Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides




Abstract
:1. Introduction
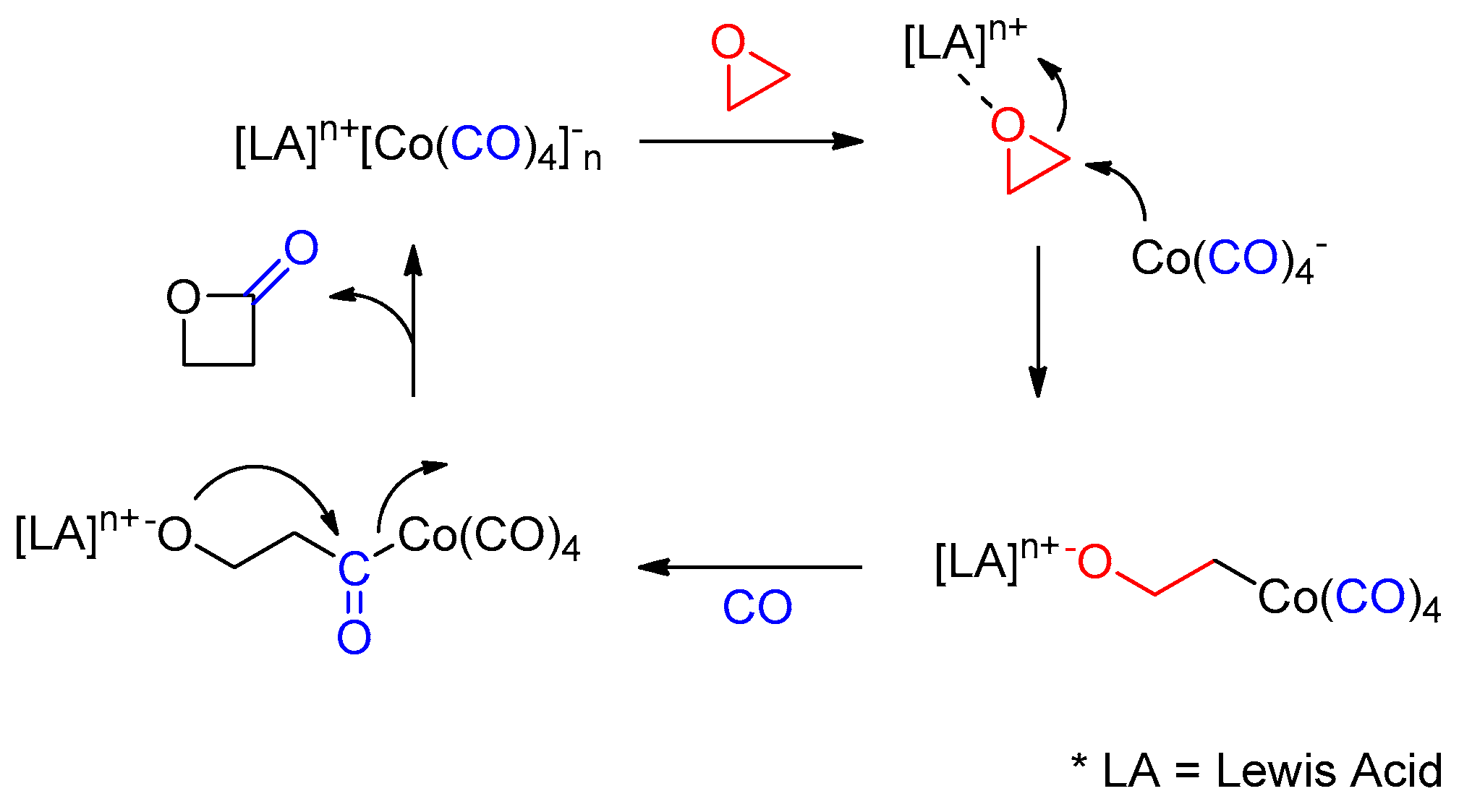
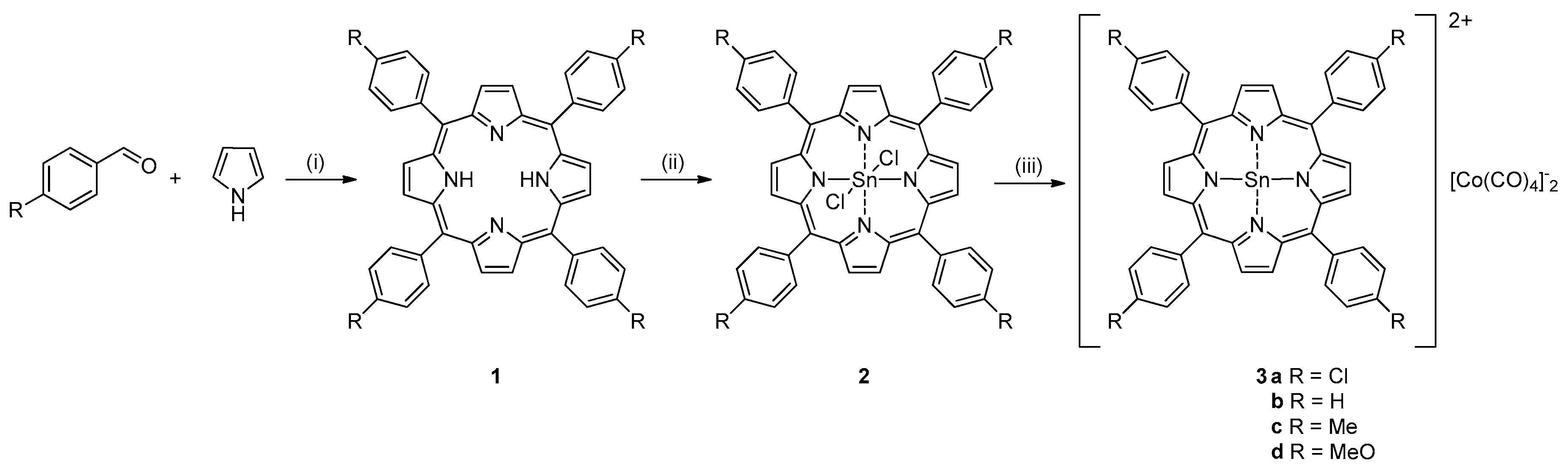
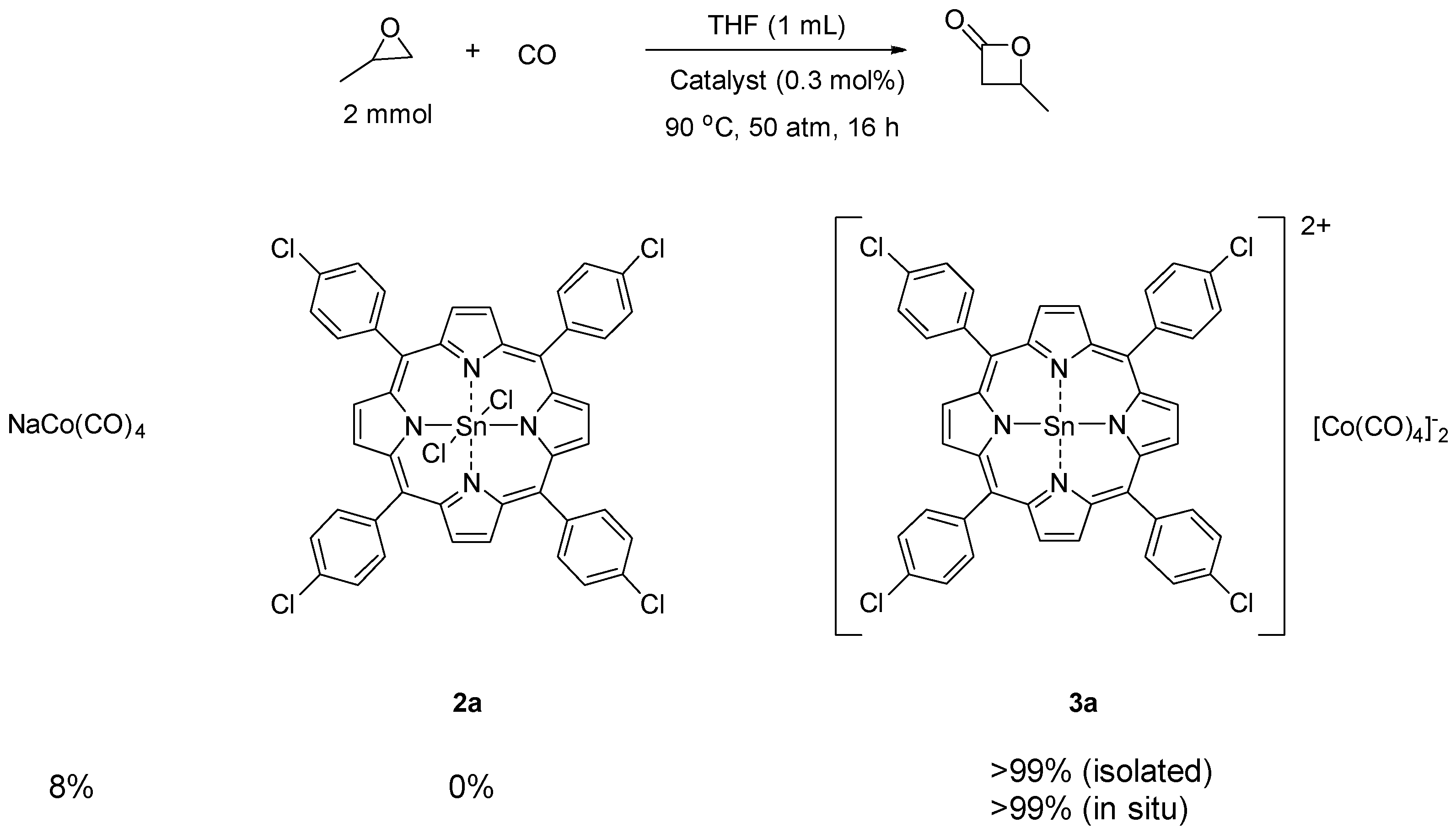
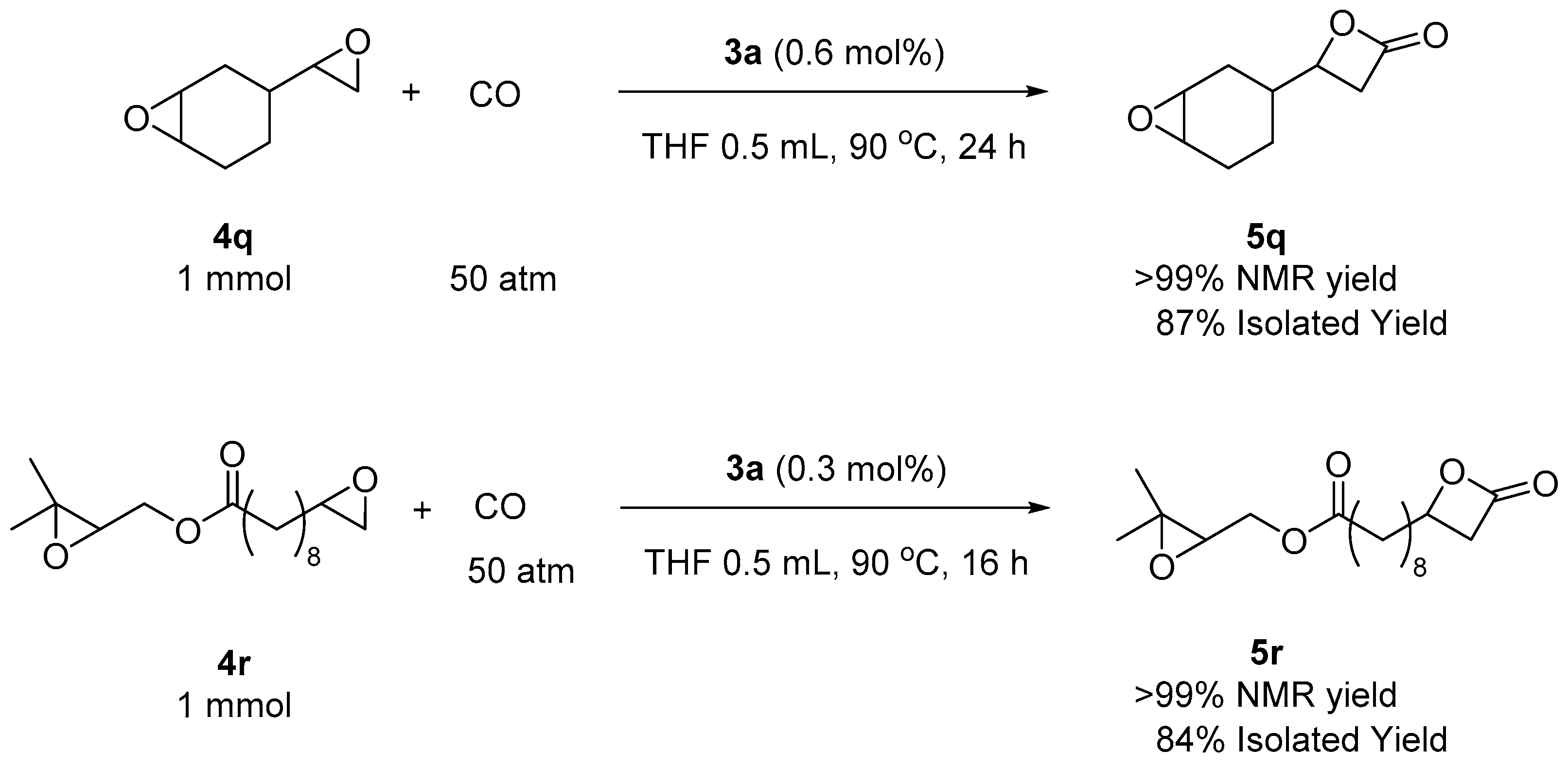
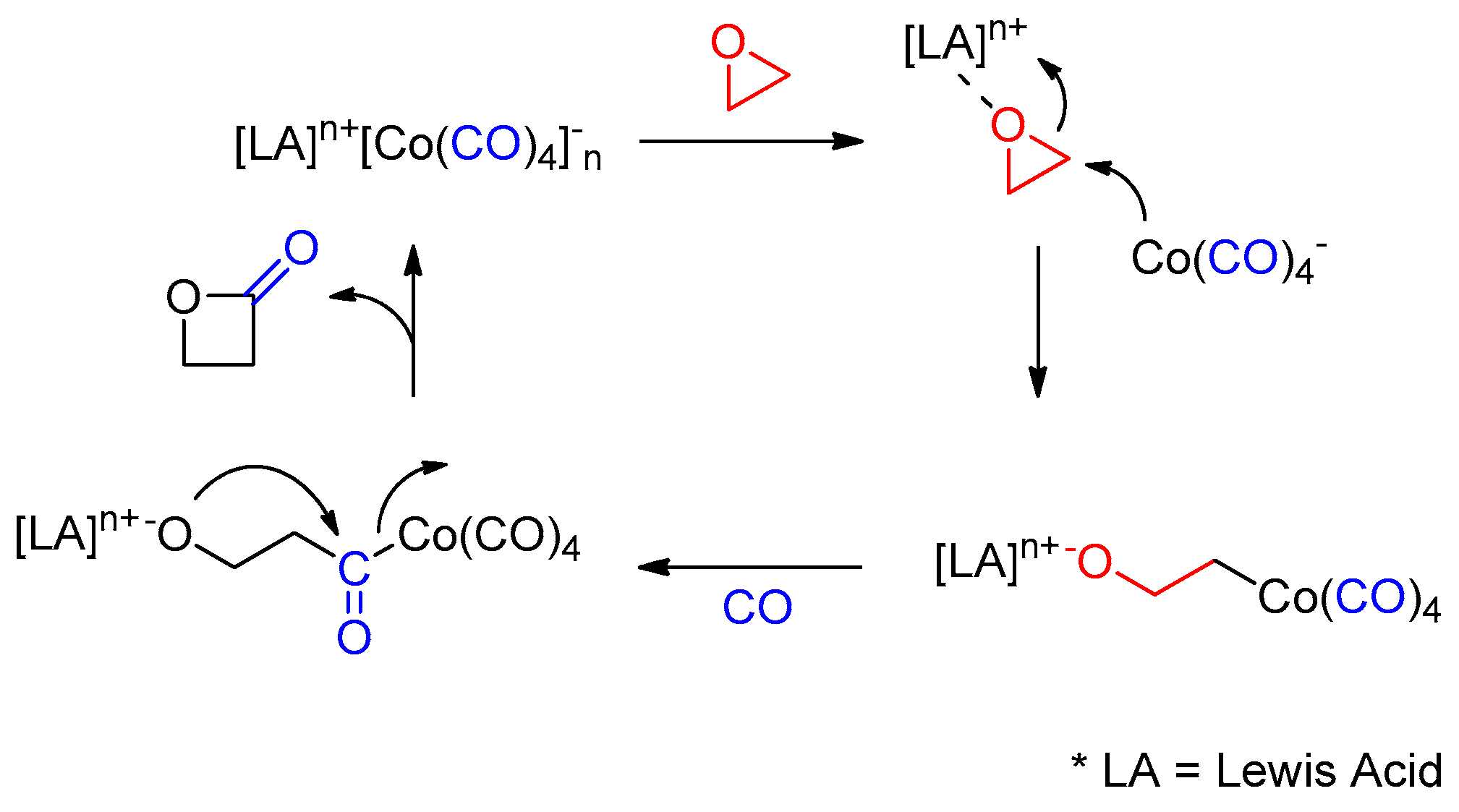
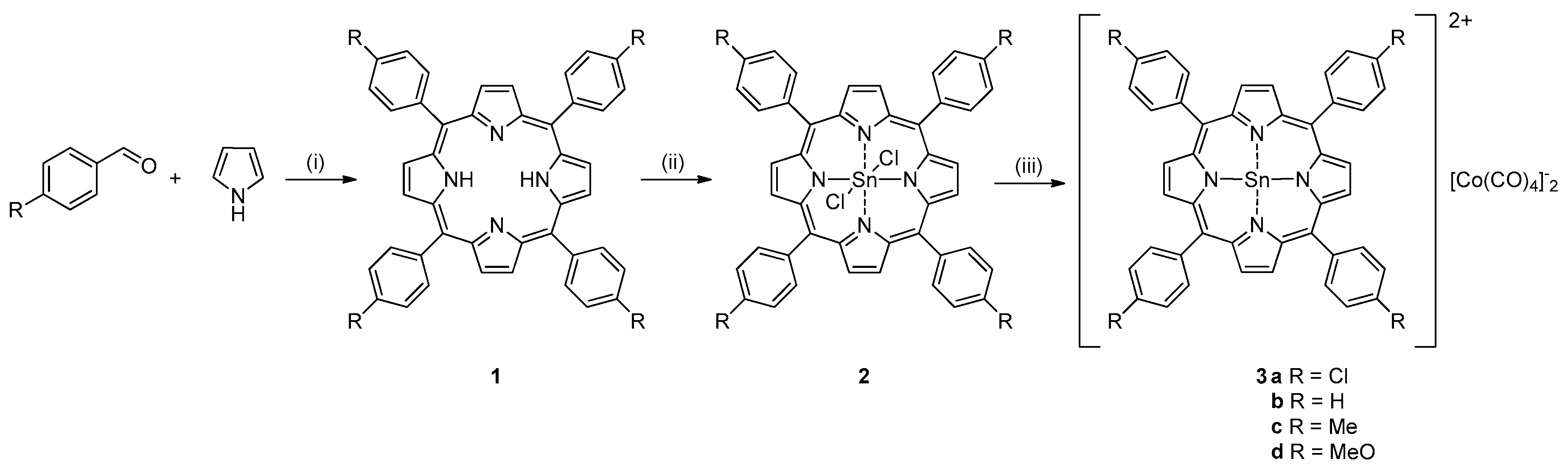
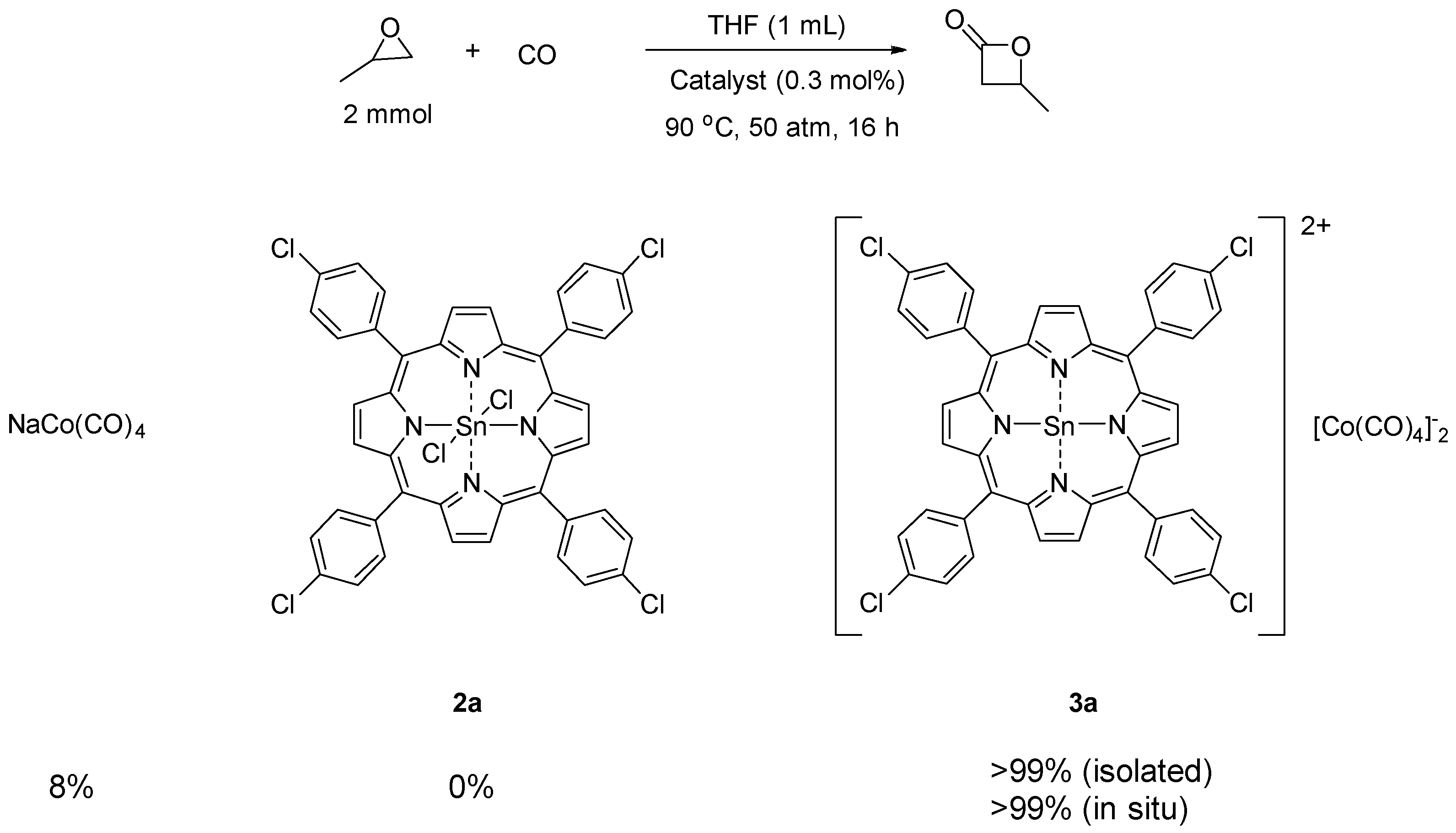
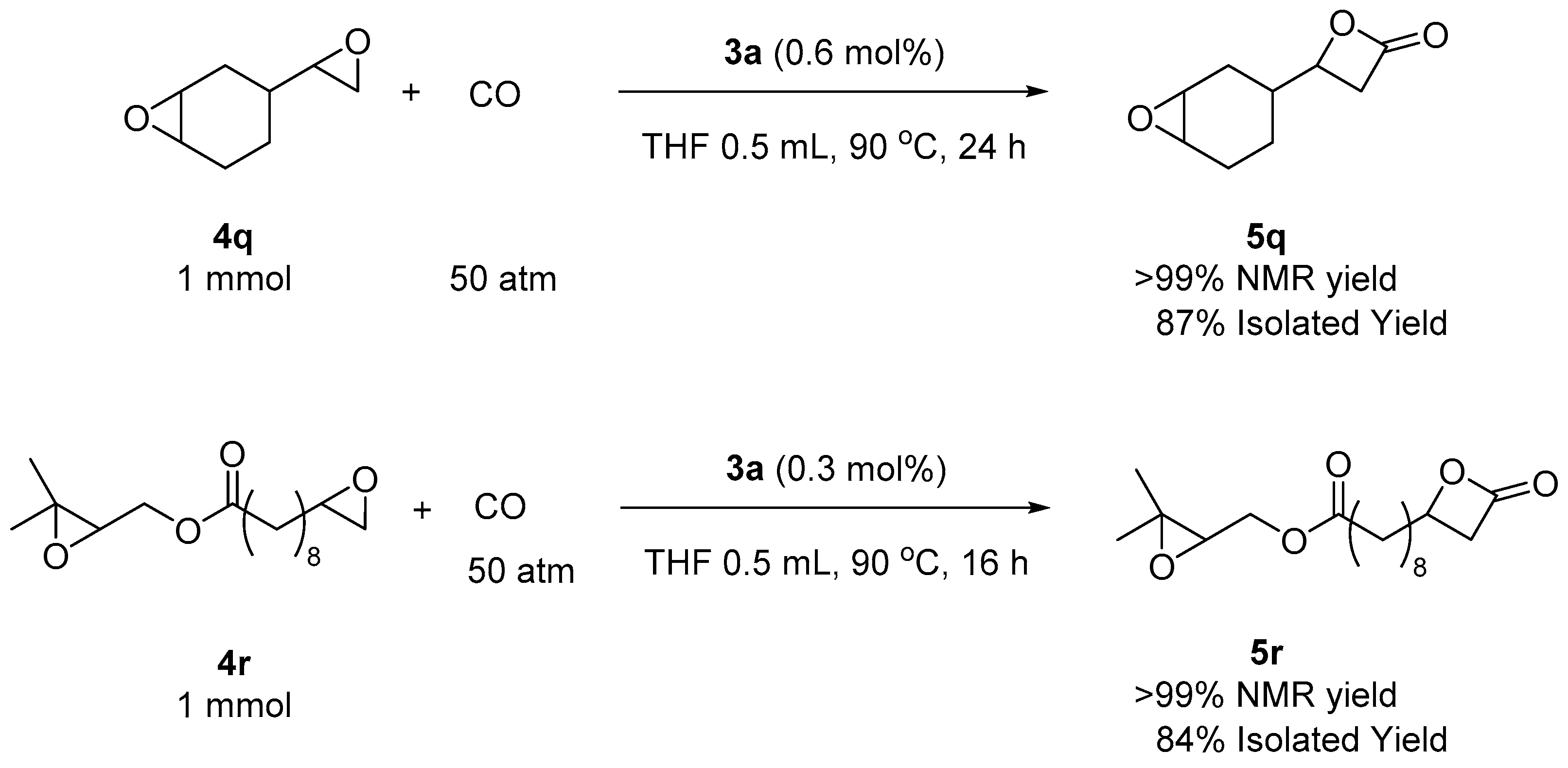
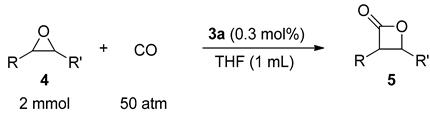
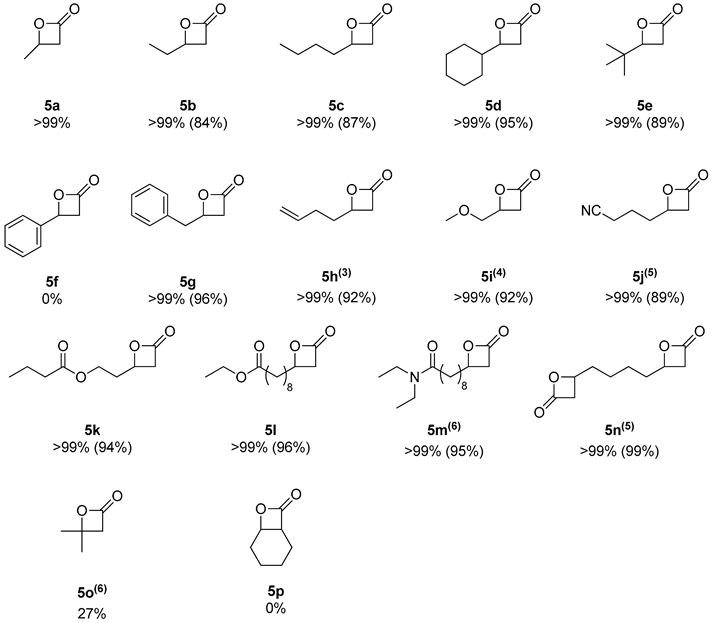
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Syntheses of Substrates
3.3. Catalyst Synthesis
3.4. General Procedure for the Carbonylation of Epoxides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kadish, K.; Smith, K.M.; Guilard, R. The Porphyrin Handbook: Chlorophylls and Bilins: Biosynthesis, Synthesis and Degradation; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Reddy, K.R.; Jiang, J.; Krayer, M.; Harris, M.A.; Springer, J.W.; Yang, E.; Jiao, J.; Niedzwiedzki, D.M.; Pandithavidana, D.; Parkes-Loach, P.S.; et al. Palette of lipophilic bioconjugatable bacteriochlorins for construction of biohybrid light-harvesting architectures. Chem. Sci. 2013, 4, 2036–2053. [Google Scholar] [CrossRef]
- Croce, R.; van Amerongen, H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Bio. 2014, 10, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Feher, G.; Allen, J.P.; Okamura, M.Y.; Rees, D.C. Structure and function of bacterial photosynthetic reaction centres. Nature 1989, 339, 111–116. [Google Scholar] [CrossRef]
- Sessler, J.L.; Seidel, D. Synthetic Expanded Porphyrin Chemistry. Angew. Chem. Int. Ed. 2003, 42, 5134–5175. [Google Scholar] [CrossRef]
- Saito, S.; Osuka, A. Expanded porphyrins: Intriguing Structures, Electronic Properties, and Reactivities. Angew. Chem. Int. Ed. 2011, 50, 4342–4373. [Google Scholar] [CrossRef]
- Jiang, L.; Engle, J.T.; Sirk, L.; Hartley, C.S.; Ziegler, C.J.; Wang, H. Triphenylene-Fused Porphyrins. Org. Lett. 2011, 13, 3020–3023. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Furuta, H.; Yoza, K.; Igarashi, S.; Osuka, A. meso-Aryl-Substituted Expanded Porphyrins. J. Am. Chem. Soc. 2001, 123, 7190–7191. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takeuchi, Y.; Matsuda, A. meso-Aryl Subporphyrins. Angew. Chem. Int. Ed. 2007, 46, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Szyszko, B.; Latos-Grażyński, L. Core Chemistry and Skeletal Rearrangements of Porphyrinoids and Metalloporphyrinoids. Chem. Soc. Rev. 2015, 44, 3588–3616. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.J.; Latos-Grażyński, L. Core Modified Porphyrins—A Macrocyclic Platform for Organometallic Chemistry. Coord. Chem. Rev. 2005, 249, 2510–2533. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.; Brocksom, T.J.; de Oliveira, K.T. Tetrabenzoporphyrins: Synthetic Developments and Applications. Chem. Soc. Rev. 2013, 42, 3302–3317. [Google Scholar] [CrossRef]
- Roznyatovskiy, V.V.; Lee, C.-H.; Sessler, J.L. π-Extended Isomeric and Expanded Porphyrins. Chem. Soc. Rev. 2013, 42, 1921–1933. [Google Scholar] [CrossRef]
- Davis, N.K.S.; Thompson, A.L.; Anderson, H.L. A Porphyrin Fused to Four Anthracenes. J. Am. Chem. Soc. 2011, 133, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Z.; Zhang, X.; Wang, Y.; Tian, J.; Huang, Y.; Ma, Y.; Zhang, X.; Chen, Y. A Graphene Hybrid Material Covalently Functionalized with Porphyrin: Synthesis and Optical Limiting Property. Adv. Mater. 2009, 21, 1275–1279. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Wathier, M.; Grinstaff, M.W. Synthesis and Properties of Supramolecular Ionic Networks. J. Am. Chem. Soc. 2008, 130, 9648–9649. [Google Scholar] [CrossRef]
- Drain, C.M.; Varotto, A.; Radivojevic, I. Self-Organized Porphyrinic Materials. Chem. Rev. 2009, 109, 1630–1658. [Google Scholar] [CrossRef]
- Vlascici, D.; Cosma, E.F.; Pica, E.M.; Cosma, V.; Bizerea, O.; Mihailescu, G.; Olenic, L. Free Base Porphyrins as Ionophores for Heavy Metal Sensors. Sensors 2008, 8, 4995–5004. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef]
- Endo, M.; Fujitsuka, M.; Majima, T. Diastereochemically Controlled Porphyrin Dimer Formation on a DNA Duplex Scaffold. J. Org. Chem. 2008, 73, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Takanami, T.; Hayashi, M.; Iso, K.; Nakamoto, H.; Suda, K. Highly Regioselective [3,3] Rearrangement of Aliphatic Allyl Vinyl Ethers Catalyzed by a Metalloporphyrin Complex, Cr (TPP) Cl. Tetrahedron 2006, 62, 9467–9474. [Google Scholar] [CrossRef]
- Brothers, P.J. Organoelement Chemistry of Main-Group Porphyrin Complexes. Adv. Organometal. Chem. 2001, 48, 289–342. [Google Scholar]
- Brothers, P.J. Recent developments in the coordination chemistry of porphyrin complexes containing non-metallic and semi-metallic elements. J. Porphyrins Phthalocyanines 2002, 6, 259–267. [Google Scholar] [CrossRef]
- Enakieva, Y.Y.; Volostnykh, M.V.; Nefedov, S.E.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y.; Bessmertnykh-Lemeune, A.G.; Stern, C.; Guilard, R. Gallium(III) and Indium(III) Complexes with meso-Monophosphorylated Porphyrins: Synthesis and Structure. A First Example of Dimers Formed by the Self-Assembly of meso-Porphyrinylphosphonic Acid Monoester. Inorg. Chem. 2017, 56, 3055–3070. [Google Scholar] [CrossRef]
- Chatterjee, C.; Chisholm, M.H. The Influence of the Metal (Al, Cr, and Co) and the Substituents of the Porphyrin in Controlling the Reactions Involved in the Copolymerization of Propylene Oxide and Carbon Dioxide by Porphyrin Metal(III) Complexes. 1. Aluminum Chemistry. Inorg. Chem. 2011, 50, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.; Menotti, A.R. Porphyrin Studies. V. The Metal Complex Salts of α,β,γ,δ-Tetraphenylporphine. J. Am. Chem. Soc. 1948, 70, 1808–1812. [Google Scholar] [CrossRef] [PubMed]
- Rafiemanzelat, F.; Abdollahi, E.; Moghadam, M.; Mirkhani, V.; Tangestaninejad, S.; Mohammadpoor-Baltork, I. Application of Tin(IV) Porphyrin Complexes as Novel catalysts for the Synthesis of New Copolyurethanes with Cyclopeptide Moiety. J. Appl. Polym. Sci. 2012, 124, 638–646. [Google Scholar] [CrossRef]
- Kumar, A.A.; Giribabu, L.; Reddy, D.R.; Maiya, B.G. New Molecular Arrays Based on a Tin(IV) Porphyrin Scaffold. Inorg. Chem. 2001, 40, 6757–6766. [Google Scholar] [CrossRef]
- Tangestaninejad, S.; Habibi, M.H.; Mirkhani, V.; Moghadam, M. Rapid and Efficient Acetylation of Alcohols and Phenols with Acetic Anhydride using Tin(IV) Porphyrin as Catalyst. Synth. Commun. 2002, 32, 1337–1343. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpour-Baltork, I.; Shaibani, R. Rapid and Efficient Acetylation of Alcohols and Phenols with Acetic Anhydride Catalyzed by Electron-deficient Tin (IV) Porphyrin. J. Mol. Catal. A: Chem. 2004, 219, 73–78. [Google Scholar] [CrossRef]
- Gharaati, S.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Kosari, F. Highly Efficient and Selective Methoxymethylation of Alcohols and Phenols Catalyzed by High-valent Tin (IV) Porphyrin. Inorg. Chim. Acta 2010, 363, 1995–2000. [Google Scholar] [CrossRef]
- Arnold, D.P.; Blok, J. The coordination chemistry of tin porphyrin complexes. Coord. Chem. Rev. 2004, 248, 299–319. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Taghavi, S.A. High-valent tin(IV) porphyrin, SnIV(TPP)(BF4)2, as an efficient catalyst for the ring-opening of epoxides. Catal. Commun. 2007, 8, 2087–2095. [Google Scholar] [CrossRef]
- Lee, J.T.; Thomas, P.J.; Alper, H. Synthesis of β-Lactones by the Regioselective, Cobalt and Lewis Acid Catalyzed Carbonylation of Simple and Functionalized Epoxides. J. Org. Chem. 2001, 66, 5424–5426. [Google Scholar] [CrossRef]
- Schmidt, J.A.R.; Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. A Readily Synthesized and Highly Active Epoxide Carbonylation Catalyst Based on a Chromium Porphyrin Framework: Expanding the Range of Available β-Lactones. Org. Lett. 2004, 6, 373–376. [Google Scholar] [CrossRef]
- Schmidt, J.A.R.; Lobkovsky, E.B.; Coates, G.W. Chromium(III) Octaethylporphyrinato Tetracarbonylcobaltate: A Highly Active, Selective, and Versatile Catalyst for Epoxide Carbonylation. J. Am. Chem. Soc. 2005, 127, 11426–11435. [Google Scholar] [CrossRef]
- Kramer, J.W.; Lobkovsky, E.B.; Coates, G.W. Practical β-Lactone Synthesis: Epoxide Carbonylation at 1 atm. Org. Lett. 2006, 8, 3709–3712. [Google Scholar] [CrossRef]
- Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. [Lewis Acid]+[Co(CO)4]− Complexes: A Versatile Class of Catalysts for Carbonylative Ring Expansion of Epoxides and Aziridines. Angew. Chem. Int. Ed. 2002, 41, 2781–2784. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A Simplified Synthesis for meso-Tetraphenylporphine. J. Org. Chem 1967, 32, 476. [Google Scholar] [CrossRef]
- Slagt, V.F.; van Leeuwen, P.W.N.M.; Reek, J.N.H. Supramolecular Bidentate Phosphorus Ligands Based on Bis-zinc (II) and Bis-tin (IV) Porphyrin Building Blocks. Dalton Trans. 2007, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Torres, R.; Alvarez, S. Coordinating ability of Anions and Solvents towards Transition Metals and Lanthanides. Dalton Trans. 2011, 40, 10742–10750. [Google Scholar] [CrossRef]
- Rowley, J.M.; Lobkovsky, E.B.; Coates, G.W. Catalytic Double Carbonylation of Epoxides to Succinic Anhydrides: Catalyst Discovery, Reaction Scope, and Mechanism. J. Am. Chem. Soc. 2007, 129, 4948–4960. [Google Scholar] [CrossRef] [PubMed]
- Coates and coworkers’ original development of Cr-porphyrin catalyst exhibited high TON up to 10000 in ref. 43. However, the use of propylene oxide was not presented.
- Edgell, W.F.; Lyford, J., IV. The Preparation of Sodium Cobalt Tetracarbonyl. Inorg. Chem. 1970, 9, 1932–1933. [Google Scholar] [CrossRef]
- Sharma, R.; Bulger, P.G.; McNevin, M.; Dormer, P.G.; Ball, R.G.; Streckfuss, E.; Cuff, J.F.; Yin, J.; Chen, C.-y. A Cascade Approach to Cyclic Aminonitrones: Reaction Discovery, Mechanism, and Scope. Org. Lett. 2009, 11, 3194–3197. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Croft, R.A.; Whittingham, W.G.; Bower, J.F. Modular Access to Substituted Azocanes via a Rhodium-Catalyzed Cycloaddition–Fragmentation Strategy. J. Am. Chem. Soc. 2015, 137, 8054–8057. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.R.; Jung, Y.; Coates, G.W. Meinwald-type Rearrangement of Monosubstituted Epoxides to Methyl Ketones using an [Al porphyrin]+[Co(CO)4]− catalyst. Org. Chem. Front. 2015, 2, 346–349. [Google Scholar] [CrossRef]
- Taber, D.F.; Paquette, C.M.; Gu, P.; Tian, W. Cyclohexanones by Rh-Mediated Intramolecular C–H Insertion. J. Org. Chem. 2013, 78, 9772–9780. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.; Wang, Z.; Jiao, L.; Wang, S. “Click” Tetradentate Ligands. Dalton Trans. 2010, 39, 2660–2666. [Google Scholar] [CrossRef]
- Arnold, D.P.; Morrison, E.A.; Hanna, J.V. Tin(IV) Porphyrin Complexes—III. NMR Trans-influences in Tin(IV) Tetraphenylporphyrin Complexes. Polyhedron 1990, 9, 1331–1336. [Google Scholar] [CrossRef]
- Sigma-Aldrich, commercial product, Cas number 3068-88-0.
- Ganji, P.; Doyle, D.J.; Ibrahim, H. In Situ Generation of the Coates Catalyst: A Practical and Versatile Catalytic System for the Carbonylation of meso-Epoxides. Org. Lett. 2011, 13, 3142–3145. [Google Scholar] [CrossRef]
- Nelson, S.G.; Spencer, K.L. Sequential Acyl Halide−Aldehyde Cyclocondensation and Enzymatic Resolution as a Route to Enantiomerically Enriched β-Lactones. J. Org. Chem. 2000, 65, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Gnanadesikan, V.; Corey, E.J. Enantioselective β-Lactone Formation from Ketene and Aldehydes Catalyzed by a Chiral Oxazaborolidine. Org. Lett. 2006, 8, 4943–4945. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Ageishi, S.; Isobe, H.; Hayashi, Y.; Yamamoto, Y. Lipase Promoted Asymmetric Transesterification of 4-Alkyl-, 3-Alkyl-and 3, 4-Dialkyloxetan-2-ones with Ring-opening. J. Chem. Soc. Perkin Trans. 2000, 71–77. [Google Scholar] [CrossRef]
- Concellón, J.M.; Concellón, C. Aldol-type Reactions of Unmasked Iodoacetic Acid with Carbonyl Compounds Promoted by Samarium Diiodide: Efficient Synthesis of Carboxylic 3-Hydroxyacids and Their Derivatives. J. Org. Chem. 2006, 71, 4428–4432. [Google Scholar] [CrossRef] [PubMed]
- Martín, C.; Belderraín, T.R.; Pérez, P.J. Rediscovering Copper-based Catalysts for Intramolecular Carbon–Hydrogen Bond Functionalization by Carbene Insertion. Org. Biomol. Chem. 2009, 7, 4777–4781. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | mol % | Temp (oC) | CO (atm) | Solvent | Yield (%)3 | TON 4 |
|---|---|---|---|---|---|---|---|
| 1 | 3a | 0.3 | 90 | 50 | THF | >99 | 333 |
| 2 | 3b | 0.3 | 90 | 50 | THF | 94 | 313 |
| 3 | 3c | 0.3 | 90 | 50 | THF | 83 | 277 |
| 4 | 3d | 0.3 | 90 | 50 | THF | 57 | 190 |
| 5 | 3a | 0.3 | 90 | 50 | 2-Me-THF | >99 | 333 |
| 6 | 3a | 0.3 | 90 | 50 | 1,4-dioxane | 89 | 297 |
| 7 | 3a | 0.3 | 90 | 50 | DMF | 0 | - |
| 8 | 3a | 0.3 | 90 | 50 | MeOH | 0 | - |
| 9 | 3a | 0.3 | 90 | 50 | o-xylene | 35 | 117 |
| 10 | 3a | 0.3 | 90 | 50 | toluene | 13 | 43 |
| 11 | 3a | 0.3 | 90 | 50 | n-hexane | 3 | 10 |
| 12 | 3a | 0.3 | 70 | 50 | THF | 61 | 203 |
| 13 | 3a | 0.3 | 50 | 50 | THF | 20 | 67 |
| 14 | 3a | 0.3 | 90 | 30 | THF | 95 | 317 |
| 15 | 3a | 0.3 | 90 | 10 | THF | 63 | 210 |
| 16 | 3a | 0.2 | 90 | 50 | THF | 91 | 455 |
| 17 | 3a (24 h) | 0.2 | 90 | 50 | THF | >99 | 500 |
| 18 | 3a (24 h) | 0.1 | 90 | 50 | THF | 80 | 800 |
| 19 | 3a (36 h) | 0.1 | 90 | 70 | THF | >99 | 1000 |
| 21 | 3a (36 h) | 0.05 | 110 | 70 | THF | 63 | 1260 |
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baral, E.R.; Kim, D.; Lee, S.; Park, M.H.; Kim, J.G. Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides. Catalysts 2019, 9, 311. https://doi.org/10.3390/catal9040311
Baral ER, Kim D, Lee S, Park MH, Kim JG. Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides. Catalysts. 2019; 9(4):311. https://doi.org/10.3390/catal9040311
Chicago/Turabian StyleBaral, Ek Raj, Dongwook Kim, Sunwoo Lee, Myung Hwan Park, and Jeung Gon Kim. 2019. "Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides" Catalysts 9, no. 4: 311. https://doi.org/10.3390/catal9040311
APA StyleBaral, E. R., Kim, D., Lee, S., Park, M. H., & Kim, J. G. (2019). Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides. Catalysts, 9(4), 311. https://doi.org/10.3390/catal9040311