Pt/C and Pt/SnOx/C Catalysts for Ethanol Electrooxidation: Rotating Disk Electrode Study

 , ,
, ,  ,
,  ,
, 

Abstract
1. Introduction
2. Results and Discussion
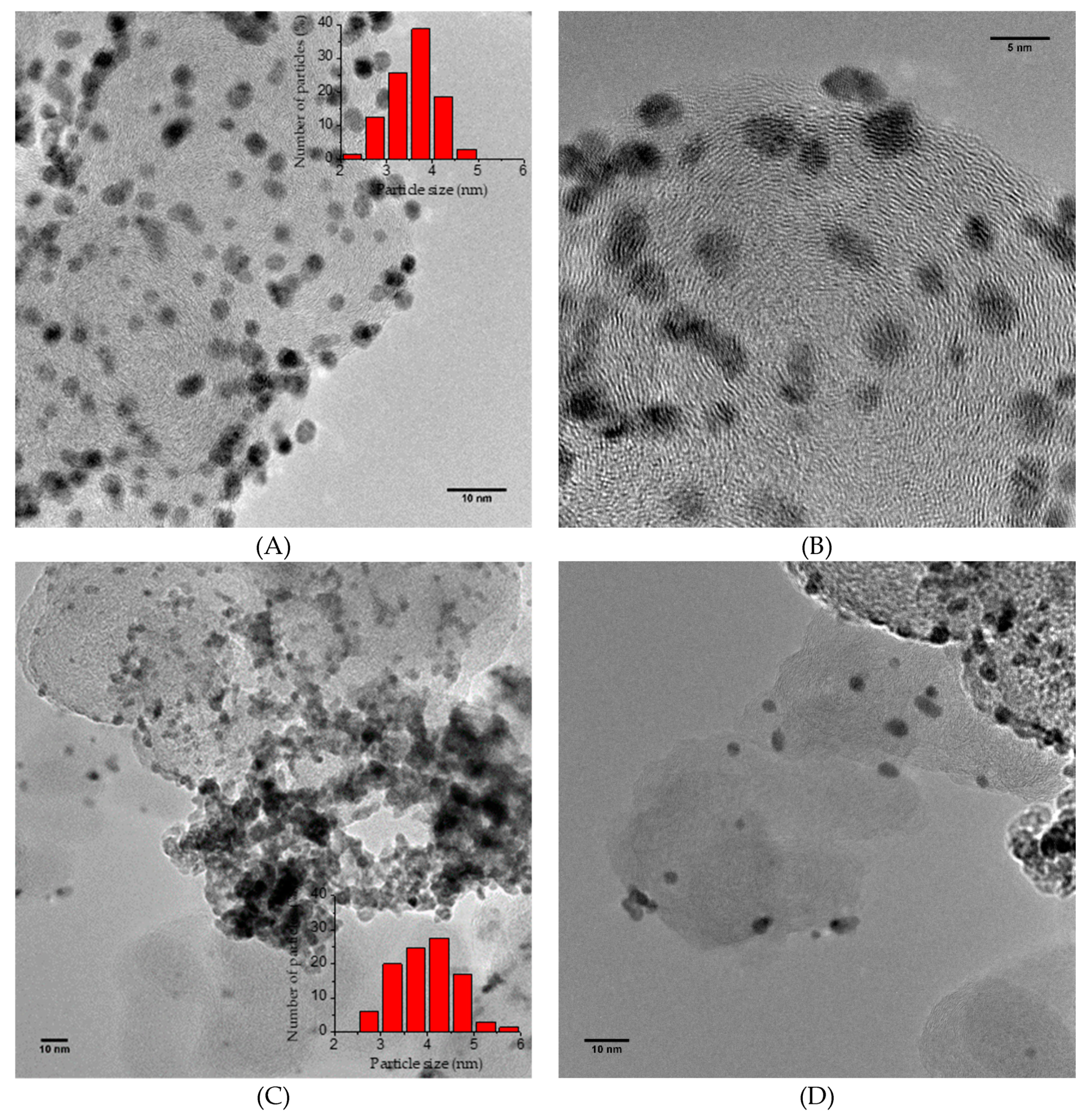
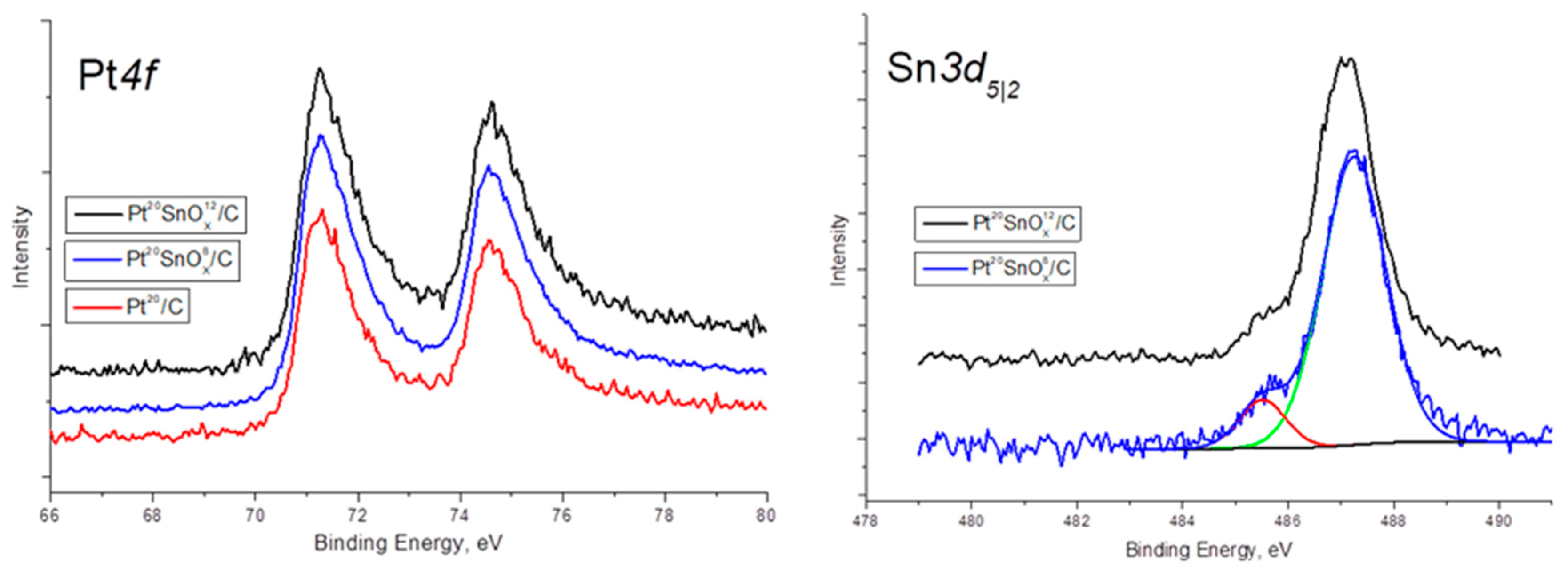
2.1. Catalyst Characterization
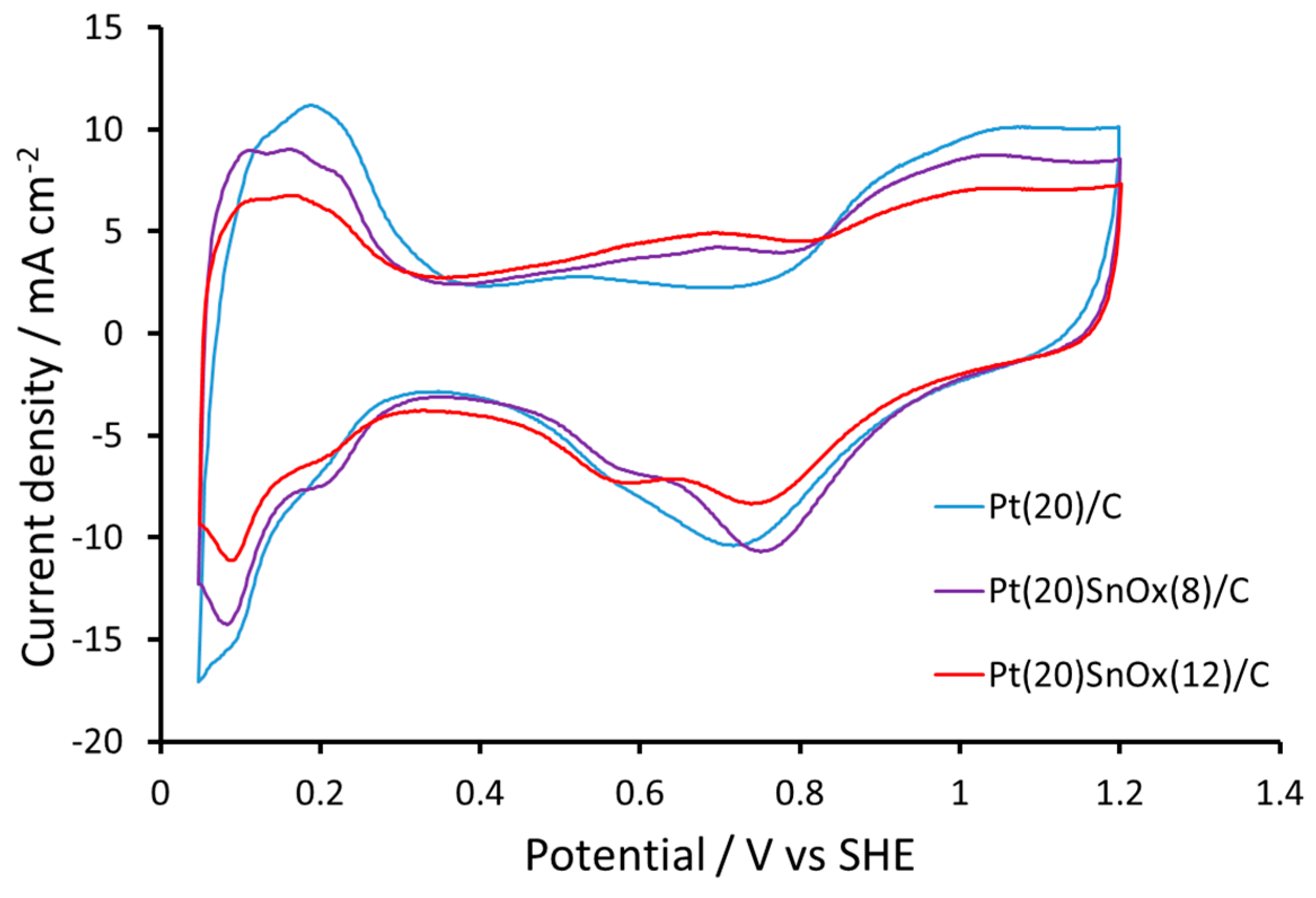
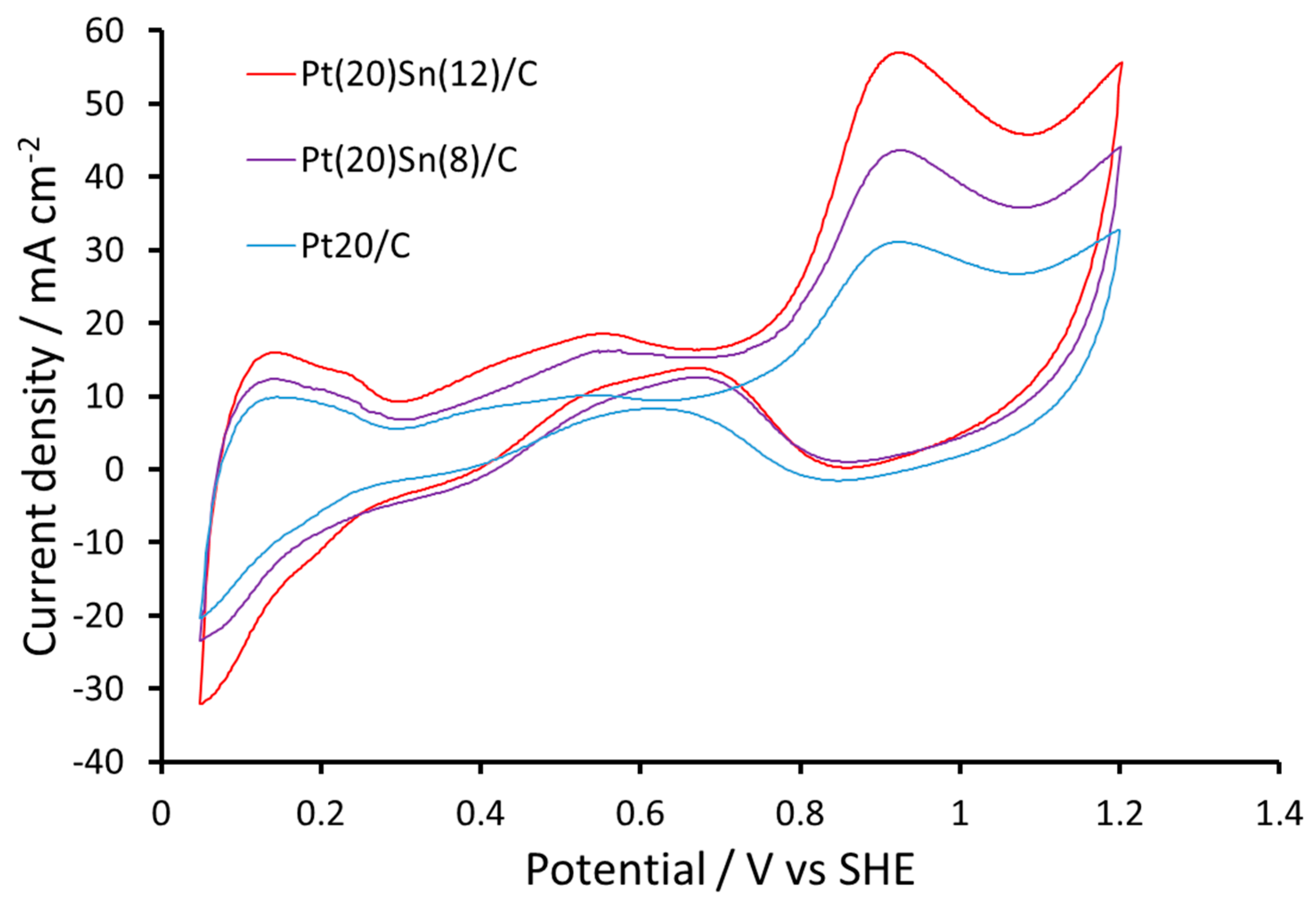
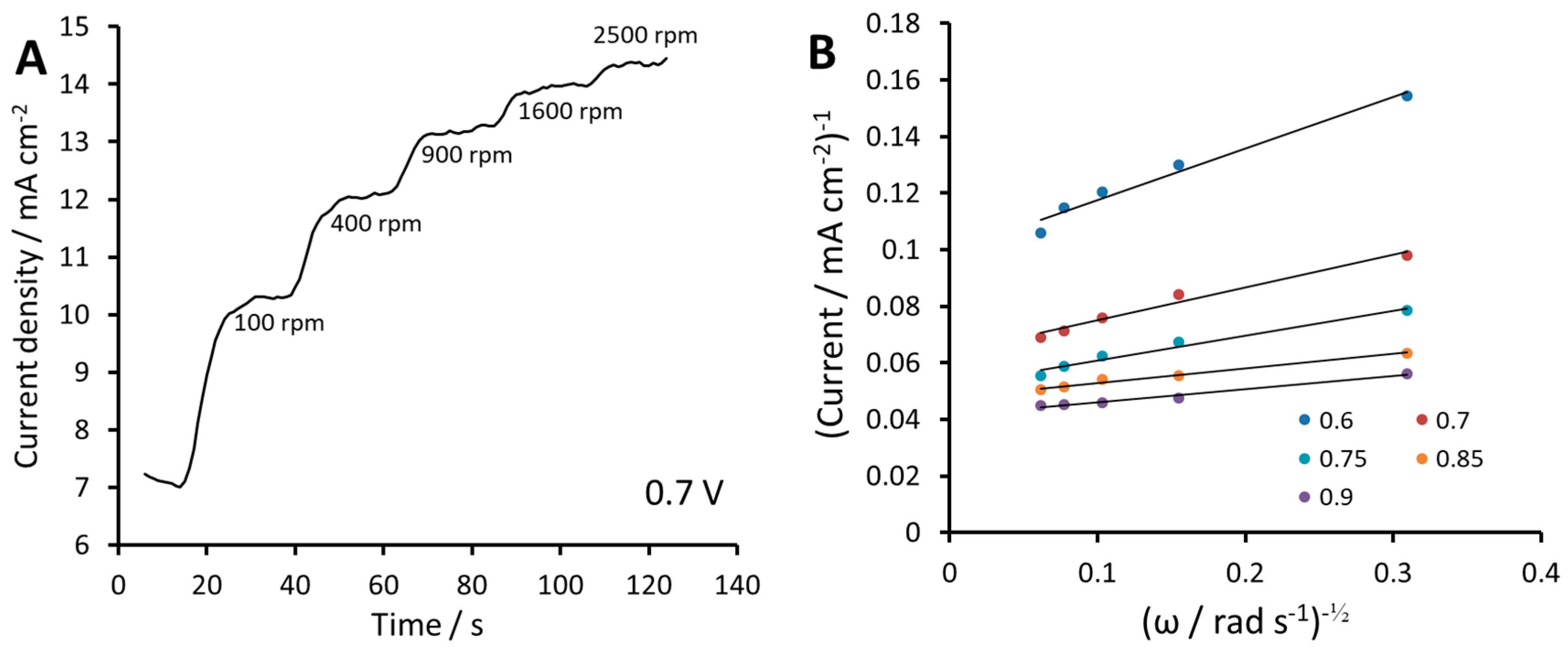
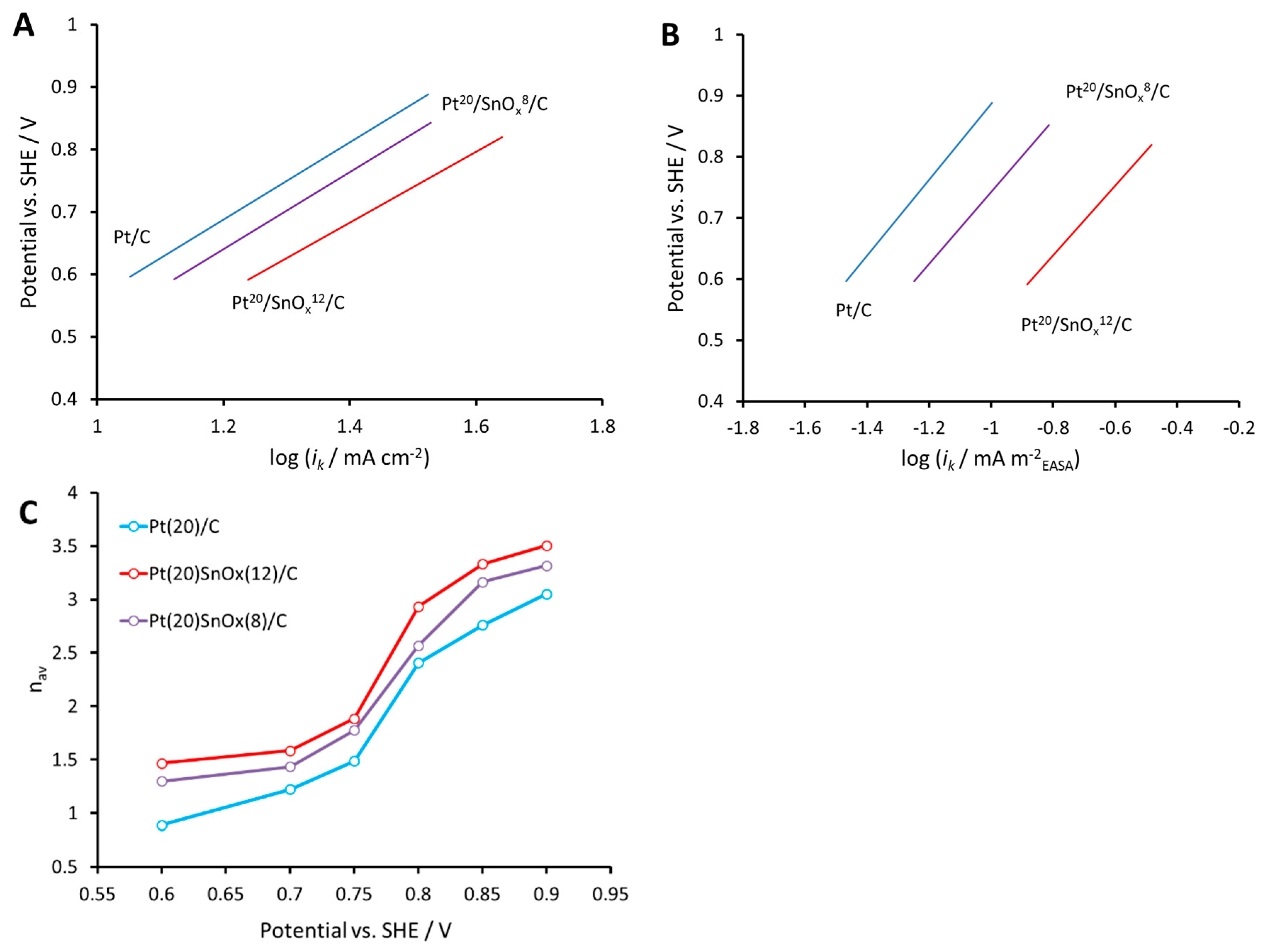
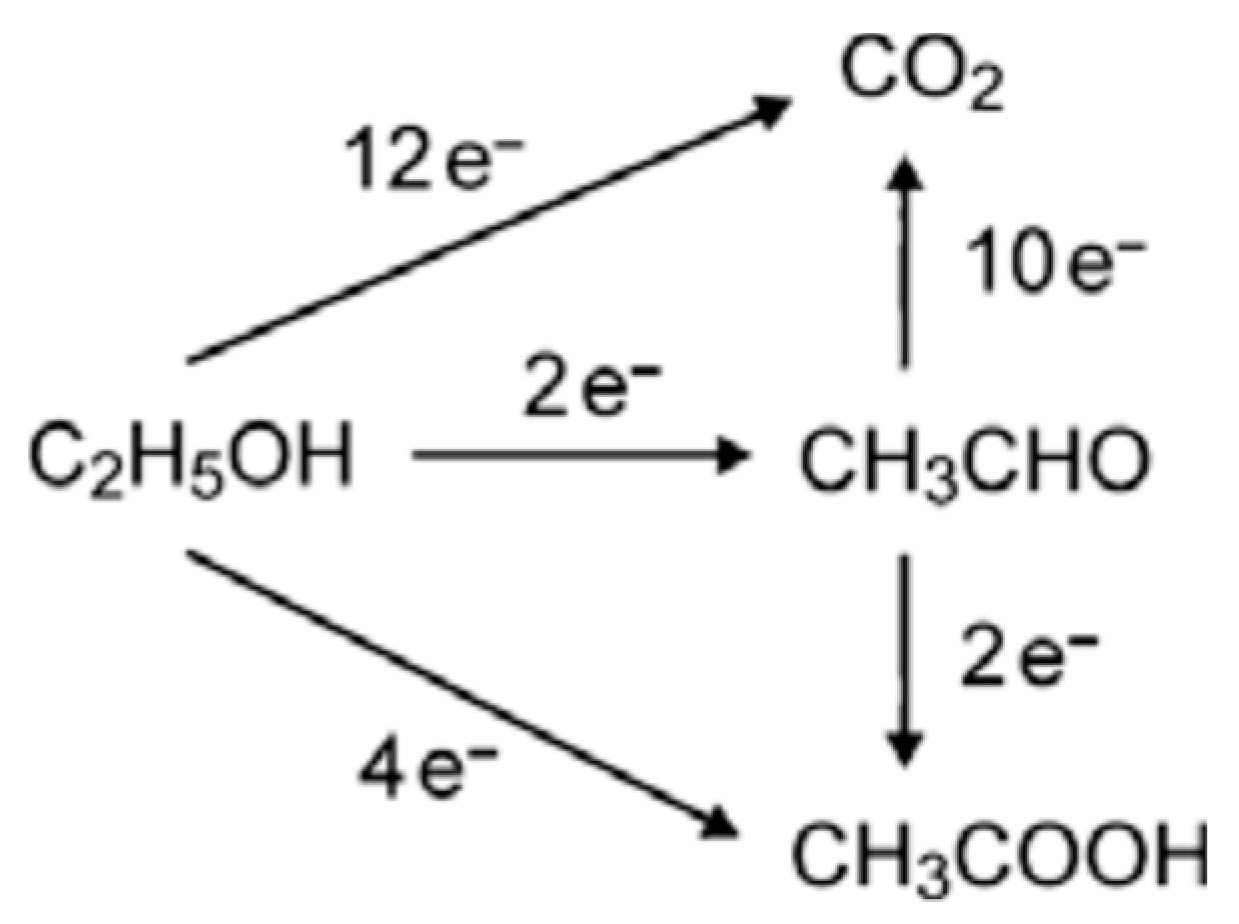
2.2. Electrochemical Measurements
3. Materials and Methods
3.1. Pt/SnOx/C Catalyst Synthesis
3.2. Electrode Preparation
3.3. Electrochemical Measurements
3.4. Catalysts Structure and Morphology Characterization
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Lamy, C.; Jaubert, T.; Baranton, S.; Coutanceau, C. Clean hydrogen generation through the electrocatalytic oxidation of ethanol in a Proton Exchange Membrane Electrolysis Cell (PEMEC): Effect of the nature and structure of the catalytic anode. J. Power Sources 2014, 245, 927–936. [Google Scholar] [CrossRef]
- Caravaca, A.; Sapountzi, F.M.M.; de Lucas-Consuegra, A.; Molina-Mora, C.; Dorado, F.; Valverde, J.L.L. Electrochemical reforming of ethanol–water solutions for pure H2 production in a PEM electrolysis cell. Int. J. Hydrogen Energy 2012, 37, 9504–9513. [Google Scholar] [CrossRef]
- Ju, H.K.; Giddey, S.; Badwal, S.P.S.; Mulder, R.J. Electro-catalytic conversion of ethanol in solid electrolyte cells for distributed hydrogen generation. Electrochim. Acta 2016, 212, 744–757. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R.J. Ethanol Electrooxidation on a Carbon-Supported Pt Catalyst: Reaction Kinetics and Product Yields. J. Phys. Chem. B 2004, 108, 19413–19424. [Google Scholar] [CrossRef]
- Li, M.; Cullen, D.A.; Sasaki, K.; Marinkovic, N.S.; More, K.; Adzic, R.R. Ternary electrocatalysts for oxidizing ethanol to carbon dioxide: Making Ir capable of splitting C-C bond. J. Am. Chem. Soc. 2013, 135, 132–141. [Google Scholar] [CrossRef]
- Pushkareva, I.V.; Pushkarev, A.S.; Grigoriev, S.A.; Lyutikova, E.K.; Akel’kina, S.V.; Osina, M.A.; Slavcheva, E.P.; Fateev, V.N. Electrochemical conversion of aqueous ethanol solution in an electrolyzer with a solid polymer electrolyte. Russ. J. Appl. Chem. 2016, 89, 2109–2111. [Google Scholar] [CrossRef]
- Camara, G.A.; Iwasita, T. Parallel pathways of ethanol oxidation: The effect of ethanol concentration. J. Electroanal. Chem. 2005, 578, 315–321. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, S.; Cai, W.-B. Recent Advances on Electro-Oxidation of Ethanol on Pt- and Pd-Based Catalysts: From Reaction Mechanisms to Catalytic Materials. Catalysts 2015, 5, 1507–1534. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. Effect of synthesis method and structural characteristics of Pt–Sn fuel cell catalysts on the electro-oxidation of CH3OH and CH3CH2OH in acid medium. Catal. Today 2011, 160, 28–38. [Google Scholar] [CrossRef]
- Ermete, A. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Li, H.; Sun, G.; Cao, L.; Jiang, L.; Xin, Q. Comparison of different promotion effect of PtRu/C and PtSn/C electrocatalysts for ethanol electro-oxidation. Electrochim. Acta 2007, 52, 6622–6629. [Google Scholar] [CrossRef]
- Kamarudin, M.Z.F.; Kamarudin, S.K.; Masdar, M.S.; Daud, W.R.W. Review: Direct ethanol fuel cells. Int. J. Hydrogen Energy 2013, 38, 9438–9453. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Perrard, A.; Belgsir, E.M.; Lamy, C. Development of anode catalysts for a direct ethanol fuel cell. J. Appl. Electrochem. 2004, 34, 439–446. [Google Scholar] [CrossRef]
- Yang, G.; Namin, L.M.; Aaron Deskins, N.; Teng, X. Influence of ∗OH adsorbates on the potentiodynamics of the CO2 generation during the electro-oxidation of ethanol. J. Catal. 2017, 353, 335–348. [Google Scholar] [CrossRef]
- James, D.D.; Moghaddam, R.B.; Chen, B.; Pickup, P.G. Ruthenium-Tin Oxide/Carbon Supported Platinum Catalysts for Electrochemical Oxidation of Ethanol in Direct Ethanol Fuel Cells. J. Electrochem. Soc. 2018, 165, F215–F219. [Google Scholar] [CrossRef]
- Chen, B.; Brueckner, T.M.; Altarawneh, R.M.; Pickup, P.G. Composition Dependence of Ethanol Oxidation at Ruthenium-Tin Oxide/Carbon Supported Platinum Catalysts. J. Electrochem. Soc. 2018, 165, J3019–J3025. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A. Rotating thin-film method for supported catalysts. In Handbook of Fuel Cells; John Wiley & Sons: Chichester, UK, 2003; pp. 316–333. ISBN 9780470974001. [Google Scholar]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Schmidt, T.J. Characterization of High-Surface-Area Electrocatalysts Using a Rotating Disk Electrode Configuration. J. Electrochem. Soc. 1998, 145, 2354. [Google Scholar] [CrossRef]
- Puthiyapura, V.K.; Lin, W.F.; Russell, A.E.; Brett, D.J.L.; Hardacre, C. Effect of Mass Transport on the Electrochemical Oxidation of Alcohols Over Electrodeposited Film and Carbon-Supported Pt Electrodes. Top. Catal. 2018, 61, 240–253. [Google Scholar] [CrossRef]
- Sayadi, A.; Pickup, P.G. Evaluation of ethanol oxidation catalysts by rotating disc voltammetry. Electrochim. Acta 2016, 215, 84–92. [Google Scholar] [CrossRef]
- Sayadi, A.; Pickup, P.G. Evaluation of methanol oxidation catalysts by rotating disc voltammetry. Electrochim. Acta 2016, 199, 12–17. [Google Scholar] [CrossRef]
- Grigoriev, S.A.; Fateev, V.N.; Pushkarev, A.S.; Pushkareva, I.V.; Ivanova, N.A.; Kalinichenko, V.N.; Presnyakov, M.Y.; Wei, X. Reduced Graphene Oxide and Its Modifications as Catalyst Supports and Catalyst Layer Modifiers for PEMFC. Materials (Basel) 2018, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.W.; Zhou, W.P.; White, M.G. Promotion of Pt surfaces for ethanol electro-oxidation by the addition of small SnO2 nanoparticles: Activity and mechanism. Appl. Catal. B Environ. 2014, 152–153, 397–402. [Google Scholar] [CrossRef]
- Lim, D.-H.; Choi, D.-H.; Lee, W.-D.; Park, D.-R.; Lee, H.-I. The Effect of Sn Addition on a Pt/C Electrocatalyst Synthesized by Borohydride Reduction and Hydrothermal Treatment for a Low-Temperature Fuel Cell. Electrochem. Solid State Lett. 2007, 10, B87. [Google Scholar] [CrossRef]
- Ruiz-Camacho, B.; Santoyo, H.H.R.H.H.R.; Medina-Flores, J.M.J.M.; Álvarez-Martínez, O.; Martínez-Álvarez, O. Platinum deposited on TiO2-C and SnO2-C composites for methanol oxidation and oxygen reduction. Electrochim. Acta 2014, 120, 344–349. [Google Scholar] [CrossRef]
- Kou, R.; Shao, Y.; Mei, D.; Nie, Z.; Wang, D.; Wang, C.; Viswanathan, V.V.; Park, S.; Aksay, I.A.; Lin, Y.; et al. Stabilization of Electrocatalytic Metal Nanoparticles at Metal−Metal Oxide−Graphene Triple Junction Points. J. Am. Chem. Soc. 2011, 133, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Li, Z.; Ye, L.; Wang, Y.; Lin, S. One-pot synthesis of Pt/SnO2/GNs and its electro-photo-synergistic catalysis for methanol oxidation. Int. J. Hydrogen Energy 2016, 41, 255–264. [Google Scholar] [CrossRef]
- Zhu, M.; Sun, G.; Xin, Q. Effect of alloying degree in PtSn catalyst on the catalytic behavior for ethanol electro-oxidation. Electrochim. Acta 2009, 54, 1511–1518. [Google Scholar] [CrossRef]
- Li, X.-L.; Lin, B.-Z.; Xu, B.-H.; Chen, Z.-J.; Wang, Q.-Q.; Kuang, J.-D.; Zhu, H. Preparation and characterization of mesoporous SnO2-pillared HTaWO6 with enhanced photocatalytic activity. J. Mater. Chem. 2010, 20, 3924. [Google Scholar] [CrossRef]
- Taylor, J.A.; Merchant, S.M.; Perry, D.L. Study of the oxidation of gold-tin preforms using x-ray photoelectron spectroscopy. J. Appl. Phys. 1995, 78, 5356–5361. [Google Scholar] [CrossRef]
- Rizo, R.; Sebastián, D.; Lázaro, M.J.; Pastor, E. On the design of Pt-Sn efficient catalyst for carbon monoxide and ethanol oxidation in acid and alkaline media. Appl. Catal. B Environ. 2017, 200, 246–254. [Google Scholar] [CrossRef]
- Silva, J.C.M.; De Souza, R.F.B.; Parreira, L.S.; Neto, E.T.; Calegaro, M.L.; Santos, M.C. Ethanol oxidation reactions using SnO2@Pt/C as an electrocatalyst. Appl. Catal. B Environ. 2010, 99, 265–271. [Google Scholar] [CrossRef]
- Lim, D.H.; Choi, D.H.; Lee, W.D.; Lee, H.I. A new synthesis of a highly dispersed and CO tolerant PtSn/C electrocatalyst for low-temperature fuel cell; its electrocatalytic activity and long-term durability. Appl. Catal. B Environ. 2009, 89, 484–493. [Google Scholar] [CrossRef]
- Arenz, M.; Stamenkovic, V.; Blizanac, B.B.; Mayrhofer, K.J.; Markovic, N.M.; Ross, P.N. Carbon-supported Pt-Sn electrocatalysts for the anodic oxidation of H2, CO, and H2/CO mixtures.: Part II: The structure-activity relationship. J. Catal. 2005, 232, 402–410. [Google Scholar] [CrossRef]
- Pozio, A.; de Francesco, M.; Cemmi, A.; Cardellini, F.; Giorgi, L. Comparison of high surface Pt/C catalysts by cyclic voltammetry. J. Power Sources 2002, 105, 13–19. [Google Scholar] [CrossRef]
- Tarasevich, M.R.; Korchagin, O.V.; Kuzov, A. V Electrocatalysis of anodic oxidation of ethanol. Russ. Chem. Rev. 2013, 82, 1047–1065. [Google Scholar] [CrossRef]
- Angerstein-Kozlowska, H.; Conway, B.E.; Sharp, W.B.A. The real condition of electrochemically oxidized platinum surfaces. J. Electroanal. Chem. Interfacial Electrochem. 1973, 43, 9–36. [Google Scholar] [CrossRef]
- Leung, L.W.H.; Weaver, M.J. Real-time FTIR spectroscopy as a quantitative kinetic probe of competing electrooxidation pathways of small organic molecules. J. Phys. Chem. 1988, 92, 4019–4022. [Google Scholar] [CrossRef]
- Fujiwara, N.; Friedrich, K.A.; Stimming, U. Ethanol oxidation on PtRu electrodes studied by differential electrochemical mass spectrometry. J. Electroanal. Chem. 1999, 472, 120–125. [Google Scholar] [CrossRef]
- Seland, F.; Foss, C.E.L.; Tunold, R.; Harrington, D. Increasing and Decreasing Mass Transport Effects in the Oxidation of Small Organic Molecules. ECS Trans. 2010, 28, 203–210. [Google Scholar] [CrossRef]
- Kuriganova, A.B.; Leontyeva, D.V.; Ivanov, S.; Bund, A.; Smirnova, N.V. Electrochemical dispersion technique for preparation of hybrid MOx–C supports and Pt/MOx–C electrocatalysts for low-temperature fuel cells. J. Appl. Electrochem. 2016, 46, 1245–1260. [Google Scholar] [CrossRef]
- Bach Delpeuch, A.; Asset, T.; Chatenet, M.; Cremers, C. Influence of the temperature for the ethanol oxidation reaction (EOR) on Pt/C, Pt-Rh/C and Pt-Rh-SnO2/C. Fuel Cells 2015, 15, 352–360. [Google Scholar] [CrossRef]
- Katikawong, P.; Ratana, T.; Veerasai, W. Temperature dependence studies on the electro-oxidation of aliphatic alcohols with modified platinum electrodes. J. Chem. Sci. 2009, 121, 329–337. [Google Scholar] [CrossRef]
- Baranova, E.A.A.; Tavasoli, A.; Amir, T. Particle Size Effect of Unsupported Pt/SnOx Nanoparticles for Ethanol Electro-Oxidation. Electrocatalysis 2011, 2, 89–95. [Google Scholar] [CrossRef]
- Jiang, L.; Colmenares, L.; Jusys, Z.; Sun, G.Q.; Behm, R.J. Ethanol electrooxidation on novel carbon supported Pt/SnOx/C catalysts with varied Pt:Sn ratio. Electrochim. Acta 2007, 53, 377–389. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, G.Q.; Jiang, L.H.; Xin, Q.; Sun, S.G.; Jiang, Y.X.; Chen, S.P.; Jusys, Z.; Behm, R.J. Adsorption and oxidation of ethanol on colloid-based Pt/C, PtRu/C and Pt3Sn/C catalysts: In situ FTIR spectroscopy and on-line DEMS studies. Phys. Chem. Chem. Phys. 2007, 9, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Bach Delpeuch, A.; Maillard, F.; Chatenet, M.; Soudant, P.; Cremers, C. Ethanol oxidation reaction (EOR) investigation on Pt/C, Rh/C, and Pt-based bi- and tri-metallic electrocatalysts: A DEMS and in situ FTIR study. Appl. Catal. B Environ. 2016, 181, 672–680. [Google Scholar] [CrossRef]
- Kavanagh, R.; Cao, X.M.; Lin, W.F.; Hardacre, C.; Hu, P. Origin of low CO2 selectivity on platinum in the direct ethanol fuel cell. Angew. Chemie Int. Ed. 2012, 51, 1572–1575. [Google Scholar] [CrossRef]
- Rousseau, S.; Coutanceau, C.; Lamy, C.; Léger, J.-M. Direct ethanol fuel cell (DEFC): Electrical performances and reaction products distribution under operating conditions with different platinum-based anodes. J. Power Sources 2006, 158, 18–24. [Google Scholar] [CrossRef]
- Wang, H.-F.; Liu, Z.-P. Comprehensive Mechanism and Structure-Sensitivity of Ethanol Oxidation on Platinum: New Transition-State Searching Method for Resolving the Complex Reaction Network. J. Am. Chem. Soc. 2008, 130, 10996–11004. [Google Scholar] [CrossRef] [PubMed]
- Asiri, H.A.; Anderson, A.B. Mechanisms for Ethanol Electrooxidation on Pt(111) and Adsorption Bond Strengths Defining an Ideal Catalyst. J. Electrochem. Soc. 2015, 162, F115–F122. [Google Scholar] [CrossRef]
- Rao, V.; Cremers, C.; Stimming, U.; Cao, L.; Sun, S.; Yan, S.; Sun, G.; Xin, Q. Electro-oxidation of Ethanol at Gas Diffusion Electrodes A DEMS Study. J. Electrochem. Soc. 2007, 154, B1138. [Google Scholar] [CrossRef]
- Altarawneh, R.M.; Majidi, P.; Pickup, P.G. Determination of the efficiency of ethanol oxidation in a proton exchange membrane electrolysis cell. J. Power Sources 2017, 351, 106–114. [Google Scholar] [CrossRef]
- Altarawneh, R.M.; Pickup, P.G. Determination of the Stoichiometry of Ethanol Oxidation from the Flow Rate Dependence of the Current in a Proton Exchange Membrane Electrolysis Cell. J. Electrochem. Soc. 2018, 165, F479–F483. [Google Scholar] [CrossRef]
- Altarawneh, R.M.; Brueckner, T.M.; Chen, B.; Pickup, P.G. Product distributions and efficiencies for ethanol oxidation at PtNi octahedra. J. Power Sources 2018, 400, 369–376. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Pt, wt % | Sn, wt % |
|---|---|---|
| Pt20/C | 19.43 | - |
| Pt20/SnOx8/C | 20.38 | 8.37 |
| Pt20/SnOx12/C | 21.24 | 11.86 |
| Catalyst | EASA/m2 gPt−1 | Tafel Slope 1/mV dec.−1 | Kinetic Current at 0.8 V/mA cm−2GEOM | Kinetic Current at 0.8 V/mA m−2EASA |
|---|---|---|---|---|
| Pt20/C | 58.1 | 619.2 | 24.2 | 0.082 |
| Pt20/SnOx8/C | 46.1 | 585.6 | 29.5 | 0.126 |
| Pt20/SnOx12/C | 26.0 | 568.4 | 36.7 | 0.277 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pushkarev, A.S.; Pushkareva, I.V.; Ivanova, N.A.; du Preez, S.P.; Bessarabov, D.; Chumakov, R.G.; Stankevich, V.G.; Fateev, V.N.; Evdokimov, A.A.; Grigoriev, S.A. Pt/C and Pt/SnOx/C Catalysts for Ethanol Electrooxidation: Rotating Disk Electrode Study. Catalysts 2019, 9, 271. https://doi.org/10.3390/catal9030271
Pushkarev AS, Pushkareva IV, Ivanova NA, du Preez SP, Bessarabov D, Chumakov RG, Stankevich VG, Fateev VN, Evdokimov AA, Grigoriev SA. Pt/C and Pt/SnOx/C Catalysts for Ethanol Electrooxidation: Rotating Disk Electrode Study. Catalysts. 2019; 9(3):271. https://doi.org/10.3390/catal9030271
Chicago/Turabian StylePushkarev, Artem S., Irina V. Pushkareva, Natalia A. Ivanova, Stephanus P. du Preez, Dmitri Bessarabov, Ratibor G. Chumakov, Vladimir G. Stankevich, Vladimir N. Fateev, Anatoly A. Evdokimov, and Sergey A. Grigoriev. 2019. "Pt/C and Pt/SnOx/C Catalysts for Ethanol Electrooxidation: Rotating Disk Electrode Study" Catalysts 9, no. 3: 271. https://doi.org/10.3390/catal9030271
APA StylePushkarev, A. S., Pushkareva, I. V., Ivanova, N. A., du Preez, S. P., Bessarabov, D., Chumakov, R. G., Stankevich, V. G., Fateev, V. N., Evdokimov, A. A., & Grigoriev, S. A. (2019). Pt/C and Pt/SnOx/C Catalysts for Ethanol Electrooxidation: Rotating Disk Electrode Study. Catalysts, 9(3), 271. https://doi.org/10.3390/catal9030271