1. Introduction
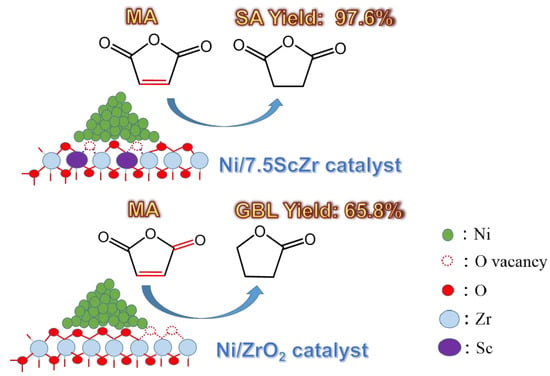
Maleic anhydride (MA)—a typical α, β-unsaturated carbonyl compound—can be hydrogenated to produce succinic anhydride (SA), γ-butyrolactone (GBL), 1,4-Butanediol (BDO), and tetrahydrofuran (THF) (
Figure 1). Among them, SA functions as an important raw material in the production of PBS (biodegradable plastic polybutylene succinate), and GBL is considered an environmentally friendly medium [
1].
MA is a five-membered ring molecule structure, with C=C, C=O, and C-O-C functional groups. The coupled molecular structure leads to a delocalization of the electron density in the C=C and C=O bonds, which makes it hard for the selective hydrogenation of the C=C bond to obtain SA or for the selective hydrogenation of C=C and C=O bonds to obtain GBL [
2]. For these reasons, the tailoring of high activity and selectivity catalysts to obtain SA or GBL, as well as an exploration of the relationship between the structure of the catalysts and their hydrogenation performances, are scientifically significant and of practical value [
3,
4].
The metal catalysts Pd, Ru, Cu, and Ni are widely accepted as being active in the hydrogenation of MA [
3,
5,
6,
7,
8]. They are usually supported on metal oxides (for example, Al
2O
3, SiO
2, diatomite, SiO
2-Al
2O
3, and zeolite) [
9,
10,
11,
12]. Yu et al. reported that adding CeO
2 to a Cu-Al
2O
3 catalyst supported the hydrogenation of MA to GBL [
13]. Hu et al. also found a similar phenomenon by adding TiO
2 to a Cu-Zn catalyst [
14]. Our research group previously investigated the maleic anhydride hydrogenation performances of Ni/TiO
2 (TiO
2 supports with different crystal structure, rutile and anatase) and Ni/CeO
2 catalysts [
15,
16], the result showed that the oxygen vacancies on surface of Ni/TiO
2 (anatase) and Ni/CeO
2 catalysts might be contributed to the higher C=O hydrogenation activity. Further, in our recent study [
17], two ZrO
2 supports, with each having monoclinic and tetragonal mixed crystalline phases but differing in tetragonal content, were prepared. In MA hydrogenation reaction, the two ZrO
2-supported nickel catalysts exhibited completely different C=O hydrogenation activities. Through the analysis of the catalysts’ structure–activity relationship, we concluded that the C=O hydrogenation activity of Ni/ZrO
2 catalysts is closely related to the electronic properties of oxygen vacancies on the surface of Ni/ZrO
2 catalysts. Relatively electron-deficient oxygen vacancies could activate the C=O group and synergized with active metal Ni
0 to complete C=O hydrogenation, while relatively electron-rich oxygen vacancies could not activate the C=O group effectively and played no part in the C=O hydrogenation.
As concerns the effect of the ZrO
2 supports on their supported metal catalysts’ hydrogenation performance, attention has been always paid to the relationship between the crystal structure of the ZrO
2 support (the bulk structure examined by XRD) and its supported metal catalysts’ hydrogenation performance. However, the reported results showed that there was no clear correlation between the two, even some of the results were contradictory [
18,
19,
20]. As for ZrO
2, the phase transformation starts from its surface region and then gradually develops into its bulk [
21]. This means the surface structure may be different from its bulk structure for a specific ZrO
2 sample. We speculate that the different catalytic performances of ZrO
2-supported metal catalysts may originate from the differences in the ZrO
2 supports’ surface structure.
In our aforementioned earlier article [
17], the Raman, H
2-TPR, and XPS characterization results hinted that there was a relationship between the electronic properties of surface oxygen vacancies on the Ni/ZrO
2 catalysts and the surface structure of the ZrO
2 supports. However, due to a comparison of only two ZrO
2-supported nickel catalysts in the previous article, it was difficult for us to reach an unambiguous conclusion concerning the relationship between the surface structure of the ZrO
2 support and the electronic properties of surface oxygen vacancies on the Ni/ZrO
2 catalysts, as well as the selective hydrogenation performance of ZrO
2-supported nickel catalyst. Therefore, it was necessary and scientifically significant to design and prepare a series of ZrO
2 supports with different surface structures, and to explore the ZrO
2-supported nickel catalysts’ selective hydrogenation performance. Through the study of the catalysts’ structure–activity relationship, it could be able to draw a clear conclusion about this.
The surface structure of ZrO
2 nanoparticles and their oxygen vacancies concentration can be regulated by doping cations, such as Sc
3+, Y
3+, Ca
2+, and Mg
2+ [
22,
23]. However, the body of research lacks information about the use of doped ZrO
2 nanoparticles as supports in the preparation of metal–supported catalysts. Nor has there been adequate investigation into the effect of the surface structure of ZrO
2-based supports on ZrO
2-supported metal catalysts’ selective hydrogenation performance.
This study aimed to fill these documented gaps in the literature, and to gain a more detailed insight into the effect of the surface structure of the ZrO2-based supports on the selective hydrogenation performances in MA hydrogenation of the ZrO2-based supported nickel catalysts. Sc3+ was doped into ZrO2 support to modify the surface structure of the ZrO2-based support, and the performances of the Ni/Sc-doped ZrO2 catalysts for MA hydrogenation were tested. XRD, Raman, H2-TPR, H2-TPD, XPS, and in situ FT-IR measurements were used to clarify the structure–activity relationship between the surface structure of the ZrO2-based support and their selective hydrogenation performances of ZrO2-supported nickel catalysts.
2. Results
The prepared Sc-doped ZrO2 support with different Sc2O3 contents denoted as xScZr, where x was the Sc2O3/(Sc2O3 + ZrO2) molar ratio. In this work, x = 0, 1.5, 2.5, 5.0, and 7.5% (mol/mol), and the corresponding xScZr supports were noted as ZrO2, 1.5ScZr, 2.5ScZr, 5.0ScZr, and 7.5ScZr, respectively.
2.1. Catalytic Performance of Ni/xScZr Catalysts in Maleic Anhydride Hydrogenation
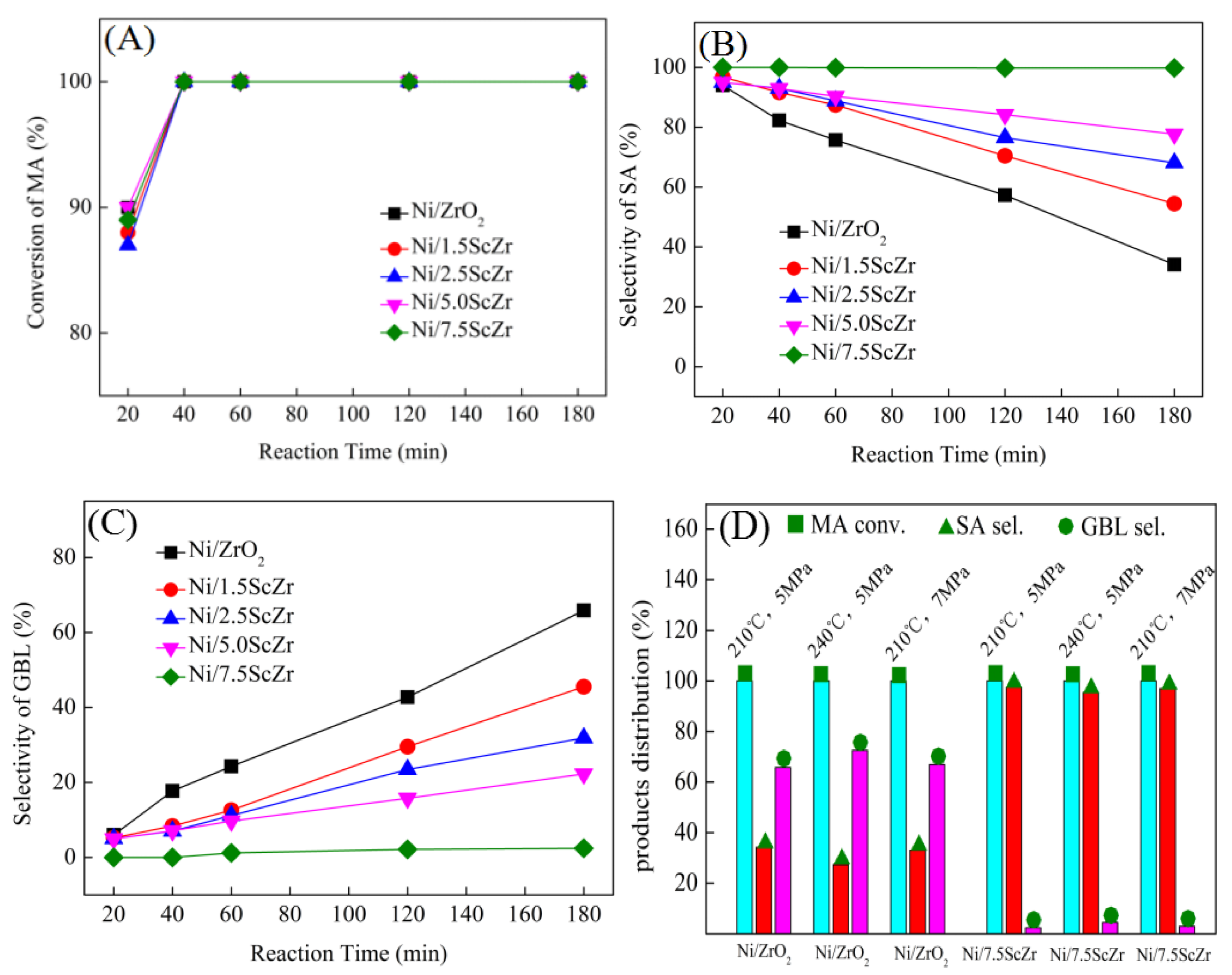
Figure 2 shows the MA conversion (A) as well as the selectivity of SA and GBL ((B) and (C)) over Ni/xScZr catalysts with different Sc
2O
3 contents at 210 °C and 5 MPa of H
2 pressure. From
Figure 2A it can be seen that Ni/xScZr catalysts, with different Sc
2O
3 contents, exhibited high catalytic activity in MA hydrogenation during the initial stage of the reaction. At a reaction time of 20 min, the MA conversion over all the Ni/xScZr catalysts was concentrated in the range of 87 to 90%. At this time, SA was the main hydrogenation product, and the selectivity towards SA was higher than 94% for all the Ni/xScZr catalysts. This result suggests that the C=C bond in the MA molecule were firstly hydrogenated and SA product was obtained. Moreover, all the Ni/xScZr catalysts showed high C=C hydrogenation activity with no significant difference.
As the reaction time prolonged, the selectivity towards SA and GBL showed different change trends over the Ni/xScZr catalysts with the different Sc2O3 contents. For the Ni/ZrO2 catalyst, SA selectivity decreased gradually and, correspondingly, GBL selectivity increased. At a reaction time of 180 minutes, the yield of GBL reached 65.8%. However, for the Sc-doped ZrO2-supported nickel catalyst, the increasing rate of GBL selectivity gradually slowed down as the Sc2O3 content increased. When the Sc2O3 content increased to 7.5 mol%, a yield of only 2.4% GBL was obtained over the Ni/7.5ScZr catalyst. GBL is produced through the further hydrogenation of the C=O group in the SA molecule. The above evaluation results indicated that the Ni/ZrO2 catalyst possessed the highest C=O hydrogenation activity and, as the Sc2O3 content increased, the C=O hydrogenation activities of Ni/xScZr catalysts decreased gradually. When the Sc2O3 content increased to 7.5 mol%, the Ni/7.5ScZr catalyst had the poorest performance with almost no C=O hydrogenation activity.
When the reaction temperature was enhanced from 210 °C to 240 °C, the selectivity towards GBL over Ni/ZrO
2 catalyst increased from 65.8% to 72.6% (
Figure 2D), also the GBL selectivity increased from 65.8% to 67.0% when the H
2 pressure increased from 5 MPa to 7 MPa. Different from the change trend of the Ni/ZrO
2 catalyst, there was almost no change in the GBL selectivity for Ni/7.5ScZr catalyst whether increasing the reaction temperature or increasing the H
2 pressure. The Ni/7.5ScZr catalyst retained its extremely low C=O hydrogenation activity, giving high yield to SA product (~97.6%).
For better understanding the regular variability in the C=O hydrogenation activity of Ni/xScZr catalysts derived from Sc3+ doping into ZrO2, a series of structural characterizations of the catalysts were conducted.
2.2. X-Ray Powder Diffraction (XRD) Patterns
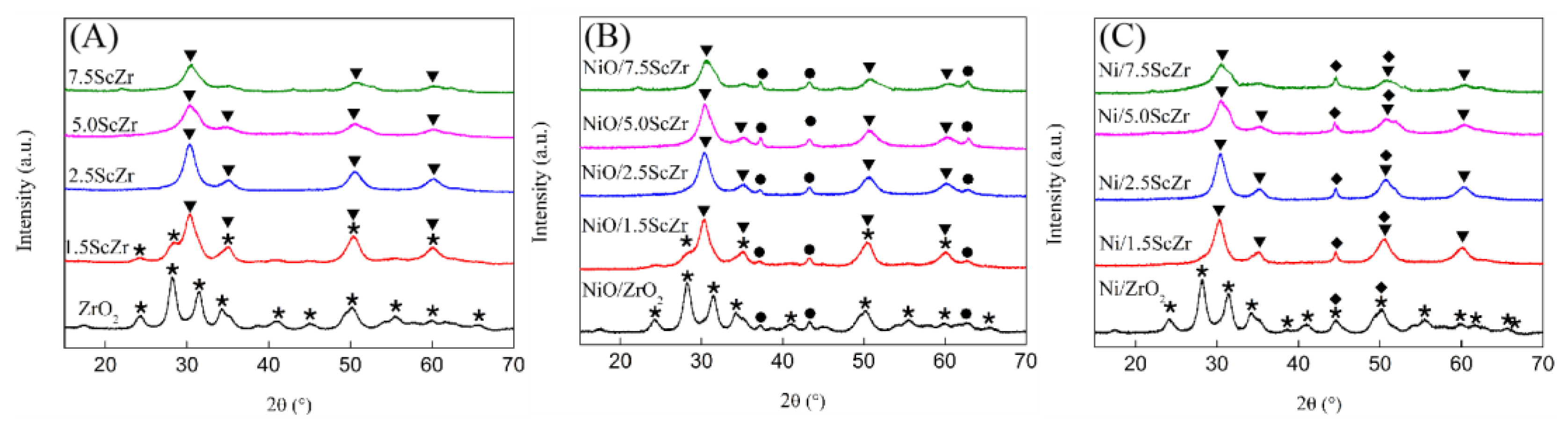
Figure 3 presents the XRD patterns of the xScZr supports and their corresponding supported nickel catalysts. It can be seen from
Figure 3A that the undoped ZrO
2 is monoclinic and single-phase. For xScZr support with 1.5 mol% Sc
2O
3 content, a new peak assigned to the tetragonal ZrO
2 (111) appeared, indicating that partially monoclinic phase transformed into tetragonal phase in 1.5ScZr support. When the Sc
2O
3 content increased to 2.5 mol%, peaks appearing in 2.5ScZr support completely converted to tetragonal phase. As the Sc
2O
3 content further increased to 5.0 and 7.5 mol%, the tetragonal phase diffraction peaks became widened. The widened diffraction peak can be indexed by a metastable tetragonal phase (t’’) or fluorite cubic phase (c) [
24]. As reported, the monoclinic phase is the stable phase for pure ZrO
2 at low temperatures, and the strong covalent nature of the Zr-O bond within the ZrO
2 lattice favors seven-fold coordination. The stabilization of tetragonal phase in the nanocrystalline ZrO
2 powder at ambient temperature appears to be due to the crystallite size effect as proposed by Garvie [
25], the tetragonal phase can be stabilized at room temperature below a critical size (30 nm), which is due to the generation of excess oxygen vacancies. As for the doped zirconia, the metastability of the tetragonal or cubic phase in the nanocrystalline ZrO
2-Sc
2O
3 powders can be observed at room temperature is due to the creation of oxygen vacancies [
26,
27,
28]. Thus, though it is difficult to distinguish the tetragonal phase from the cubic phase in the nano-ZrO
2 system by XRD due to the crystallite size effect [
24], it can be inferred that as the Sc
2O
3 content increases, the number of oxygen vacancies in the xScZr supports continued to increase, leading to the evolution of xScZr supports crystal phase structure.
Figure 3B presents the XRD patterns of the NiO/xScZr catalysts. Compared with the xScZr supports, the ZrO
2 crystalline phase in NiO/xScZr remained unchanged, and the characteristic diffraction peaks assigned to NiO appeared. In
Figure 3C, a diffraction peak at 44.5°, which is assigned to the (111) reflection of Ni, appeared in the Ni/xScZr catalysts’XRD patterns, meanwhile the NiO diffraction peaks disappeared. For the Ni/1.5ScZr catalyst, the monoclinic phase diffraction peaks disappeared compared to the NiO/1.5ScZr’s XRD pattern, which implies that the ZrO
2-based support structure could be changed because of the interaction between the nickel species and the ZrO
2-based support.
Table 1 lists the calculated mean crystalline size of NiO in the NiO/xScZr samples and Ni in the Ni/xScZr catalysts. The NiO crystalline size in NiO/xScZr samples concentrated at ~16 nm, and the Ni crystalline size in the Ni/xScZr catalysts concentrated at ~21 nm. However, the Ni crystalline size in Ni/ZrO
2 catalysts cannot be accurately calculated because of the partial overlap between the monoclinic ZrO
2 (211) diffraction peak and the Ni (111) diffraction peak. Combining the results that the NiO crystalline size in NiO/ZrO
2 was close to that in the Sc-doped NiO/xScZr samples, and the diffraction peak at 44.5° of the Ni/ZrO
2 catalyst showed no apparent difference with that of the Sc-doped Ni/xScZr catalyst (based on the (211) diffraction peak of monoclinic ZrO
2, which was very weak), then the Ni crystalline size in the Ni/ZrO
2 catalyst can be estimated to be close in size to the Sc-doped Ni/xScZr catalysts.
Table 1 also lists the specific surface area of the xScZr supports and Ni/xScZr catalysts. The xScZr supports, with different Sc
2O
3 contents, had a similar specific surface area, and their supported nickel catalysts’ specific surface areas were also close.
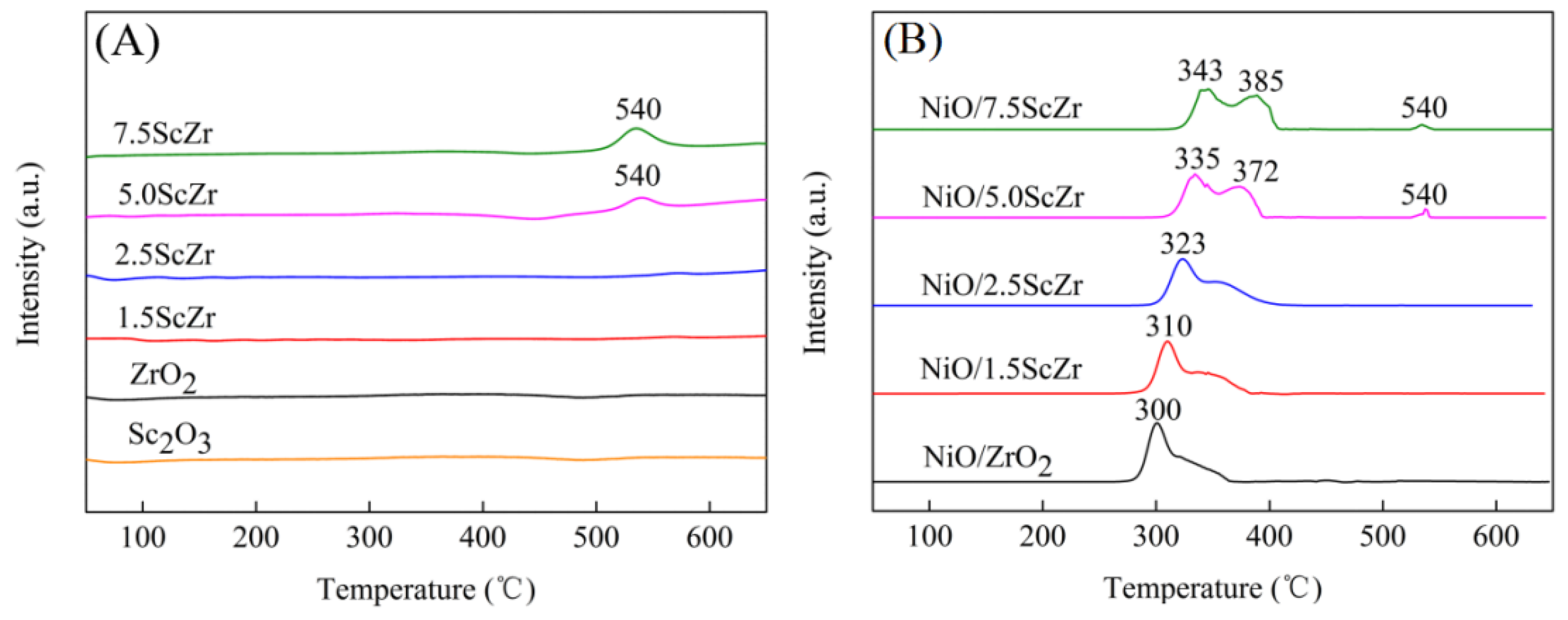
2.3. H2 Temperature-Programmed Reduction (H2-TPR)
H
2 temperature-programmed reduction was carried out to characterize the reducibility of the xScZr and NiO/xScZr samples (
Figure 4).
Figure 4A showed that pure ZrO
2 and Sc
2O
3 exhibited no reduction signal within the test temperature range. The 5ScZr support displayed a broad reduction peak, with low intensity at 540 °C. However this reduction peak became more intense in the H
2-TPR profile of the 7.5ScZr support, which suggested that the addition of the Sc
3+ into the ZrO
2 enhanced the reducibility of the xScZr supports. Previous research has suggested that it is easier for the reduction of ZrO
2 which has more surficial low-coordinated oxygen ions and oxygen vacancies [
29]. In line with this view, we can deduce that the introducing Sc
3+ into ZrO
2 support promoted the formation of oxygen vacancies as well as low-coordinated oxygen ions. As the Sc
2O
3 content increased, the concentration of surface oxygen vacancies and low coordination oxygen ions increased gradually, also the xScZr supports became more easily reduced.
Figure 4B shows the H
2-TPR profiles of the NiO/xScZr samples. All of the NiO/ xScZr samples (0 ≤ x ≤ 7.5) had two reduction peaks in the 300 to 400 °C region, indicating that there were two forms of nickel species in the NiO/xScZr samples. The NiO/ZrO
2 sample exhibited a strong reduction peak which was centered at 300 °C, and there was a minor shoulder peak. The reduction peak at 300 °C was assigned to the reduction of the nickel species that were similar to the bulk NiO, while the shoulder peak could be explained by the reduction of the nickel species, which had a weak interaction with the ZrO
2 support [
30]. As the Sc
2O
3 content increased, the initial peak temperature and maximum peak temperature of the reduction peak shifted to higher temperature, and the peak’s area in the lower temperature region decreased, while the peak’s area in higher temperature region increased gradually. When the Sc
2O
3 content increased to 7.5 mol%, the NiO/7.5ScZr sample showed two continuous reduction peaks, which were centered at 343 and 385 °C, respectively. The peak at 385 °C could be attributed to the reduction of the nickel species which had a relatively strong interaction with the 7.5ScZr support. In addition, both the NiO/5.0ScZr and NiO/7.5ScZr samples exhibited weak reduction peaks at 540 °C, which was consistent with their corresponding supports’ reduction peaks.
According to the analysis of the above results, it can be summarized that the interaction between the nickel species and the xScZr supports was highly dependent on the Sc2O3 content. As the Sc2O3 content increased, the interaction between the nickel species and the xScZr supports enhanced gradually.
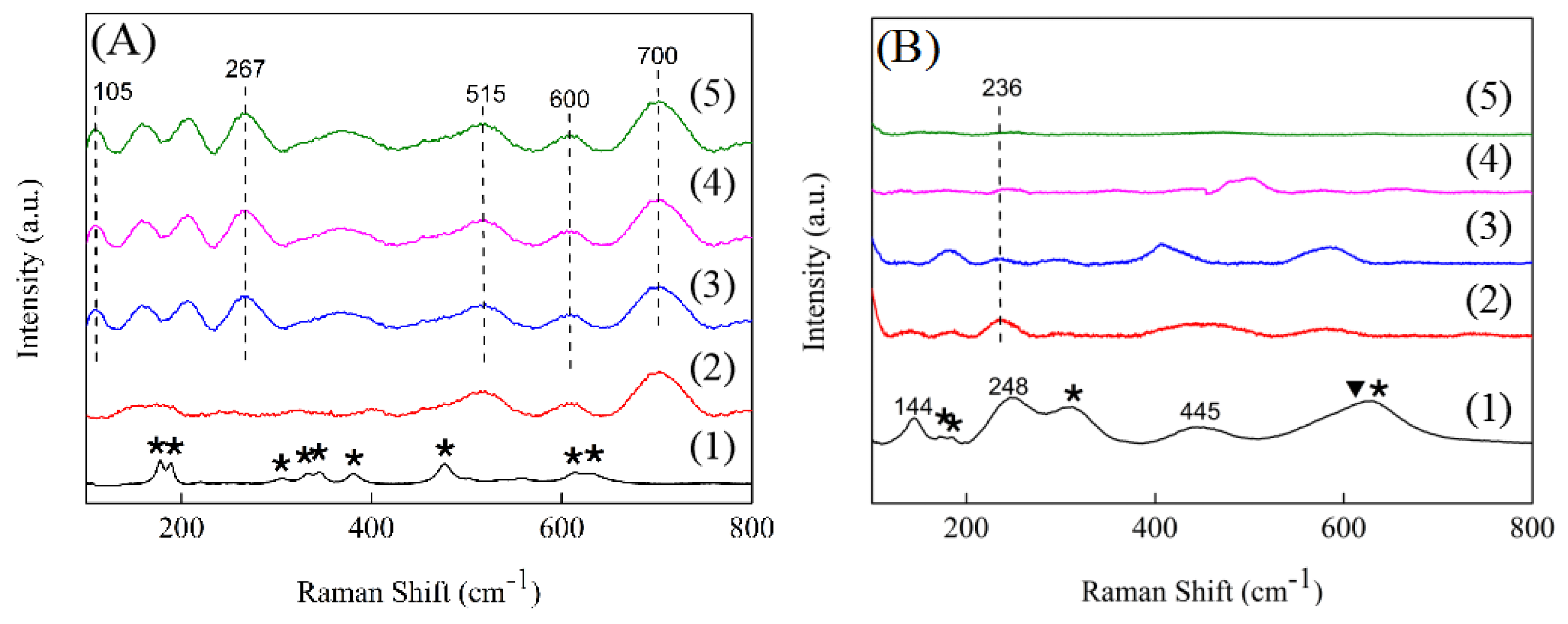
2.4. Raman Spectra
Compared with XRD characterization, Raman is more sensitive to the oxygen displacement and intermediate range order structure, thus Raman is used to further investigate the microstructure changes of the samples [
24]. As
Figure 5A showed, the Raman spectra of the pure ZrO
2 support had all the characteristics of a monoclinic ZrO
2 [
31]. However, for the 1.5ScZr support, the peaks that were assigned to the monoclinic ZrO
2 disappeared and three additional, broadened peaks appeared, located at 515, 600, and 700 cm
−1, respectively. Among them, the peak centered at 515 cm
−1 was assigned to monoclinic ZrO
2 and the peak located at 600 cm
−1 was ascribed to tetragonal ZrO
2, while the peak at 700 cm
−1 was induced by its surface oxygen vacancies [
32], suggesting that the introduction of Sc
3+ promoted the oxygen vacancies generated. When the Sc
2O
3 content increased to 2.5 mol%, additional new peaks appeared, centered at 105 and 267 cm
−1, which were also attributed to the peaks induced by oxygen vacancies [
32]. The Raman peaks induced by oxygen vacancies can also be observed in the 5.0ScZr and 7.5ScZr supports, also with the Sc
2O
3 content increasing, these peaks intensity showed an increasing trend, indicating that the concentration of surface oxygen vacancies continually increased with the Sc
2O
3 content increasing, which is consistent with the inference from XRD. In addition, from the Raman spectra of the 5.0ScZr and 7.5ScZr supports, it can be judged that the crystal phase structure of the 5.0ScZr and 7.5ScZr supports were metastable tetragonal phase structure [
24].
Figure 5B presents the Raman spectra of the Ni/xScZr catalysts. Compared to the pure ZrO
2 support’s Raman spectra, some of the peaks assigned to monoclinic ZrO
2 disappeared and three new peaks appeared in Ni/ZrO
2 Raman spectra, located at 144, 248, and 445 cm
−1, respectively. As previously reported [
31], the bulk tetragonal ZrO
2 Raman peaks were centered at 147, 267, and 459 cm
-1. The three new peaks in the Ni/ZrO
2 spectra exhibited a shift to lower frequencies compared with those of bulk tetragonal ZrO
2, which could be ascribed to a decrease in the symmetry of the tetragonal phase structure induced by more oxygen vacancies [
33,
34]. This suggests that there is an increase in the number of surface oxygen vacancies after ZrO
2 loading nickel. However, for the Sc-doped ZrO
2-supported nickel catalysts, the Raman spectra showed a significantly different change. For the Ni/1.5ScZr catalyst, only a weak peak appeared, located at 236 cm
−1. Although this peak was also attributed to a peak induced by oxygen vacancies, the stronger peak at 700 cm
−1 disappeared, indicating that the concentration of surface oxygen vacancies would potentially show a decrease trend after the 1.5ScZr loading nickel. As the Sc
2O
3 content further increased, no Raman peak was observed in the Ni/xScZr spectra (x = 2.5, 5.0, and 7.5). This strongly suggests that all of the Sc-doped ZrO
2-supported nickel catalysts were exhibiting a significant decrease in the concentration of surface oxygen vacancies after being loaded with nickel. Also with Sc
2O
3 content increasing, the surface oxygen vacancy concentration on Ni/xScZr catalysts decreased gradually.
For pure monoclonal ZrO
2 support, after its loading nickel, more new oxygen vacancies generated on surface of its supported nickel catalyst, while for Sc-doped ZrO
2-based supports, a significant decrease in oxygen vacancy concentration occurred after their loading nickel. This indicated that the nickel species had different interactions with the pure ZrO
2 support and the Sc-doped ZrO
2-based supports. Puigdollers et al. stated that the ZrO
2 support with more low-coordinated oxygen ions and oxygen vacancies has a stronger interaction with deposited metal species [
29]. The density functional theory calculation results of Tosoni et al. showed that [
35] Ni adsorption was stronger on the ZrO
2 surface, which was rich of oxygen vacancies (−4.28 eV), than on the stoichiometric surface (−2.31 eV). Moreover, when the oxygen vacancies were distributed on the surface of ZrO
2, the most stable arrangement is with the Ni atom adsorbed directly on top of the oxygen vacancy. Based on the above theoretical calculation results, combining our H
2-TPR and Raman results, it can be concluded that the introduction of Sc
3+ into ZrO
2 support would promote the low-coordinated oxygen ions and oxygen vacancies generated on the surface of the ZrO
2-based support. As the Sc
2O
3 content increased, the concentration of low-coordinated oxygen ions and oxygen vacancies increased, and the interaction between nickel species and ZrO
2-based support enhanced gradually. The stronger interaction further resulted in the decrease of the oxygen vacancies concentration on surface of ZrO
2-based supported nickel catalysts, which probably because a small amount of nickel species entered into oxygen vacancies. This result is consistent with our previous study [
17], in that the strong interaction between nickel species and ZrO
2 support would lead to a small amount of nickel species entering into the tetragonal ZrO
2 lattice, and in HRTEM it had been clearly seen that the lattice spacing of the tetragonal ZrO
2 increased from 0.29 nm to 0.30 nm, thereby leading to a significant decrease of the surface oxygen vacancies concentration.
Based on the above results, it can be concluded that the introduction of Sc3+ into ZrO2 changes the surface structure of ZrO2-based supports. The different surface structure of ZrO2-based support, led to the different degree of interaction between nickel species and the ZrO2-based support. Also, the different degree of interaction further changes the surface oxygen vacancy concentration of ZrO2-based supported nickel catalysts.
2.5. XPS Characterization
XPS was carried out to further probe the properties of the oxygen species.
Figure 6 showed the O 1s XPS spectra of ZrO
2 and 7.5ScZr supports and their corresponding supported nickel catalysts. An analysis of the O1s spectra of the samples was conducted, and the spectra was fitted into four peaks [
29,
36]. They were lattice oxygen (O’) on ZrO
2 (including the oxygen vacancies with two electrons), singly-charged oxygen vacancies (O’’) on ZrO
2 (with one electron remaining in the oxygen vacancy), doubly-charged oxygen vacancies (O’’’) on ZrO
2 (without any electron in the oxygen vacancy), and the hydroxyl or/and carbonates groups (O’’’’) on ZrO
2, respectively. The details about the binding energy and the surface atomic concentration which was calculated on the basis of different oxygen species peak areas are summarized in
Table 2.
For the pure ZrO
2 support, the singly-charged oxygen vacancies and doubly-charged oxygen vacancies centered at 530.8 (BE
2) and 531.7 eV (BE
3), respectively. The total oxygen vacancies concentration was 24.2%. When introducing Sc
3+ into ZrO
2, the total oxygen vacancies concentration of 7.5ScZr support increased to 32.6%, illustrating that the introduction of Sc
3+ promoted the additional oxygen vacancies generated, which was consistent with the XRD, H
2-TPR and Raman results. It is worth noting that the binding energy of two forms of oxygen vacancies varied at the same time. For 7.5ScZr support, the singly-charged oxygen vacancies and doubly-charged oxygen vacancies centered at 531.3 (BE
2) and 532.2 eV (BE
3). Compared with those of ZrO
2 support, the binding energy of oxygen vacancies on 7.5ScZr support both shifted to higher binding energy, indicating that average charge density of oxygen vacancies decreased and exhibited relatively electron-deficient properties. This is probably because the promoted oxygen vacancies possessed lower charge density, thus the average charge density detected and analyzed by XPS showed a decreased trend. The theoretical calculation results of Cadi-Essadek et al. showed that [
37] nickel will preferentially adsorb on the electron-deficient ZrO
2 surface and had strong interact with it. Combined with the calculation results of Puigdollers [
29] and Tosoni et al. [
35] mentioned above and the H
2-TPR and Raman results in this study, it can be well understood why nickel species have different degrees of interaction with ZrO
2-based supports which were doped with different scandium amounts. As the Sc
2O
3 content increased, the concentration of relatively electron-deficient oxygen vacancies and low-coordinated oxygen ions increased. The nickel species are more preferentially adsorb on surface relatively electron-deficient oxygen vacancies and low-coordinated oxygen ions and interact with them strongly. Therefore, the interaction between nickel species and ZrO
2-based supports enhanced gradually with the Sc
2O
3 content increasing.
From the further investigation of the surface oxygen vacancy properties of ZrO2- and 7.5ScZr-supported nickel catalysts, it can be found that the oxygen vacancy concentration and their electronic properties showed the different changes after ZrO2 and 7.5ScZr supports loading nickel. For pure ZrO2 support, the total oxygen vacancies concentration increased from 24.2% to 26.8% after its loading with nickel. This suggest that the interaction between nickel species and ZrO2 support promoted the additional oxygen vacancies generated, which is consistent with Raman result. Furthermore, the average charge density of oxygen vacancies on Ni/ZrO2 catalyst decreased (the singly-charged oxygen vacancies and doubly-charged oxygen vacancies shifted from 530.8, 531.7 eV to 531.1, 532.0 eV). This is similar with the decrease of average charge density of ZrO2 support with introducing Sc3+, and suggest that the oxygen vacancies promoted by the interaction between nickel species and ZrO2 support possessed lower charge density and exhibited relatively electron-deficient properties.
On the contrary, after nickel loaded on the 7.5ScZr support, the total oxygen vacancies concentration decreased significantly, from 32.6% to 29.2%. Moreover, the singly-charged oxygen vacancies and doubly-charged oxygen vacancies shifted from 531.3, 532.2 eV to 530.9, 531.8 eV, the average charge density of oxygen vacancies increased. This is consistent with our previous study [
17], the strong interaction between nickel species and ZrO
2-based support would lead to a small amount of nickel species incorporation into the tetragonal ZrO
2 lattice or oxygen vacancies and form the Ni–O–Zr structure. Thus a decrease of the oxygen vacancies concentration was observed. Also in the Ni–O–Zr structure, the Ni was electron-donating, the oxygen vacancies bore extra charges for the charge balance [
37], therefore the average charge density of oxygen vacancies on Ni/7.5ScZr catalyst increased and the oxygen vacancies exhibited relatively electron-rich properties.
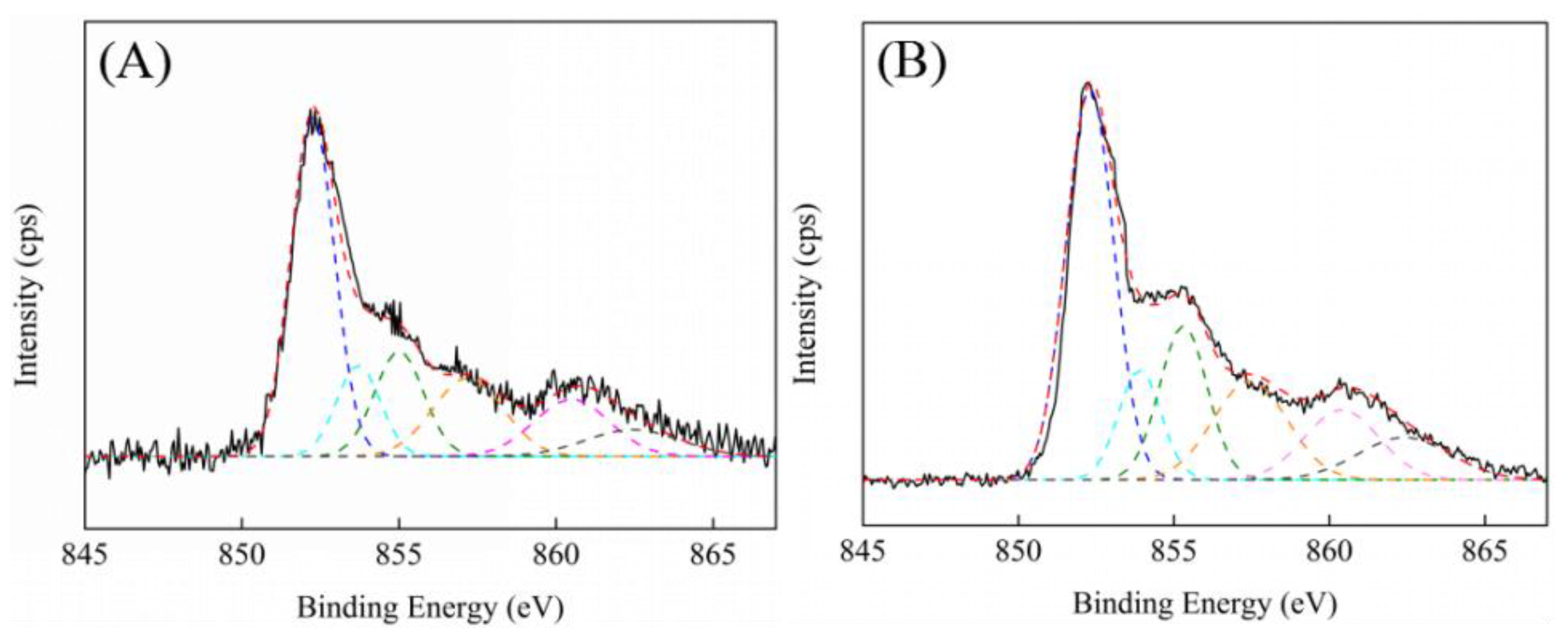
Figure 7 showed the Ni 2p XPS spectra of the Ni/ZrO
2 and Ni/7.5ScZr catalysts. The details of the binding energy and the surface atomic concentration which was calculated on the basis of different nickel species peak areas, are summarized in
Table 3. It can be seen that the nickel species mainly existed in the form of Ni
0 for both Ni/ZrO
2 and Ni/7.5ScZr catalysts. Furthermore, the binding energy of Ni
0 on two catalysts were same, both at 852.2 eV, indicating that the surface properties of Ni
0 were similar.
Except for Ni
0, there were two other nickel species that existed in Ni/ZrO
2 and Ni/7.5ScZr catalysts. However, their corresponding binding energy on two catalysts were different. For the Ni/ZrO
2 catalyst, the peak at 853.7 eV was ascribed to the Ni
2+ in the form of NiO, while the peak at 855.0 eV was assigned to the Ni
2+ in the form of oxide or hydroxide phases [
38]. For the Ni/7.5ScZr catalyst, the binding energy of two nickel species was higher, centered at 853.9 and 855.3 eV, respectively. Cadi-Essadek et al. [
37] systematically studied the electron structure of Ni atom over the heteroatom doped ZrO
2 surface using spin polarized density functional theory (DFT) and identified charge-transfer between Ni and the heteroatom-doped ZrO
2 surface by using Bader analysis, the density of states (DOS), and the electron-density difference calculation, the result showed that after the Ni adsorption on the surface, the oxidation of metallic Ni occurred and the Ni transfer electron to the surface. For the surface with the promoted oxygen vacancies, the electronic cloud is less located, and the properties of metal Ni for use as an electron donor were enhanced. This calculation result is consistent with our experimental result, and further confirmed the stronger interaction between nickel species and 7.5ScZr support would change the electronic structure at the interface where nickel and ZrO
2-based close contact. Furthermore, the higher binding energy of the two nickel species in the Ni/7.5ScZr catalyst is consistent with the lower binding energy of their oxygen vacancies.
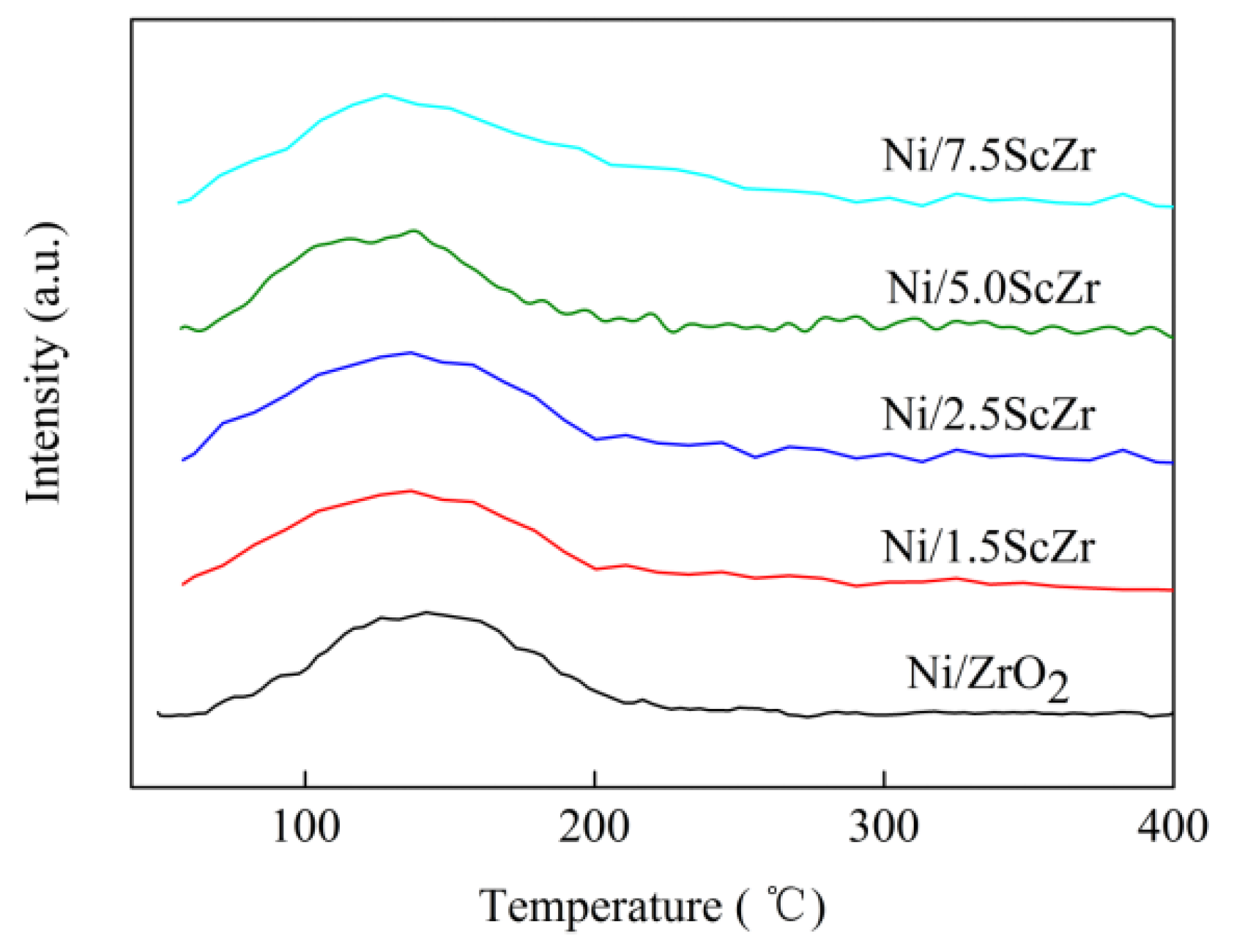
2.6. H2 Temperature-Programmed Desorption (H2-TPD)
H
2 temperature-programmed desorption (H
2-TPD) was conducted to examine the ability of the catalysts in activating hydrogen. As shown in
Figure 8, all of the Ni/xScZr catalysts exhibited one hydrogen desorption peak in the 50 to 250 °C temperature region, which could be assigned to the desorption of active hydrogen from Ni surface [
38]. As previously reported [
30], the different hydrogen desorption temperature indicated the hydrogen had different interaction with Ni or nickel species. This also implied that the different hydrogen desorption temperature originated from the different surface properties of Ni or nickel species. In this study, the hydrogen desorption peak for all of the Ni/xScZr catalysts appeared in the same temperature region, suggesting that the Ni surface properties for all of the Ni/xScZr catalysts were similar. The amounts of desorbed hydrogen calculated from the area of the peaks were also similar, showing that the number of activating hydrogen sites was similar for all of the Ni/xScZr catalysts. This is in line with the assumption that similar Ni crystalline sizes should possess similar accessible catalytic active sites.
2.7. In Situ FT-IR Spectra
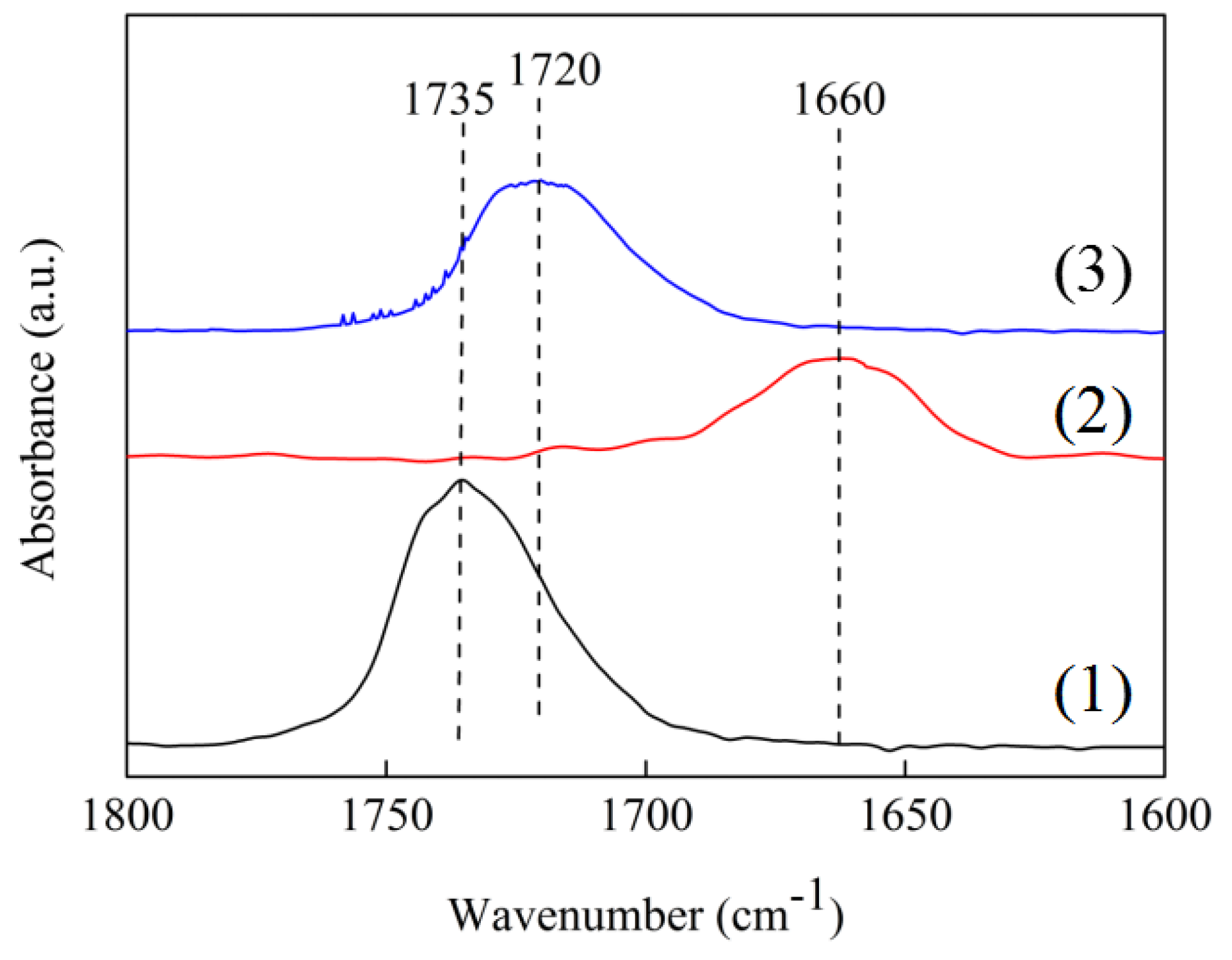
In situ FT-IR using cyclohexanone as probe molecule was carried out to investigate the activating C=O ability of the Ni/ZrO
2 and Ni/7.5ScZr catalysts. As shown in
Figure 9, the peak centered at 1735 cm
−1 was ascribed to the C=O stretching vibration of pure cyclohexanone. In comparison with the peak of pure cyclohexanone, a red-shift of the C=O stretching vibration peaks could be found over the Ni/ZrO
2 and Ni/7.5ScZr catalysts. The C=O stretching vibration for the Ni/ZrO
2 catalyst centered at 1660 cm
−1, while that was located at 1720 cm
−1 for the Ni/7.5SrZr catalyst. The much larger shift of the C=O stretching vibration peak for the Ni/ZrO
2 (75 cm
−1), indicated that the C=O bonds had been weakened and were activated on the surface of the Ni/ZrO
2 catalyst [
39]. The C=O stretching vibration peak on Ni/7.5ScZr catalyst showed only a slight shift (15 cm
−1) compared with the intrinsic C=O stretching vibration peak of pure cyclohexanone, although the peak area of the Ni/7.5ScZr catalyst was slightly higher than that of the Ni/ZrO
2 catalyst. This suggests that the Ni/7.5ScZr catalyst possessed slightly more adsorbed C=O sites than the Ni/ZrO
2 catalyst, whereas its activation ability towards C=O groups was much weaker than that of the Ni/ZrO
2 catalyst.
3. Discussion
Generally speaking, supported catalyst’s catalytic performance is associated with the active metal site and its support, as well as the metal–support interaction. In terms of the hydrogenation reaction, H
2 could be dissociated into active hydrogen atoms or hydrogen ions on metal surface, and subsequently a hydrogenation process towards the unsaturated functional groups of reactant molecules was performed [
40]. In this view, the catalysts with similar metal crystalline size should possess similar accessible catalytic active sites and show the similar hydrogenation activity. In this study, the surface properties of Ni in Ni/xScZr catalysts with different Sc
2O
3 content were similar (XPS and H
2-TPD results); also the similar Ni crystalline size of Ni/xScZr catalysts possessed the similar active surface area. Therefore, all of the Ni/xScZr catalysts exhibited the similar activation hydrogenation ability (H
2-TPD results). In the MA hydrogenation, all the Ni/xScZr catalysts exhibited the similar C=C bond hydrogenation activity, which is consistent with the understanding given above. However, the C=O hydrogenation activity of ZrO
2-based supported nickel catalysts decreased gradually with the increasing of the Sc
2O
3 content in the ZrO
2 support. This suggests the C=O hydrogenation of the ZrO
2-based supported nickel catalysts is not solely related with the activation hydrogenation ability of the catalysts, other factors also participate in the C=O hydrogenation process and play an important role in determining the C=O hydrogenation activity of the Ni/xScZr catalysts.
In situ FT-IR result suggested that the Ni/ZrO
2 catalyst had strong activation C=O ability. Ni/7.5ScZr catalyst showed more adsorbed C=O sites, yet its activation abilities towards C=O groups were extremely weak. Combined with the C=O hydrogenation activity result of the catalysts in the MA hydrogenation reaction, it can be inferred that the high C=O hydrogenation activity of Ni/ZrO
2 catalyst is ascribed to the effective activation C=O groups of SA molecular; while the Ni/7.5ScZr catalyst could not effectively activate C=O groups of SA molecular, thereby showing almost no C=O hydrogenation activity. In the dimethyl succinate (DMS) hydrogenation study by Hu et al. [
41], the similar redshift occurred in in situ FT-IR after Mn-containing spinel-supported copper catalyst adsorbed dimethyl succinate molecular, combined with the intermediate species analysis of the same Mn-containing spinel-supported copper catalyst adsorbing carbon dioxide, the author inferred the active sites in adsorbing and activating the C=O groups of dimethyl succinate molecular were the catalyst’s surface oxygen vacancies. This suggest the surface oxygen vacancies of the catalyst promote the C=O hydrogenation through activating C=O groups. Similarly, Zhou et al. [
42] and Manyar et al. [
43] found that the surface oxygen vacancies on CeO
2-supported nickel catalyst and TiO
2-supported Pt catalyst contributed to the C=O hydrogenation in CO
2 methanation and carboxylic acids’ hydrogenation reaction. Further, in our recently study [
17], it was found that not all the surface oxygen vacancies could activate the C=O groups of the SA molecules and promote the hydrogenation of SA to GBL for the Ni/ZrO
2 catalyst system. Only relatively electron-deficient oxygen vacancies could effectively activate the C=O groups of the SA molecular, and thereby hydrogenated the C=O groups with the synergy of the neighboring Ni
0. In this study, the XPS result suggested the surface oxygen vacancies on Ni/ZrO
2 catalyst showed relatively electron-deficient properties, while the surface oxygen vacancies on Ni/7.5ScZr catalyst exhibited relatively electron-rich properties. Compared with the relatively electron-rich oxygen vacancies, the relatively electron-deficient oxygen vacancies are more likely to activate the C=O bond by accepting a lone pair of electrons from the oxygen atom of the C=O bonds, thereby weakening the C=O bonds, and the active hydrogen species activated by the neighboring Ni
0 attack the activated C=O bonds, thus complete C=O bond hydrogenation. Therefore, Ni/ZrO
2 catalyst exhibited high C=O hydrogenation activity in MA hydrogenation reaction. Whereas for Ni/7.5ScZr catalyst, though it possessed more surface oxygen vacancies and showed more adsorb C=O groups sites, it exhibited almost no C=O hydrogenation activity in MA hydrogenation reaction because its relatively electron-rich oxygen vacancies could not effectively activate the C=O group of the SA molecular. This is consistent with our previous study.
In this study, the more important finding is that there is an interdependence between the surface structure of ZrO
2-based support and the C=O hydrogenation performance of the ZrO
2-based supported nickel catalysts. By controlling the Sc
2O
3 content in the ZrO
2-based support, the surface structures of ZrO
2-based support could be regulated effectively. The different surface structure of ZrO
2-based supports resulted in the different degree of interaction between the nickel species and ZrO
2-based supports; furthermore, the different interaction led to the different surface oxygen vacancies electron properties of ZrO
2-based supported nickel catalysts and the C=O hydrogenation activity of the catalysts. For pure monoclinic ZrO
2 support, there were more relatively electron-rich oxygen vacancies and stable oxygen ions on its surface (H
2-TPR, XPS). Nickel species had weaker interaction with pure monoclinic ZrO
2 support and the interaction promoted the additional relatively electron-deficient oxygen vacancies generated on surface of Ni/ZrO
2 catalyst (H
2-TPR, Raman and XPS). Therefore, the synergism between the surface relatively electron-deficient oxygen vacancies and Ni
0 catalyzes C=O hydrogenation [
17], in which the contribution of the surface relatively electron-deficient oxygen vacancies on Ni/ZrO
2 catalyst is to effectively activate the C=O groups of the SA molecular (in situ FT-IR). When a dopant Sc
3+ was introduced into ZrO
2, relatively electron-deficient oxygen vacancies and low coordination oxygen ions were promoted dut to the charge balance. Also, as the Sc
2O
3 content increased, the concentration of relatively electron-deficient oxygen vacancies and low coordination oxygen ions on surface of ZrO
2-based supports increased (H
2-TPR, Raman, and XPS) and, correspondingly, the ZrO
2-based supports structure transformed from monoclinic phase structure to tetragonal phase structure and then to metastable tetragonal phase structure (XRD and Raman). Compared with the relatively electron-rich oxygen vacancies and stable oxygen ions, nickel species preferentially adsorb on the relatively electron-deficient oxygen vacancies and low coordination oxygen ions and interact strongly with them. Therefore, the interaction between nickel species and ZrO
2-based supports enhanced gradually with the Sc
2O
3 content increasing (H
2-TPR). The strong interaction would lead to a small amount of nickel species entering into the tetragonal ZrO
2 lattice or oxygen vacancies, and thereby forming the Ni–O–Zr structure [
17], thus the number of oxygen vacancies decreased compared with their corresponding support (Raman and XPS). In the Ni–O–Zr structure, or the interface where nickel species interact strongly with ZrO
2-based supports, the strong interaction enhances the electron-donating property of nickel [
37], the oxygen vacancies bore extra charges for the charge balance, thereby resulting in the increase of the average charge density of surface oxygen vacancies. Thus the number of the active sites in activating C=O bonds decreased and the ability in activating C=O bonds became weaker as the Sc
2O
3 content increased. When the Sc
2O
3 content in the ZrO
2 support was increased to 7.5 mol%, the surface oxygen vacancies exhibited extremely weak ability in activating C=O groups, and could not effectively activate the C=O groups of the SA molecular, therefore the Ni/7.5ScZr catalyst exhibited almost no C=O hydrogenation activity in MA hydrogenation reaction.
Based on the above results, it can be concluded that there is an interdependence between the surface structure of the ZrO2-based support and the C=O hydrogenation activity of the ZrO2-based supported nickel catalysts. Through regulating the surface structure of ZrO2-based support, the C=O hydrogenation activity of ZrO2-based supported nickel catalysts can be modulated, thereby, the product distribution in the MA hydrogenation reaction could be effectively manipulated. With the Ni/ZrO2 catalyst, the GBL product could be selectively obtained; while with Ni/7.5ScZr catalyst, the SA product could be selectively acquired.
4. Materials and Methods
4.1. Catalysts Preparation
4.1.1. Preparation of Sc-doped ZrO2 Supports
Sc-doped ZrO
2 supports were prepared with the adoption of the hydrothermal method [
44]. They were synthesized at the temperature of 140 °C for 14 h in a Teflon-lined, stainless steel autoclave (100 mL). For the autoclave containing solutions (80 mL), the molar ratio of ZrO(NO
3)
2·2H
2O (Beijing Chemicals, Beijing, China) to Sc(NO
3)
3 (Xiya Reagent, Linyi, China) was (1−x):2x, the total cation concentration was 0.2 M, and the molar ratio of urea to total cation was 10:1. The precipitates were washed with absolute alcohol and the washing stopped when their pH value reached 7. Then, the precipitates were dried at 110 °C for 12 h before they were calcined at 400 °C for 4 h. The above x denotes the Sc
2O
3/(Sc
2O
3 + ZrO
2) molar ratio. In this work, x = 0, 1.5, 2.5, 5.0, and 7.5% (mol/mol), and the corresponding Sc-doped ZrO
2 supports were noted as ZrO
2, 1.5ScZr, 2.5ScZr, 5.0ScZr, and 7.5ScZr, respectively.
4.1.2. Preparation of Ni/Sc-doped ZrO2 Catalysts
The NiO/Sc-doped ZrO2 samples were prepared with the use of a conventional impregnation method. One gram of xScZr support was poured into an aqueous solution which contained 0.5476 g of nickel nitrate and 2.2 mL H2O. Then, the solution was stirred vigorously. After that, the sample was dried at 120 °C for 12 h. Then, it was calcined in air at 450 °C for 3 h, and the samples were denoted as NiO/xScZr. After that, the samples were reduced at 400 °C for 3 h in an H2 flow (30 mL/min), denoted as Ni/xScZr.
4.2. Structure Characterizations
The specific surface areas of the xScZr supports and the Ni/xScZr catalysts were measured with the use of an ASAP-2020 instrument (Micromeritics, Atlanta, GA, USA) at −196 °C.
XRD was performed with the use of an X-ray diffractometer (Bruker D8 Advance, Karlsruhe, Germany). Cu Kα radiation (λ = 1.54056 Å) was used and the operating voltage and current were set at 40 kV and 40 mA, respectively.
Laser Raman spectra were gained with the use of a Lab RAM HR Evolution Raman microscope (Horiba Scientific, Paris, France) which used Ar+ (532 nm) as its excitation source.
H2 temperature-programmed reduction (H2-TPR) was then carried out. A Micromeritics Auto Chem II 2920 (Atlanta, GA, USA), which had a thermal conductivity detector, was used to determine catalyst reducibility. The followings procedures were carried out. First, 30 mg of the NiO/xScZr sample were treated in Ar at 300 °C for 1 h. Then, the sample was cooled to 50 °C. The H2-TPR profiles were recorded when the samples in H2/Ar (10% v/v) with 50 mL/min of gas flow were being heated with the temperature ranging from 50 °C to 700 °C at a ramp of 10 °C /min.
H2 temperature-programmed desorption (H2-TPD) measurements were done on the apparatus that had been used for the H2-TPR (Micromeritics Auto Chem II 2920, Atlanta, GA, USA). The following procedures were carried out. First, a 100 mg NiO/xScZr sample was reduced in situ at 400 °C for 3 h in pure H2. Then, the sample was cooled until the temperature reached 50 °C. The sample was then purged with Ar. The purging lasted for 1 h at the temperature of 50 °C. The reason for carrying it out in this way lies in the purpose of removing excess hydrogen. Then, the hydrogen was adsorbed on the surface and H2/Ar (10% v/v) was injected at 50 mL/min until specific saturation was gained. Ar was used to flush the sample until the baseline was stable. H2-TPD profiles were recorded up to 700 °C at a heating rate of 10 °C /min.
X-ray photoelectron spectroscopy (XPS) measurements were adopted with the use of a Kratos AXIS Ultra DLD spectrometer (Manchester, UK) with a monochromatic Al Kα (1486.6 eV) irradiation source. The X-ray gun was operated at 150 W. The survey spectra were recorded, the pass energy was set at 160 eV, and the high-resolution spectra were recorded with pass energy of 40 eV. The area of the sampling area was 300 × 700 μm2. The binding energy was corrected by setting the C1s peak at 284.6 eV. In terms of the xScZr samples’ test, these samples were placed into an XPS sample cell. Before the spectra were recorded, the XPS sample cell was pumped down to 10−8 Pa. In terms of Ni/xScZr samples’ test, the NiO/xScZr samples were first put in an XPS sample cell. Then, the samples were reduced at 400 °C for 3 h. After that, it was cooled down to the room temperature in H2 flow (30 mL/min). The sample cell was subsequently pumped down to 10−8 Pa. After that, the spectra were recorded.
In-situ FT-IR spectra of cyclohexanone were collected on a spectrometer (Nicolet IS 50, Thermo Fisher Scientific, Waltham, MA, USA). 0.02 g NiO/xScZr sample was put into an IR cell. Before the adsorption of cyclohexanone, the sample was reduced at 400 °C for 3 h in H2 flow (30 mL/min). The sample was then cooled down until the temperature reached 210 °C. The IR cell that contained samples was pumped down to <6 × 10−3 Pa. What should be mentioned is that a spectrum was recorded as the background. After that, gas cyclohexanone molecules were put into the IR cell for adsorption. The process lasted for 60 min. The cell was then desorbed via vacuum pumping down to <6 × 10−3 Pa. The spectra were recorded and the resolution was 2 cm−1.
4.3. Catalytic Activity Tests
Ni/xScZr catalysts’ catalytic performances in the hydrogenation of MA were recorded in a batch reactor (100 mL). The reaction conditions of maleic anhydride hydrogenation are as follows; the reaction temperature was 210 °C, the pressure of H2 was 5 MPa, and the agitator operated at 400 rpm. Prior to the test, some procedures were carried out. The catalysts were prereduced with a stream of H2 (30 mL/min) in a quartz tube for 3 h and the temperature was set at 400 °C. Then, they were cooled until their temperature reached the room one. At the same time, the MA (4.9 g) and THF (the purity of THF ≥ 99.99%, H2O ≤ 20 ppm) were charged into the autoclave. After that, the reduced catalyst (0.3 g) (40–60 mesh) was charged into the autoclave under N2 protection.
Solvents including 1,4-dioxane and cyclohexane were also under investigation. According to the results, the hydrogenation products were SA and GBL. THF or other products were not detected. The carbon balance ranged from 95 to 105%. When THF was adopted as the solvent, the products, SA, and GBL were detected. The carbon balance was calculated based on the totality of the SA and GBL, ranging from 95 to 105%. This means that when THF was used as the solvent for the present Ni/ZrO2 catalysts system, products such as THF or BDO that were obtained through deeply hydrogenation were not produced.
The samples obtained from the reactor went through analysis with the use of a gas chromatograph (GC, Agilent, 7890B) equipped with a DB-5 capillary column and an FID detector. The conversion of MA and the selectivity of the products were calculated with the use of the following equations:
where
MAin,
MAout and
Producti,out represent the molar concentration of inlet reactant, outlet reactant and outlet products
i, respectively.
5. Conclusions
In summary, this work found the interdependence between the surface structure of ZrO2-based support and the selective hydrogenation performance of ZrO2-supported nickel catalysts, presented an effective strategy for manipulating the selective hydrogenation performance of the catalyst in catalyzing MA to SA or GBL. Pure monoclinic ZrO2 support, with relatively electron-rich oxygen vacancies and stable oxygen ions on surface, had weaker interaction with the nickel species, also the interaction promoted the additional relatively electron-deficient oxygen vacancies generated on surface of Ni/ZrO2 catalyst. The relatively electron-deficient oxygen vacancies could activate the C=O group effectively, thereby C=O group could be hydrogenated with the synergy of neighboring Ni0. Therefore, in the MA hydrogenation reaction, the Ni/ZrO2 catalyst exhibited high C=O hydrogenation activity and gave a high yield (65.8%) of GBL. However, when introducing Sc3+ into ZrO2, the surface structure of the ZrO2-based support changed. Furthermore, the degree of interaction between the nickel species and the ZrO2-based support, the electronic properties of the oxygen vacancies on surface of the Ni/xScZr catalyst and its C=O hydrogenation performance, have changed subsequently. As the Sc2O3 content in the ZrO2 support increased, the concentration of relatively electron-deficient oxygen vacancies and low coordination oxygen ions increased on surface of xScZr supports, and the interaction between nickel species and ZrO2-based support enhanced gradually. The strong interaction resulted in the average charge density of surface oxygen vacancies on Ni/xScZr catalyst increased, also the number of oxygen vacancies decreased, thereby the ability in activating C=O bonds became weaker and the active site numbers in activating C=O bonds decreased, correspondingly the C=O hydrogenation activity of the catalyst decreased gradually. Therefore, in the MA hydrogenation reaction, the yield of GBL decreased gradually with the increasing of the Sc2O3 content. When the Sc2O3 content was increased to 7.5 mol%, the Ni/7.5ScZr catalyst exhibited almost no C=O hydrogenation activity and the yield of GBL was only 2.4%. Correspondingly, we obtained the high yield of SA product upon Ni/7.5ScZr catalyst. The results of this study provide a new idea for the design and preparation of high-efficiency MA selective hydrogenation catalysts, and also provide reference for designing selective hydrogenation catalysts for other unsaturated carbonyl compounds.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}