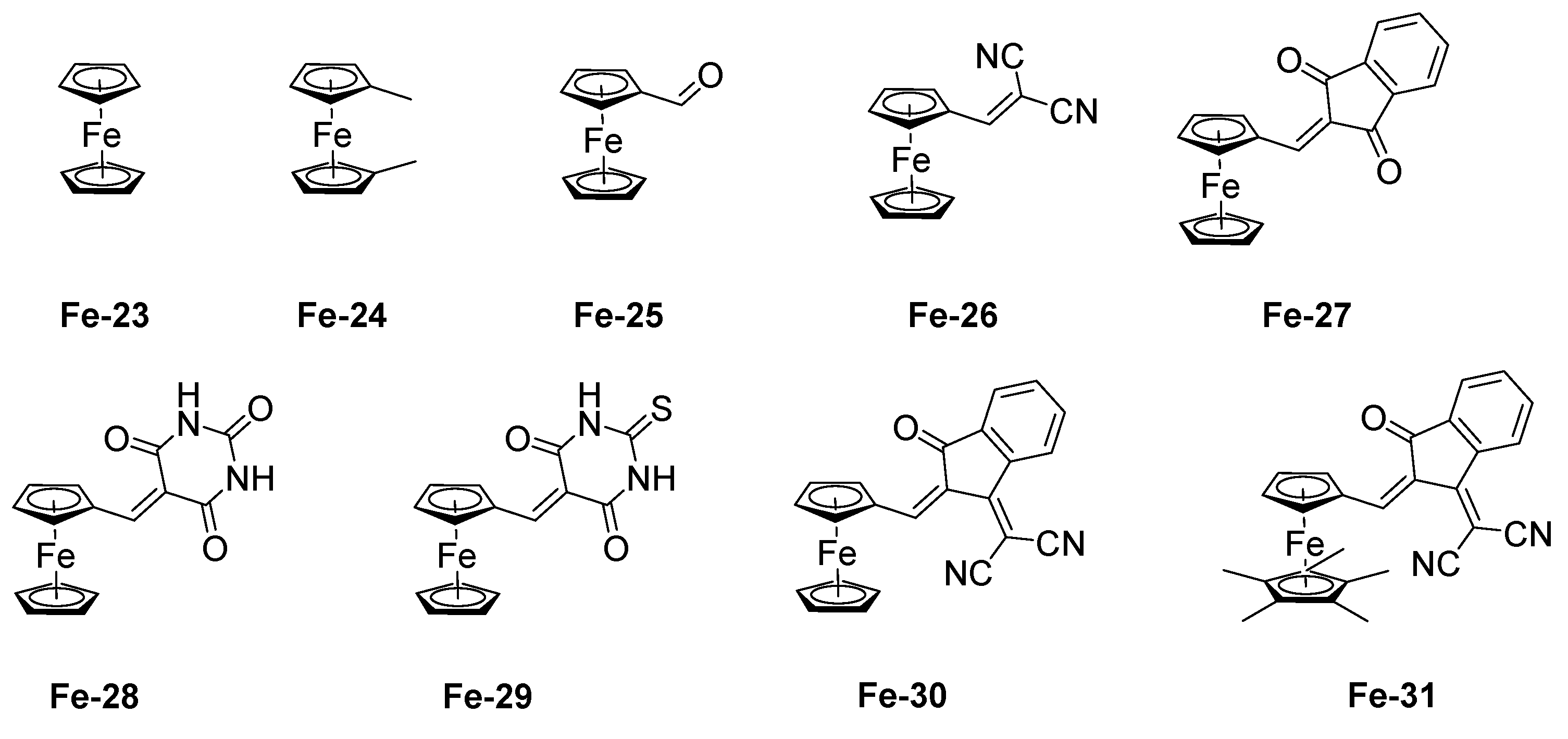
Scheme 8.
Synthetic route to Fe-2-Fe-31.
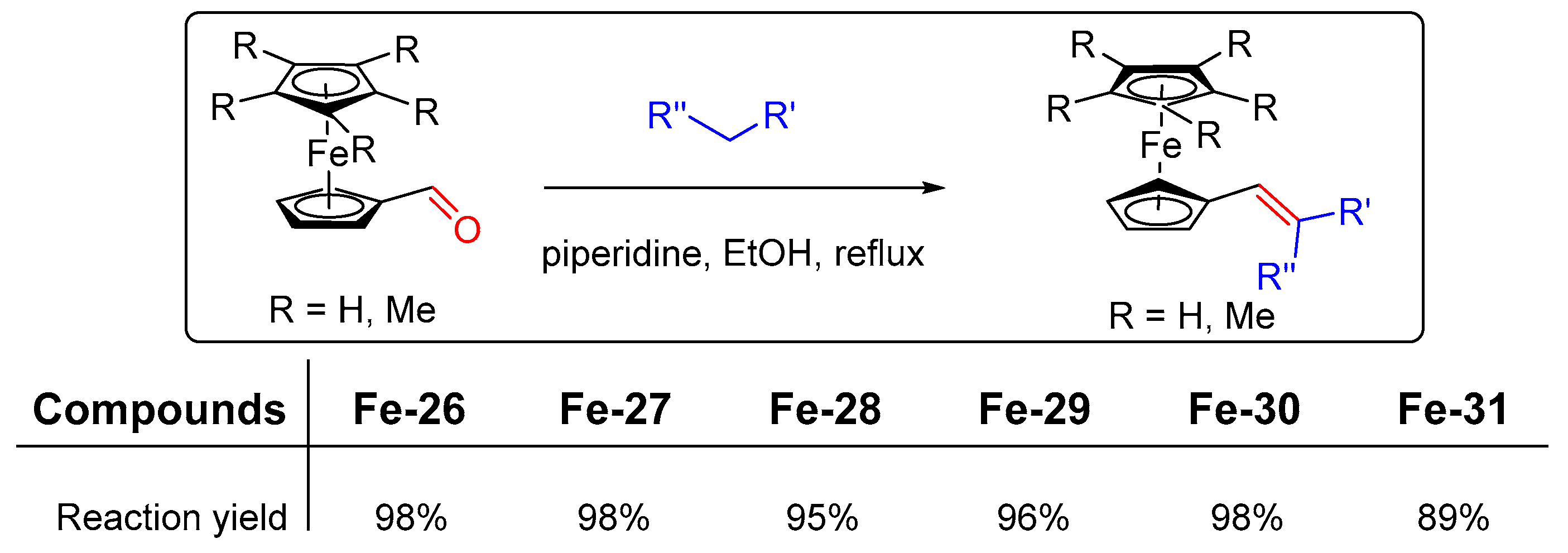
Figure 29.
Chemical structures of the different monomers and additives used with Fe-30 for redox and photoactivated redox polymerization under NIR light.
Figure 29.
Chemical structures of the different monomers and additives used with Fe-30 for redox and photoactivated redox polymerization under NIR light.
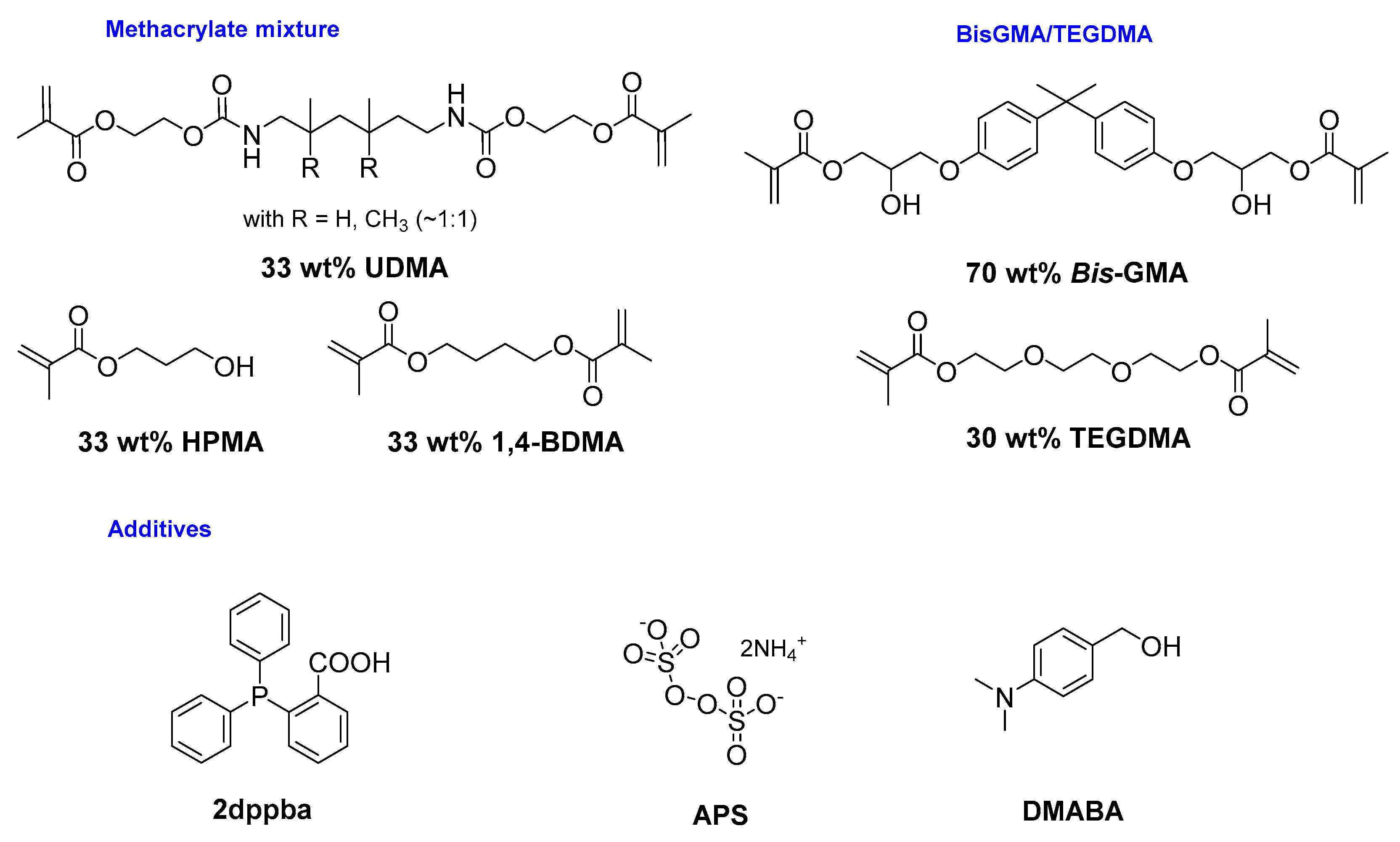
Figure 30.
(A) Monitoring the polymerization reaction of a methacrylate mixture (1,4-BDMA/ HPMA/UDMA) by optical pyrometric measurements (e) Fe-26/2-dppba/APS (f) Fe-28/2-dppba/APS (g) Fe-27/2-dppba/APS; (h) Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w). Adapted from [153] with permission from The Royal Society of Chemistry. (B) Specificities of photopolymerization, redox polymerization, and photoactivated redox polymerization. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society. (C) (a) polymerization with the three-component system Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w) (without light), (b) photoactivated redox polymerization with the four-component system Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w) (+1.0 wt% (tert-Bu)Ph2I+) under irradiation with a LED at 785 nm. Adapted from [153] with permission from The Royal Society of Chemistry.
Figure 30.
(A) Monitoring the polymerization reaction of a methacrylate mixture (1,4-BDMA/ HPMA/UDMA) by optical pyrometric measurements (e) Fe-26/2-dppba/APS (f) Fe-28/2-dppba/APS (g) Fe-27/2-dppba/APS; (h) Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w). Adapted from [153] with permission from The Royal Society of Chemistry. (B) Specificities of photopolymerization, redox polymerization, and photoactivated redox polymerization. Adapted with permission from Garra et al. [102]. Copyright 2016 American Chemical Society. (C) (a) polymerization with the three-component system Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w) (without light), (b) photoactivated redox polymerization with the four-component system Fe-30/2-dppba/APS (0.1%/1.5%/2%, w/w/w) (+1.0 wt% (tert-Bu)Ph2I+) under irradiation with a LED at 785 nm. Adapted from [153] with permission from The Royal Society of Chemistry.

Scheme 9.
The two catalytic cycles involved in the photoactivated redox polymerization of a methacrylate mixture (1,4-BDMA/ HPMA/UDMA) using Fe-30 as the catalyst in the four-component system Fe-30/2-dppba/APS/(tert-Bu)Ph2I+ (0.1%/1.5%/2%/1%, w/w/w/w).
Scheme 9.
The two catalytic cycles involved in the photoactivated redox polymerization of a methacrylate mixture (1,4-BDMA/ HPMA/UDMA) using Fe-30 as the catalyst in the four-component system Fe-30/2-dppba/APS/(tert-Bu)Ph2I+ (0.1%/1.5%/2%/1%, w/w/w/w).

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dumur, F. Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity. Catalysts 2019, 9, 736. https://doi.org/10.3390/catal9090736
AMA Style
Dumur F. Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity. Catalysts. 2019; 9(9):736. https://doi.org/10.3390/catal9090736
Chicago/Turabian StyleDumur, Frédéric. 2019. "Recent Advances on Visible Light Metal-Based Photocatalysts for Polymerization under Low Light Intensity" Catalysts 9, no. 9: 736. https://doi.org/10.3390/catal9090736
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.