Multi-Catalytic Route for the Synthesis of (S)-Tembamide

 ,
,  , and
, and
Abstract

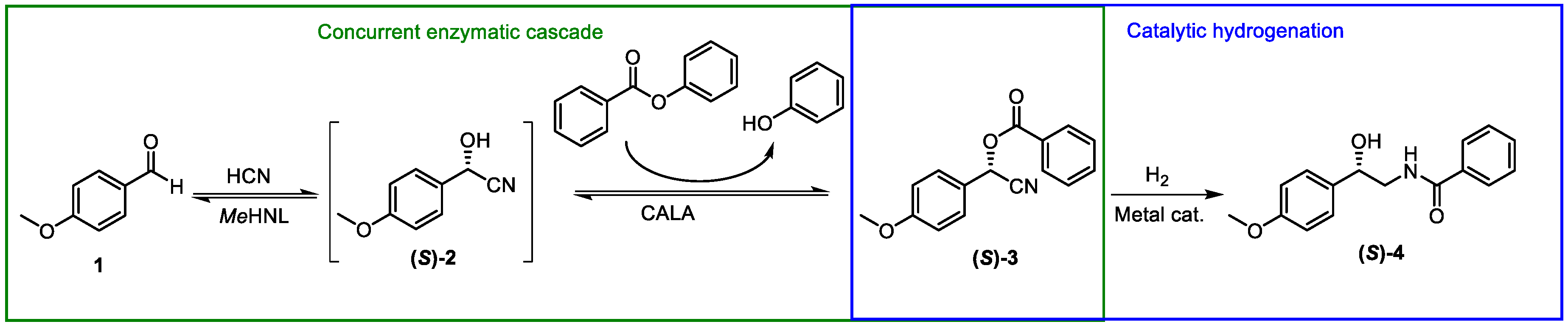
1. Introduction
2. Results and Discussion
2.1. Catalyst Screening
2.2. Selection of Catalyst and Optimization of Reaction Temperature
2.3. Identification of Side Products and Further Optimization of Reaction Parameters
2.4. Preparative Synthesis of (S)-Tembamide
3. Materials and Methods
3.1. Enzymes
3.1.1. Enzyme Immobilization
3.1.2. Enzyme Activity Measurements
3.2. Chemicals
3.2.1. Synthesis of (±)-4-Methoxymandelonitrile, (±)-2
3.2.2. Synthesis of (±)-4-Methoxymandelonitrile Benzoate, (±)-3
3.2.3. Synthesis of (R)-4-methoxymandelonitrile benzoate, (R)-3
3.3. Hydrogenation Reactions
3.3.1. Pretreatment of Catalysts
3.3.2. General Procedure for the Hydrogenation of (±)-3 at Room Temperature Under 1 Bar of H2 - Initial Catalyst Screening
3.3.3. General Procedure for the Hydrogenation of (±)-3 in Autoclave Reactor
3.3.4. Stability of Tembamide Under Hydrogenating Conditions in Autoclave Reactor
3.3.5. Hydrogenation of (R)-3 in Autoclave Reactor
3.3.6. Evaluation of the Effect of HCN on the Hydrogenation of (±)-3 in Autoclave Reactor
3.4. Preparative Synthesis of (S)-Tembamide
3.4.1. Biocatalytic Cascade Synthesis of (S)-3
3.4.2. Catalytic Hydrogenation of (S)-3
3.5. HPLC Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reetz, M.T. New Approaches to the Use of Amino Acids as Chiral Building Blocks in Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1991, 30, 1531–1546. [Google Scholar] [CrossRef]
- Bergmeier, S.C. The Synthesis of Vicinal Amino Alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar] [CrossRef]
- Klingler, F.D. Asymmetric Hydrogenation of Prochiral Amino Ketones to Amino Alcohols for Pharmaceutical Use. Acc. Chem. Res. 2007, 40, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Métro, T.X.; Duthion, B.; Gomez Pardo, D.; Cossy, J. Rearrangement of β-amino alcohols via aziridiniums: A review. Chem. Soc. Rev. 2010, 39, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.; Zhang, H.; Xiong, X.; Lu, X.; Zhou, Y. Evolution of epoxides to synthesize β-amino alcohols. Asian J. Chem. 2014, 26, 3761–3768. [Google Scholar] [CrossRef]
- Li, G.; Chang, H.T.; Barry Sharpless, K. Catalytic Asymmetric Aminohydroxylation (AA) of Olefins. Angew. Chem. Int. Ed. Engl. 1996, 35, 451–454. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Callens, C.K.A.; Flores, A.; Lacy, A.R.; Rathi, A.H. Recent developments in methodology for the direct oxyamination of olefins. Chem. Eur. J. 2011, 17, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Burchak, O.N.; Py, S. Reductive cross-coupling reactions (RCCR) between CN and CO for β-amino alcohol synthesis. Tetrahedron 2009, 65, 7333–7356. [Google Scholar] [CrossRef]
- Ye, C.X.; Melcamu, Y.Y.; Li, H.H.; Cheng, J.T.; Zhang, T.T.; Ruan, Y.P.; Zheng, X.; Lu, X.; Huang, P.Q. Dual catalysis for enantioselective convergent synthesis of enantiopure vicinal amino alcohols. Nat. Commun. 2018, 9, 410. [Google Scholar] [CrossRef]
- Cheng, M.-J.; Tsai, I.-L.; Chen, I.-S. Chemical Constituents from the Root Bark of Formosan Zanthoxylum Ailanthoides. J. Chin. Chem. Soc. 2003, 50, 1241–1246. [Google Scholar] [CrossRef]
- Johns, S.R.; Lamberton, J.A.; Tweeddale, H.J.; Willing, R.I. Alkaloids of Zanthoxylum Conspersipunctatum (Rutaceae): The Structure of a New Alkaloid Isomeric with Protopine. Aust. J. Chem. 1969, 22, 2233–2236. [Google Scholar] [CrossRef]
- Shoeb, A.; Kapil, R.S.; Popli, S.P. Coumarins and alkaloids of Aegle marmelos. Phytochemistry 1973, 12, 2071–2072. [Google Scholar] [CrossRef]
- Somanathan, R.; Aguilar, H.R.; Ventura, G.R.; Smith, K.M. Syntheses of natural hydroxyamides using trimethylsilyl cyanide. Synth. Commun. 1983, 13, 273–280. [Google Scholar] [CrossRef]
- Cheng, M.J.; Lee, K.H.; Tsai, I.L.; Chen, I.S. Two new sesquiterpenoids and anti-HIV principles from the root bark of Zanthoxylum ailanthoides. Bioorg. Med. Chem. 2005, 13, 5915–5920. [Google Scholar] [CrossRef] [PubMed]
- Albonico, S.M.; Kuck, A.M.; Deulofeu, V. Tembamide from Fagara hyemalis (St. Hill.) Engler. J. Chem. Soc. 1967, 22, 1327–1328. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Coccia, F.; Kara, S.; Grischek, B.; Kroutil, W.; D’Alessandro, N.; Hollmann, F. One-pot combination of enzyme and Pd nanoparticle catalysis for the synthesis of enantiomerically pure 1,2-amino alcohols. Green Chem. 2013, 15, 3318–3331. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, P.T.; Nanda, S.; Bhaskar Rao, A. Stereoselective synthesis of (R)-(-)-denopamine, (R)-(-)-tembamide and (R)-(-)-aegeline via asymmetric reduction of azidoketones by Daucus carota in aqueous medium. Tetrahedron Asymmetry 2002, 12, 3381–3385. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.A.; Sandbhor, M.; Malik, M.S. Chemoenzymatic synthesis of (R)- and (S)-tembamide, aegeline and denopamine by a one-pot lipase resolution protocol. Tetrahedron Asymmetry 2004, 15, 3939–3944. [Google Scholar] [CrossRef]
- Buchanan, D.J.; Dixon, D.J.; Scott, M.S.; Lainé, D.I. Short asymmetric syntheses of bioactive β-aryl ethanolamine derivatives via the highly diastereoselective delta lactol oxy-Michael addition. Tetrahedron Asymmetry 2004, 15, 195–197. [Google Scholar] [CrossRef]
- Sadyandy, R. An asymmetric dihydroxylation route to (R)-(-)-octopamine, (R)-(-)-tembamide and (R)-(-)-aegeline. Arkivoc 2005, 2005, 36–43. [Google Scholar]
- Liardo, E.; González-Fernández, R.; Ríos-Lombardía, N.; Morís, F.; García-Álvarez, J.; Cadierno, V.; Crochet, P.; Rebolledo, F.; González-Sabín, J. Strengthening the Combination between Enzymes and Metals in Aqueous Medium: Concurrent Ruthenium-Catalyzed Nitrile Hydration-Asymmetric Ketone Bioreduction. ChemCatChem 2018, 10, 4676–4682. [Google Scholar] [CrossRef]
- Cortez, N.A.; Aguirre, G.; Parra-Hake, M.; Somanathan, R. Synthesis of (R)-tembamide and (R)-aegeline via asymmetric transfer hydrogenation in water. Tetrahedron Asymmetry 2013, 24, 1297–1302. [Google Scholar] [CrossRef]
- Tae Cho, B.; Kyu Kang, S.; Hye Shin, S. Application of optically active 1,2-diol monotosylates for synthesis of β-azido and β-amino alcohols with very high enantiomeric purity. Synthesis of enantiopure (R)-octopamine, (R)-tembamide and (R)-aegeline. Tetrahedron Asymmetry 2002, 13, 1209–1217. [Google Scholar] [CrossRef]
- Lee, D.M.; Lee, J.C.; Jeong, N.; Lee, K.I. Asymmetric transfer hydrogenation of 2-tosyloxy-1-(4-hydroxyphenyl)ethanone derivatives: Synthesis of (R)-tembamide, (R)-aegeline, (R)-octopamine, and (R)-denopamine. Tetrahedron Asymmetry 2007, 18, 2662–2667. [Google Scholar] [CrossRef]
- Zhu, C.; Xia, Q.; Chen, X.; Liu, Y.; Du, X.; Cui, Y. Chiral metal–Organic framework as a platform for cooperative catalysis in asymmetric cyanosilylation of aldehydes. ACS Catal. 2016, 6, 7590–7596. [Google Scholar] [CrossRef]
- Aguirre, G.; Salgado-Rodríguez, A.; Flores-López, L.Z.; Parra-Hake, M. Asymmetric Synthesis of Naturally Occurring β-Hydroxyamides (R)-Tembamide and (R)-Aegeline. J. Mex. Chem. Soc. 2001, 45, 21–24. [Google Scholar]
- Brown, R.F.C.; Donohue, A.C.; Jackson, W.R.; McCarthy, T.D. Synthetic applications of optically active cyanohydrins. Enantioselective syntheses of the hydroxyamides tembamide and aegeline, the cardiac drug denopamine, and some analogues of the bronchodilator salbutamol. Tetrahedron 1994, 50, 13739–13752. [Google Scholar] [CrossRef]
- Baeza, A.; Nájera, C.; Sansano, J.M.; Saá, J.M. Binolam-AlCl: A two-centre catalyst for the synthesis of enantioenriched cyanohydrin O-phosphates. Chem. Eur. J. 2005, 11, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Choudhary, M.K.; Kureshy, R.I.; Roy, T.; Khan, N.U.H.; Abdi, S.H.R.; Bajaj, H.C. Enantioselective Henry and aza-Henry reaction in the synthesis of (R)-tembamide using efficient, recyclable polymeric CuII complexes as catalyst. ChemPlusChem 2014, 79, 1138–1146. [Google Scholar] [CrossRef]
- Gröger, H.; Hummel, W. Chemoenzymatic Multistep One-Pot Processes. In Cascade Biocatalysis: Integrating Stereoselective and Environmentally Friendly Reactions; Riva, S., Fessner, W.D., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 427–456. [Google Scholar]
- Gröger, H.; Hummel, W. Combining the “two worlds” of chemocatalysis and biocatalysis towards multi-step one-pot processes in aqueous media. Curr. Opin. Chem. Biol. 2014, 19, 171–179. [Google Scholar] [CrossRef]
- Schaaf, P.; Bayer, T.; Koley, M.; Schnürch, M.; Bornscheuer, U.T.; Rudroff, F.; Mihovilovic, M.D. Biocompatible metal-assisted C-C cross-coupling combined with biocatalytic chiral reductions in a concurrent tandem cascade. Chem. Commun. 2018, 54, 12978–12981. [Google Scholar] [CrossRef] [PubMed]
- Srinivasamurthy, V.S.T.; Böttcher, D.; Bornscheuer, U.T. A multi-enzyme cascade reaction for the production of 6-hydroxyhexanoic acid. Z. Nat.-Sect. C J. Biosci. 2019, 74, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.W.H.; Bassut, J.; de Souza, R.O.M.A.; Bornscheuer, U.T. Combination of the Suzuki–Miyaura Cross-Coupling Reaction with Engineered Transaminases. Chem. Eur. J. 2018, 24, 16009–16013. [Google Scholar] [CrossRef]
- Leemans, L.; van Langen, L.; Hollmann, F.; Schallmey, A. Bienzymatic Cascade for the Synthesis of an Optically Active O-benzoyl Cyanohydrin. Catalysts 2019, 9, 522. [Google Scholar] [CrossRef]
- Veum, L.; Pereira, S.R.M.; Van Der Waal, J.C.; Hanefeld, U. Catalytic hydrogenation of cyanohydrin esters as a novel approach to N-acylated β-amino alcohols—Reaction optimisation by a design of experiment approach. Eur. J. Org. Chem. 2006, 1664–1671. [Google Scholar] [CrossRef]
- Saavedra, J.Z.; Resendez, A.; Rovira, A.; Eagon, S.; Haddenham, D.; Singaram, B. Reaction of InCl 3 with various reducing agents: InCl 3-NaBH 4-mediated reduction of aromatic and aliphatic nitriles to primary amines. J. Org. Chem. 2012, 77, 221–228. [Google Scholar] [CrossRef]
- Haddenham, D.; Pasumansky, L.; DeSoto, J.; Eagon, S.; Singaram, B. Reductions of aliphatic and aromatic nitriles to primary amines with diisopropylaminoborane. J. Org. Chem. 2009, 74, 1964–1970. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.T.; Westcott, S.A. Hydroboration and diboration of imines. In Modern Reduction Methods; Anderson, P., Munslow, I.M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 305–307. ISBN 9783527317240. [Google Scholar]
- Freifelder, M. A Low Pressure Process for the Reduction of Nitriles. Use of Rhodium Catalyst. J. Am. Chem. Soc. 1960, 82, 2386–2389. [Google Scholar] [CrossRef]
- Enthaler, S.; Junge, K.; Addis, D.; Erre, G.; Beller, M. A practical and benign synthesis of primary amines through ruthenium-catalyzed reduction of nitriles. ChemSusChem 2008, 1, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Segobia, D.J.; Trasarti, A.F.; Apesteguía, C.R. Hydrogenation of nitriles to primary amines on metal-supported catalysts: Highly selective conversion of butyronitrile to n-butylamine. Appl. Catal. A Gen. 2012, 445, 69–75. [Google Scholar] [CrossRef]
- Vilches-Herrera, M.; Werkmeister, S.; Junge, K.; Börner, A.; Beller, M. Selective catalytic transfer hydrogenation of nitriles to primary amines using Pd/C. Catal. Sci. Technol. 2014, 4, 629–632. [Google Scholar] [CrossRef]
- Schärringer, P.; Müller, T.E.; Lercher, J.A. Investigations into the mechanism of the liquid-phase hydrogenation of nitriles over Raney-Co catalysts. J. Catal. 2008, 253, 167–179. [Google Scholar] [CrossRef]
- Krupka, J.; Pasek, J. Nitrile Hydrogenation on Solid Catalysts—New Insights into the Reaction Mechanism. Curr. Org. Chem. 2012, 16, 988–1004. [Google Scholar] [CrossRef]
- Bagal, D.B.; Bhanage, B.M. Recent advances in transition metal-catalyzed hydrogenation of nitriles. Adv. Synth. Catal. 2015, 357, 883–900. [Google Scholar] [CrossRef]
- Lévay, K.; Hegedűs, L. Selective Heterogeneous Catalytic Hydrogenation of Nitriles to Primary Amines. Period. Polytech. Chem. Eng. 2018, 62, 476–488. [Google Scholar] [CrossRef]
- Braun, J.; Blessing, G.; Zobel, F. Katalytische Hydrierungen unter Druck bei Gegenwart von Nickelsalzen, VI.: Nitrile. Ber. Dtsch. Chem. Ges. 1923, 36, 1988–2001. [Google Scholar] [CrossRef]
- Greenfield, H. Hydrogenation of Benzonitrile to Dibenzylamine. Ind. Eng. Chem. Prod. Res. Dev. 1976, 15, 156–158. [Google Scholar] [CrossRef]
- Hartung, W.H. Catalytic reduction of nitriles and oximes. J. Am. Chem. Soc. 1928, 50, 3370–3374. [Google Scholar] [CrossRef]
- Rosenmund, K.W.; Schindler, H. Über die katalytische Reduktion von Mandelsäuren. Arch. Pharm. (Weinh.) 1928, 432, 281–283. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, G.; Li, Y.; Yan, X.; Chen, L. Racemization of R-2-amino-1-butanol catalyzed by a fixed-bed Raney cobalt catalyst. J. Mol. Catal. A Chem. 2006, 255, 269–274. [Google Scholar] [CrossRef]
- Hertzberg, R.; Dinér, P.; Moberg, C. Palladium-Catalyzed C(sp3)–C(sp2) Cross-Couplings of O-(α-Bromoacyl) Cyanohydrins with Boronic Acids: An Entry to Enantio-enriched N-Acylated β-Amino Alcohols. Synthesis (Stuttg.) 2016, 48, 3175–3182. [Google Scholar] [CrossRef]
- Hertzberg, R.; Monreal Santiago, G.; Moberg, C. Synthesis of the β3-adrenergic receptor agonist Solabegron and Analogous N-(2-Ethylamino)-β-amino alcohols from O-acylated cyanohydrins—Expanding the scope of minor enantiomer recycling. J. Org. Chem. 2015, 80, 2937–2941. [Google Scholar] [CrossRef]
- Gomez, S.; Peters, J.A.; Maschmeyer, T. The Reductive Animation of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037–1057. [Google Scholar] [CrossRef]
- Banwell, M.G.; Jones, M.T.; Reekie, T.A.; Schwartz, B.D.; Tan, S.H.; White, L.V. RANEY® cobalt-an underutilised reagent for the selective cleavage of C-X and N-O bonds. Org. Biomol. Chem. 2014, 12, 7433–7444. [Google Scholar] [CrossRef]
- Bhor, M.D.; Bhanushali, M.J.; Nandurkar, N.S.; Bhanage, B.M. Highly efficient chemoselective catalytic hydrogenation of diaryl substituted α,β-unsaturated nitriles/carbonyls using homogeneous Pd(OAc)2/PPh3 catalyst. Catal. Commun. 2007, 8, 2064–2068. [Google Scholar] [CrossRef]
- Volf, J.; Pasek, J. Hydrogenation of Nitriles. In Studies in Surface Science and Catalysis: Catalytic Hydrogenation; Cerveny, L., Ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1986; Volume 27, pp. 105–144. ISBN 0444426825. [Google Scholar]
- Huang, Y.; Adeeva, V.; Sachtler, W.M.H. Stability of supported transition metal catalysts in the hydrogenation of nitriles. Appl. Catal. A Gen. 2000, 196, 73–85. [Google Scholar] [CrossRef]
- Fureby, A.M.; Virto, C.; Adlercreutz, P.; Mattiasson, B. Acyl group migrations in 2-monoolein. Biocatal. Biotransform. 1996, 14, 89–111. [Google Scholar] [CrossRef]
- Hsiao, M.K.; Lo, W.T.; Wang, J.H.; Chen, H.L. Hydrogenation of Hydrogen Cyanide to Methane and Ammonia by a Metal Catalyst: Insight from First-Principles Calculations. J. Phys. Chem. C 2016, 120, 22946–22956. [Google Scholar] [CrossRef]
- Besson, P.; Thirion, P. Process for Manufacture of Methylamines. US3468953A, 23 September 1969. [Google Scholar]
- Malona, J.A.; Cariou, K.; Spencer, W.T.; Frontier, A.J. Total synthesis of (±)-rocaglamide via oxidation-initiated nazarov cyclization. J. Org. Chem. 2012, 77, 1891–1908. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Catalyst Loading (g/g) 1 | Solvent | Conversion (%) | Tembamide Yield (%) 2 | Selectivity (%) 3 |
|---|---|---|---|---|---|---|
| 1 | Pd (C) 4 | 0.25 | dioxane | 100 | 0 | 0 |
| 2 | Raney Co 86% slurry | 2.2 | dioxane | 23 | 0.1 | 0.2 |
| 3 | Ni@Al2O3/SiO2 65% | 1 | dioxane | 19 | 0 | 0 |
| 4 | Raney Ni 50% slurry | 2 | dioxane | 92 | 4.4 | 4.8 |
| 5 | Rh (C) 5% | 1 | dioxane | 100 | 4.5 | 4.5 |
| 6 | Rh@SiO2 1% | 3 | dioxane | 38 | 1.1 | 2.9 |
| 7 | Rh@Al2O3 5% | 1 | dioxane | 53 | 0.3 | 0.6 |
| 8 | RhCl3·3H2O | 1 | dioxane | 47 | 0 | 0 |
| 9 | RuO2·H2O | 1 | dioxane | 37 | 0 | 0 |
| 10 | Rh@SiO2 1% | 3 | iPr2O | 45 | 4 | 8.9 |
| Entry | Catalyst | Catalyst Loading (g/g) 1 | Conversion (%) | Tembamide Yield (%) 2 | Selectivity (%) 3 |
|---|---|---|---|---|---|
| 1 | Ni@Al2O3/SiO2 65% 4 | 1 | 94 | 12 | 13 |
| 2 | Raney Ni 50% slurry | 3.5 | 100 | 25 | 25 |
| 3 | Raney Co 86% slurry | 3.5 | 69 | 13 | 19 |
| 4 | Rh (C) 5% | 1 | 100 | 0 | 0 |
| 5 | Rh@SiO2 1% | 2.5 | 100 | 0 | 0 |
| 6 | Rh@Al2O3 5% 4 | 1 | 100 | 0 | 0 |
| Entry | Catalyst Loading (g/g) 1 | (±)-3 (mM) | Additive | Reaction Time (h) | Conversion (%) | Tembamide Yield (%) 2 | Selectivity (%) 3 |
|---|---|---|---|---|---|---|---|
| 1 | 3.5 | 19 | 4 Å molecular sieves 4 | 2.5 | 100 | 21 | 21 |
| 2 | 3.5 | 19 | 2.5% MilliQ water | 2.5 | 100 | 6 | 6 |
| 3 | 3.5 | 19 | - | 2 | 100 | 24 | 24 |
| 4 | 0.5 | 19 | - | 1 | 100 | 27 | 27 |
| 5 | 0.25 | 19 | - | 1 | 96 | 20 | 21 |
| 6 | 0.5 | 112 | - | 2 | 98 | 16 | 16 |
| 7 | 0.5 | 84 | - | 2 | 95 | 17 | 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leemans, L.; Walter, M.D.; Hollmann, F.; Schallmey, A.; van Langen, L.M. Multi-Catalytic Route for the Synthesis of (S)-Tembamide. Catalysts 2019, 9, 822. https://doi.org/10.3390/catal9100822
Leemans L, Walter MD, Hollmann F, Schallmey A, van Langen LM. Multi-Catalytic Route for the Synthesis of (S)-Tembamide. Catalysts. 2019; 9(10):822. https://doi.org/10.3390/catal9100822
Chicago/Turabian StyleLeemans, Laura, Marc D. Walter, Frank Hollmann, Anett Schallmey, and Luuk M. van Langen. 2019. "Multi-Catalytic Route for the Synthesis of (S)-Tembamide" Catalysts 9, no. 10: 822. https://doi.org/10.3390/catal9100822
APA StyleLeemans, L., Walter, M. D., Hollmann, F., Schallmey, A., & van Langen, L. M. (2019). Multi-Catalytic Route for the Synthesis of (S)-Tembamide. Catalysts, 9(10), 822. https://doi.org/10.3390/catal9100822