On the Mechanism of Carbon Dioxide Reduction on Sn-Based Electrodes: Insights into the Role of Oxide Surfaces

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
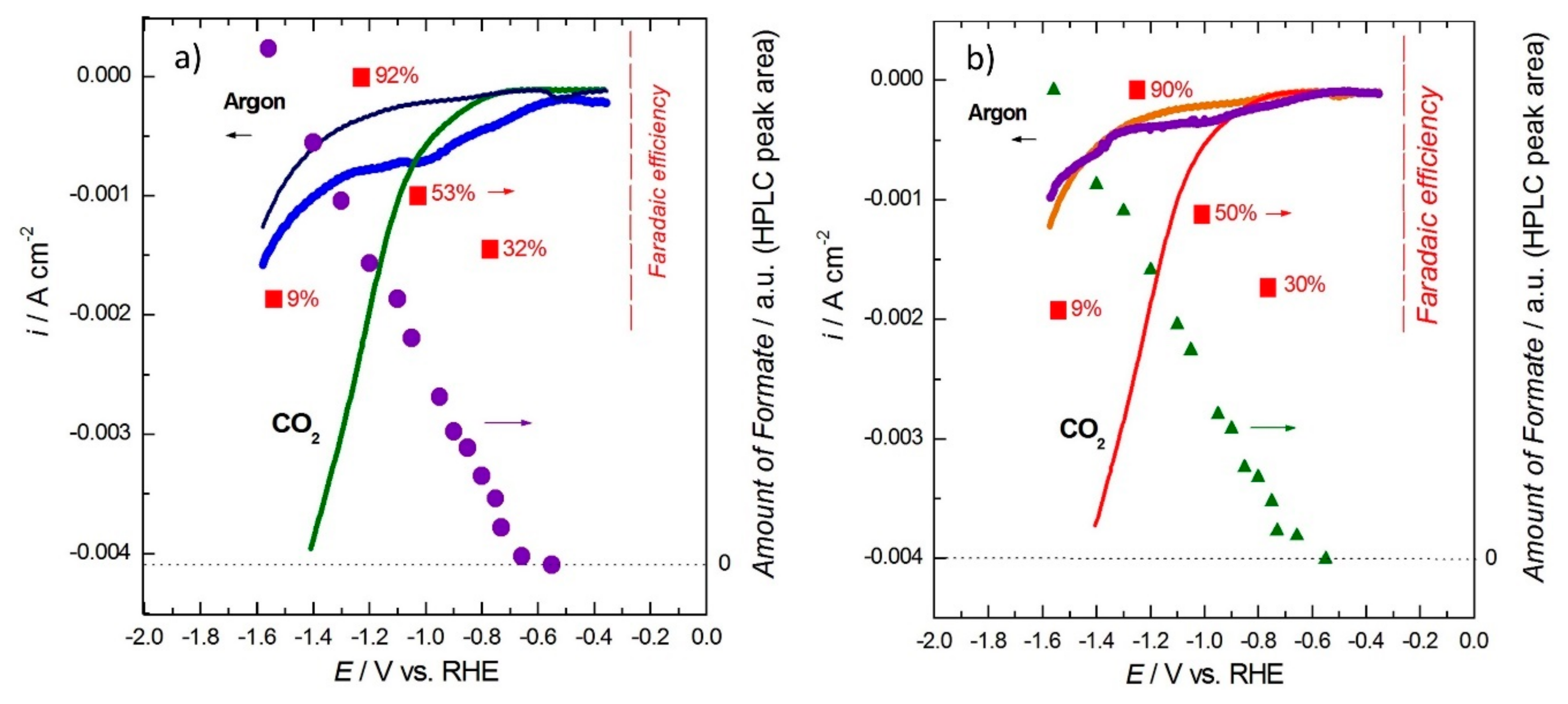
2.1. Experimental Results on Tin-Based Electrodes
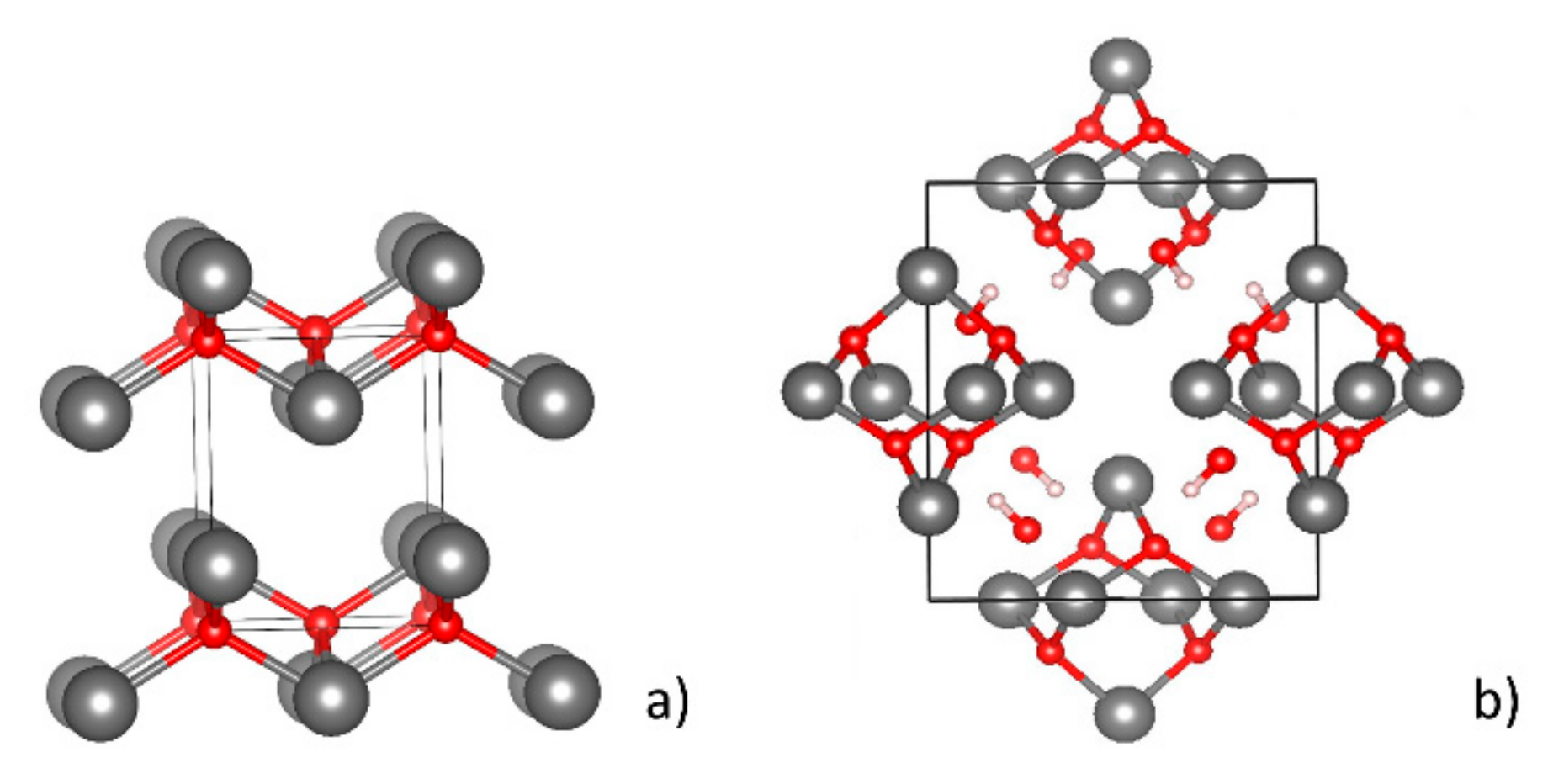
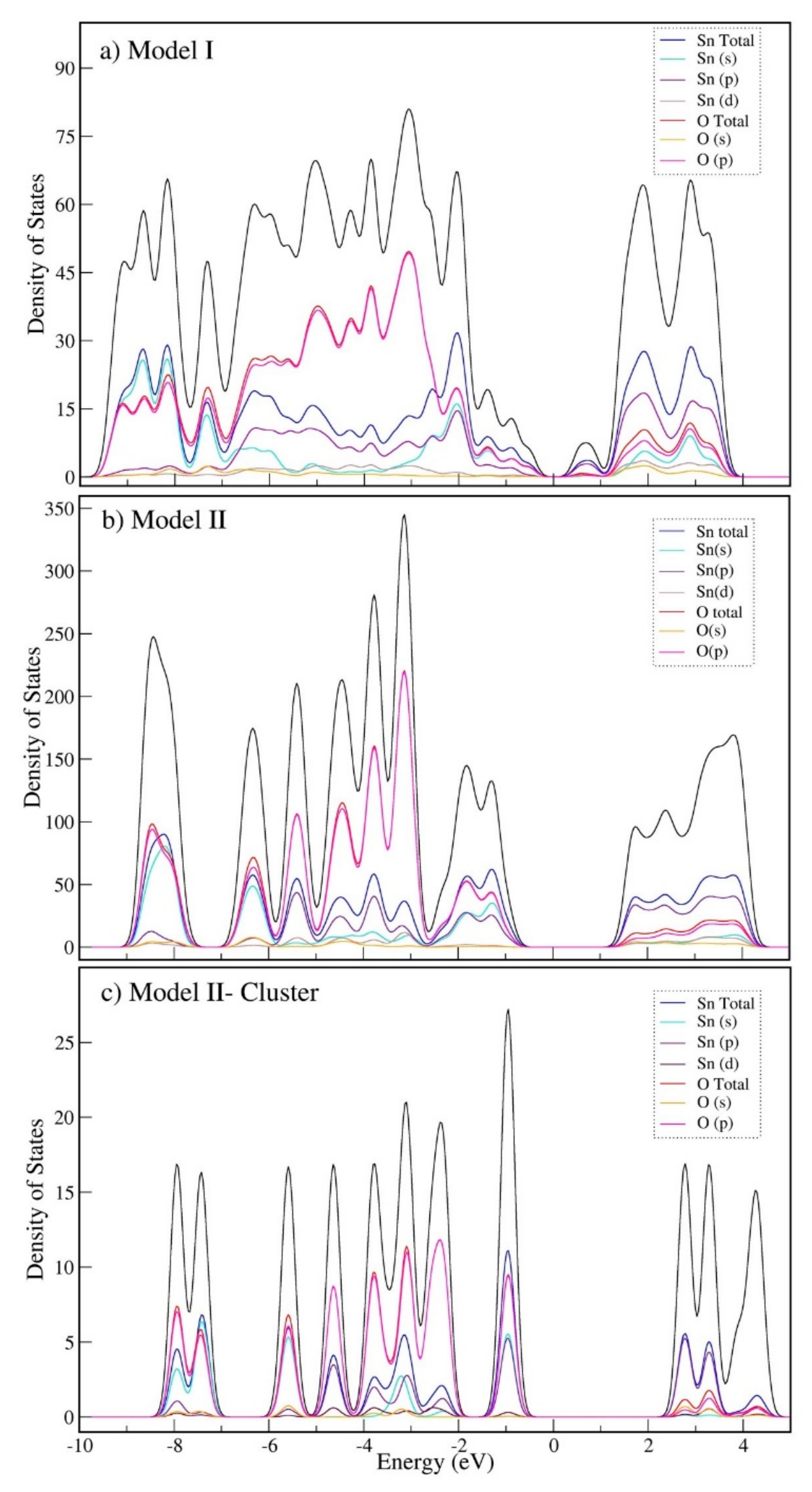
2.2. Bulk Calculations
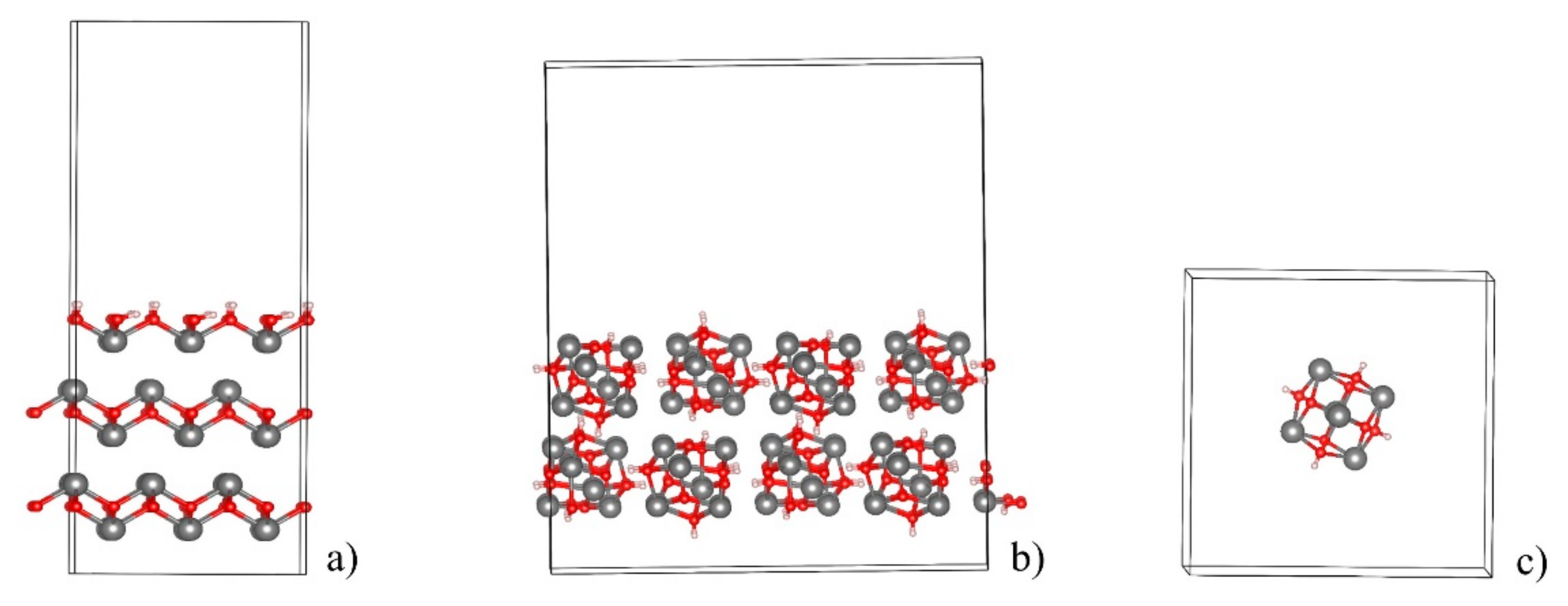
2.3. Surface Energies and Slab Structures
2.4. Insertion of CO2
2.5. Reduction Reaction
3. Materials and Methods
3.1. Measurements of Carbon Dioxide Electrochemical Reduction
3.2. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WMO. WMO Greenhouse Gas Bulletin|World Meteorological Organization. WMO Bull. 2016, 2015, 1–4. [Google Scholar]
- Baruch, M.F.; Pander, J.E.; White, J.L.; Bocarsly, A.B. Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy. ACS Catal. 2015, 5, 3148–3156. [Google Scholar] [CrossRef]
- Hu, J.; Liu, H. Advances in CO2 Conversion and Utilization; American Chemical Society: Houghton, MI, USA, 2010; Volume 1056, ISBN 0-8412-2596-6. [Google Scholar]
- Wang, Q.; Dong, H.; Yu, H. Development of rolling tin gas diffusion electrode for carbon dioxide electrochemical reduction to produce formate in aqueous electrolyte. J. Power Sources 2014, 271, 278–284. [Google Scholar] [CrossRef]
- Cui, C.; Wang, H.; Zhu, X.; Han, J.; Ge, Q. A DFT study of CO2 electrochemical reduction on Pb(211) and Sn(112). Sci. China Chem. 2015, 58, 607–613. [Google Scholar] [CrossRef]
- Leitner, W. The coordination chemistry of carbon dioxide and its relevance for catalysis: A critical survey. Coord. Chem. Rev. 1996, 153, 257–284. [Google Scholar] [CrossRef]
- Pérez, E.R.; Santos, R.H.A.; Gambardella, M.T.P.; De Macedo, L.G.M.; Rodrigues-Filho, U.P.; Launay, J.C.; Franco, D.W. Activation of carbon dioxide by bicyclic amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.K.S.; Viva, F.A.; Olah, G.A. Electrochemical reduction of CO2 over Sn-Nafion® coated electrode for a fuel-cell-like device. J. Power Sources 2013, 223, 68–73. [Google Scholar] [CrossRef]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Abe, H.; Kodiyath, R.; Ueda, S.; Srinivasan, N.; Yamaguchi, A.; Miyauchi, M. Selective electro- or photo-reduction of carbon dioxide to formic acid using a Cu–Zn alloy catalyst. J. Mater. Chem. A 2017, 5, 12113–12119. [Google Scholar] [CrossRef]
- Jiménez, C.; García, J.; Camarillo, R.; Martínez, F.; Rincón, J. Electrochemical CO2 Reduction to Fuels Using Pt/CNT Catalysts Synthesized in Supercritical Medium. Energy Fuels 2017, 31, 3038–3046. [Google Scholar] [CrossRef]
- Bei, J.; Zhang, R.; Chen, Z.; Lv, W.; Wang, W. Efficient reduction of CO2 to formate using in situ prepared nano-sized Bi electrocatalyst. Int. J. Electrochem. Sci. 2017, 12, 2365–2375. [Google Scholar] [CrossRef]
- White, J.L.; Bocarsly, A.B. Enhanced Carbon Dioxide Reduction Activity on Indium-Based Nanoparticles. J. Electrochem. Soc. 2016, 163, H410–H416. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, A.; Alvarez-Guerra, M.; Solla-Gullón, J.; Sáez, A.; Montiel, V.; Irabien, A. Sn nanoparticles on gas diffusion electrodes: Synthesis, characterization and use for continuous CO2 electroreduction to formate. J. CO2 Util. 2017, 18, 222–228. [Google Scholar] [CrossRef]
- Chen, Y.; Kanan, M.W. Tin oxide dependence of the CO2 reduction efficiency on tin electrodes and enhanced activity for tin/tin oxide thin-film catalysts. J. Am. Chem. Soc. 2012, 134, 1986–1989. [Google Scholar] [CrossRef] [PubMed]
- Sahara, G.; Ishitani, O. Efficient Photocatalysts for CO2 Reduction. Inorg. Chem. 2015, 54, 5096–5104. [Google Scholar] [CrossRef] [PubMed]
- Barton Cole, E.; Lakkaraju, P.S.; Rampulla, D.M.; Morris, A.J.; Abelev, E.; Bocarsly, A.B. Using a one-electron shuttle for the multielectron reduction of CO2 to methanol: Kinetic, mechanistic, and structural insights. J. Am. Chem. Soc. 2010, 132, 11539–11551. [Google Scholar] [CrossRef] [PubMed]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Feaster, J.T.; Shi, C.; Cave, E.R.; Hatsukade, T.; Abram, D.N.; Kuhl, K.P.; Hahn, C.; Nørskov, J.K.; Jaramillo, T.F. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 2017, 7, 4822–4827. [Google Scholar] [CrossRef]
- Cui, C.; Han, J.; Zhu, X.; Liu, X.; Wang, H.; Mei, D.; Ge, Q. Promotional effect of surface hydroxyls on electrochemical reduction of CO2 over SnOx/Sn electrode. J. Catal. 2016, 343, 257–265. [Google Scholar] [CrossRef]
- Hoflund, G.B.; Corallo, G.R. Electron-energy-loss study of the oxidation of polycrystalline tin. Phys. Rev. B 1992, 46, 7110–7120. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yu, J.; Kang, S.K.; Shih, D.Y. Oxidation study of pure tin and its alloys via electrochemical reduction analysis. J. Electron. Mater. 2005, 34, 635–642. [Google Scholar] [CrossRef]
- Dutta, A.; Kuzume, A.; Rahaman, M.; Vesztergom, S.; Broekmann, P. Monitoring the Chemical State of Catalysts for CO2 Electroreduction: An In Operando Study. ACS Catal. 2015, 5, 7498–7502. [Google Scholar] [CrossRef]
- Dutta, A.; Kuzume, A.; Kaliginedi, V.; Rahaman, M.; Sinev, I.; Ahmadi, M.; Roldán Cuenya, B.; Vesztergom, S.; Broekmann, P. Probing the chemical state of tin oxide NP catalysts during CO2 electroreduction: A complementary operando approach. Nano Energy 2018, 53, 828–840. [Google Scholar] [CrossRef]
- Lee, C.H.; Kanan, M.W. Controlling H+ vs CO2 Reduction Selectivity on Pb Electrodes. ACS Catal. 2015, 5, 465–469. [Google Scholar] [CrossRef]
- Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.; Jiao, F. Ag-sn bimetallic catalyst with a core-shell structure for CO2 reduction. J. Am. Chem. Soc. 2017, 139, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Li, F.; Easton, C.D.; Li, J.; Bond, A.M.; Zhang, J. Direct Detection of Electron Transfer Reactions Underpinning the Tin-Catalyzed Electrochemical Reduction of CO2 using Fourier-Transformed ac Voltammetry. ACS Catal. 2017, 7, 4846–4853. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Xin, H. Insights into electrochemical CO2 reduction on tin oxides from first-principles calculations. Green Energy Environ. 2017, 2, 168–171. [Google Scholar] [CrossRef]
- Lee, C.W.; Cho, N.H.; Yang, K.D.; Nam, K.T. Reaction Mechanisms of the Electrochemical Conversion of Carbon Dioxide to Formic Acid on Tin Oxide Electrodes. ChemElectroChem 2017, 4, 2130–2136. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions Version 2; National Association of Corrosion Engineers: Houston, TX, USA, 2016; Volume 52, ISBN 0915567989. [Google Scholar]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Gu, J.; Héroguel, F.; Luterbacher, J.; Hu, X. Densely Packed, Ultra Small SnO Nanoparticles for Enhanced Activity and Selectivity in Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 2943–2947. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Risalvato, F.G.; Ke, F.-S.; Pellechia, P.J.; Zhou, X.-D. Electrochemical Reduction of Carbon Dioxide I. Effects of the Electrolyte on the Selectivity and Activity with Sn Electrode. J. Electrochem. Soc. 2012, 159, F353–F359. [Google Scholar] [CrossRef]
- Zhang, R.; Lv, W.; Lei, L. Role of the oxide layer on Sn electrode in electrochemical reduction of CO2 to formate. Appl. Surf. Sci. 2015, 356, 24–29. [Google Scholar] [CrossRef]
- Won, D.H.; Choi, C.H.; Chung, J.; Chung, M.W.; Kim, E.H.; Woo, S.I. Rational Design of a Hierarchical Tin Dendrite Electrode for Efficient Electrochemical Reduction of CO2. ChemSusChem 2015, 8, 3092–3098. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Risalvato, F.G.; Ma, S.; Zhou, X.D. Electrochemical reduction of carbon dioxide III. the role of oxide layer thickness on the performance of Sn electrode in a full electrochemical cell. J. Mater. Chem. A 2014, 2, 1647–1651. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Li, J.-W.; Hou, Y.-F.; Yu, X.-Y.; Su, C.-Y.; Kuang, D.-B. Hierarchical tin oxide octahedra for highly efficient dye-sensitized solar cells. Chemistry 2010, 16, 8620–8625. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.; Watson, G.W. Electronic structures of rocksalt, litharge, and herzenbergite SnO by density functional theory. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 70, 235114. [Google Scholar] [CrossRef]
- Tao, J.; Guan, L. Tailoring the electronic and magnetic properties of monolayer SnO by B, C, N, O and F adatoms. Sci. Rep. 2017, 7, 44568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, Y.; Liu, C.; Ren, W.; Li, Y.; Wang, Q.; Su, N.; Li, Y.; Ma, B.; Ma, Y.; et al. Electrical Transport Properties of SnO under High Pressure. J. Phys. Chem. C 2011, 115, 20710–20715. [Google Scholar] [CrossRef]
- Abrahams, I.; Grimes, S.M.; Johnston, S.R.; Knowles, J.C. Tin(II) oxyhydroxide by X-ray powder diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 286–288. [Google Scholar] [CrossRef]
- Watson, G.W. Origin of the electron distribution in SnO. J. Chem. Phys. 2001, 114, 758–763. [Google Scholar] [CrossRef]
- Krishna, M.V.R.; Friesner, R.A. Quantum confinement effects in semiconductor clusters. J. Chem. Phys. 1991, 95, 8309–8322. [Google Scholar] [CrossRef]
- Azizi, O.; Jafarian, M.; Gobal, F.; Heli, H.; Mahjani, M.G. The investigation of the kinetics and mechanism of hydrogen evolution reaction on tin. Int. J. Hydrog. Energy 2007, 32, 1755–1761. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Vitos, L.; Ruban, A.V.; Skriver, H.L.; Kollár, J. The surface energy of metals. Surf. Sci. 1998, 411, 186–202. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef]
- Deiss, E. Average Voltage, Energy Density, and Specific Energy of Lithium-Ion Batteries. J. Electrochem. Soc. 1997, 144, 3877–3881. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damas, G.B.; Miranda, C.R.; Sgarbi, R.; Portela, J.M.; Camilo, M.R.; Lima, F.H.B.; Araujo, C.M. On the Mechanism of Carbon Dioxide Reduction on Sn-Based Electrodes: Insights into the Role of Oxide Surfaces. Catalysts 2019, 9, 636. https://doi.org/10.3390/catal9080636
Damas GB, Miranda CR, Sgarbi R, Portela JM, Camilo MR, Lima FHB, Araujo CM. On the Mechanism of Carbon Dioxide Reduction on Sn-Based Electrodes: Insights into the Role of Oxide Surfaces. Catalysts. 2019; 9(8):636. https://doi.org/10.3390/catal9080636
Chicago/Turabian StyleDamas, Giane B., Caetano R. Miranda, Ricardo Sgarbi, James M. Portela, Mariana R. Camilo, Fabio H. B. Lima, and C. Moyses Araujo. 2019. "On the Mechanism of Carbon Dioxide Reduction on Sn-Based Electrodes: Insights into the Role of Oxide Surfaces" Catalysts 9, no. 8: 636. https://doi.org/10.3390/catal9080636
APA StyleDamas, G. B., Miranda, C. R., Sgarbi, R., Portela, J. M., Camilo, M. R., Lima, F. H. B., & Araujo, C. M. (2019). On the Mechanism of Carbon Dioxide Reduction on Sn-Based Electrodes: Insights into the Role of Oxide Surfaces. Catalysts, 9(8), 636. https://doi.org/10.3390/catal9080636