Single-Atom X/g-C3N4(X = Au1, Pd1, and Ru1) Catalysts for Acetylene Hydrochlorination: A Density Functional Theory Study

Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Geometrical Structure of the Reactants
3.2. Adsorption of C2H2 and HCl onto SACs
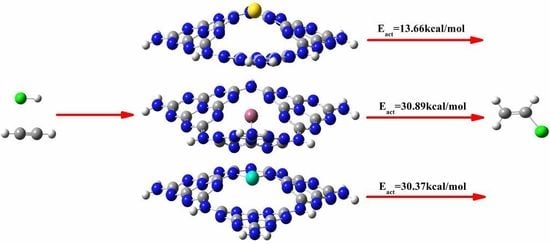
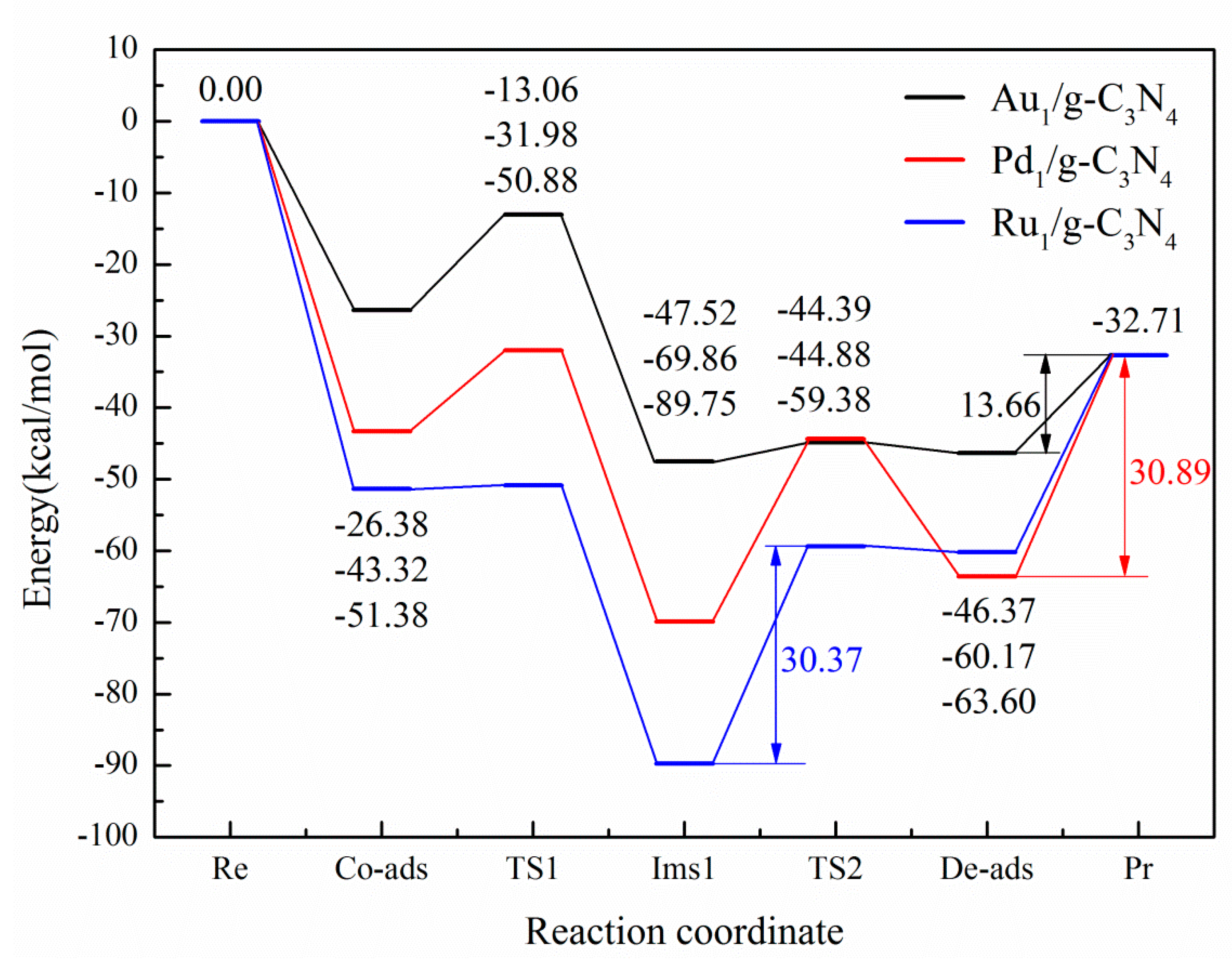
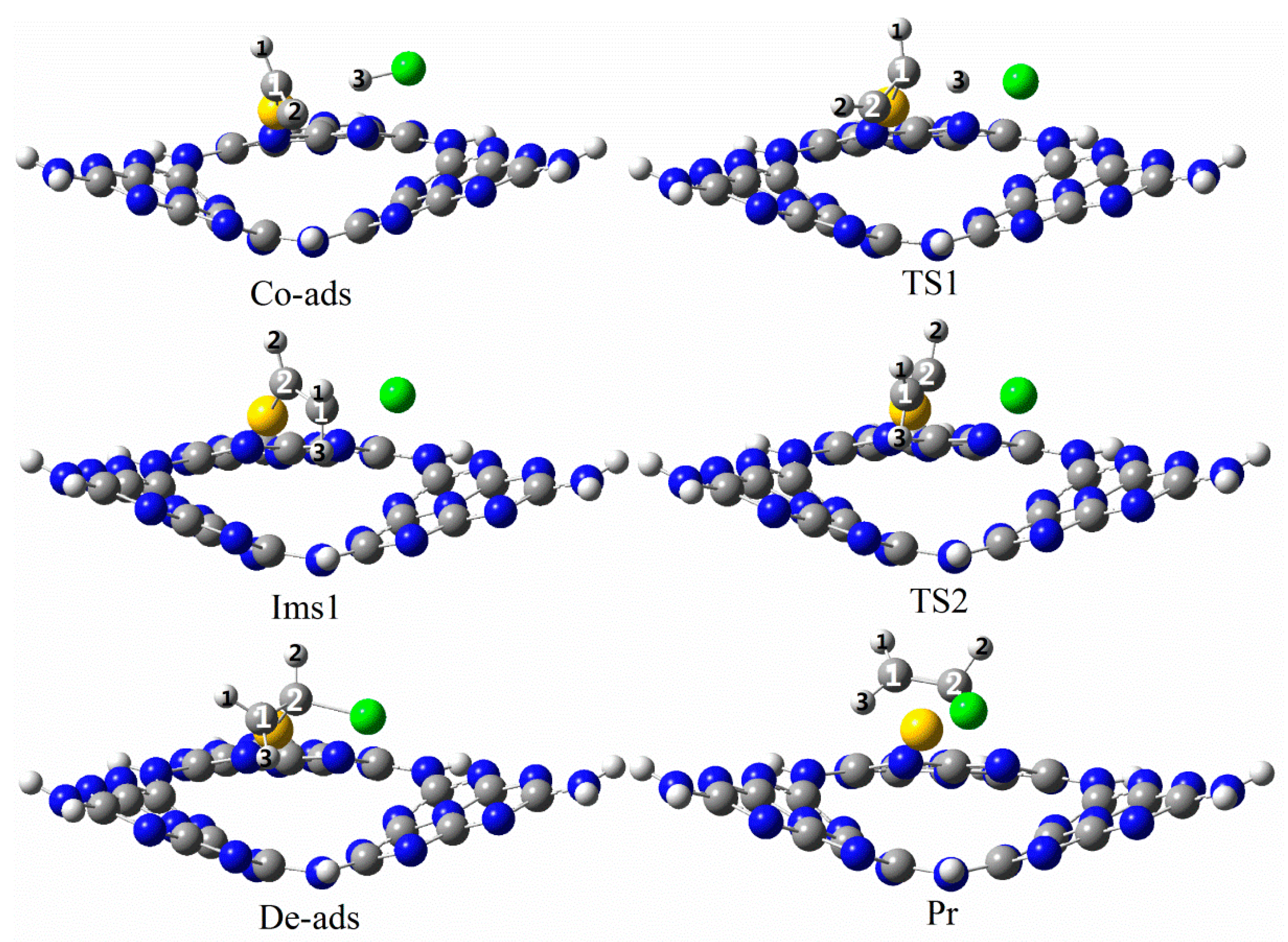
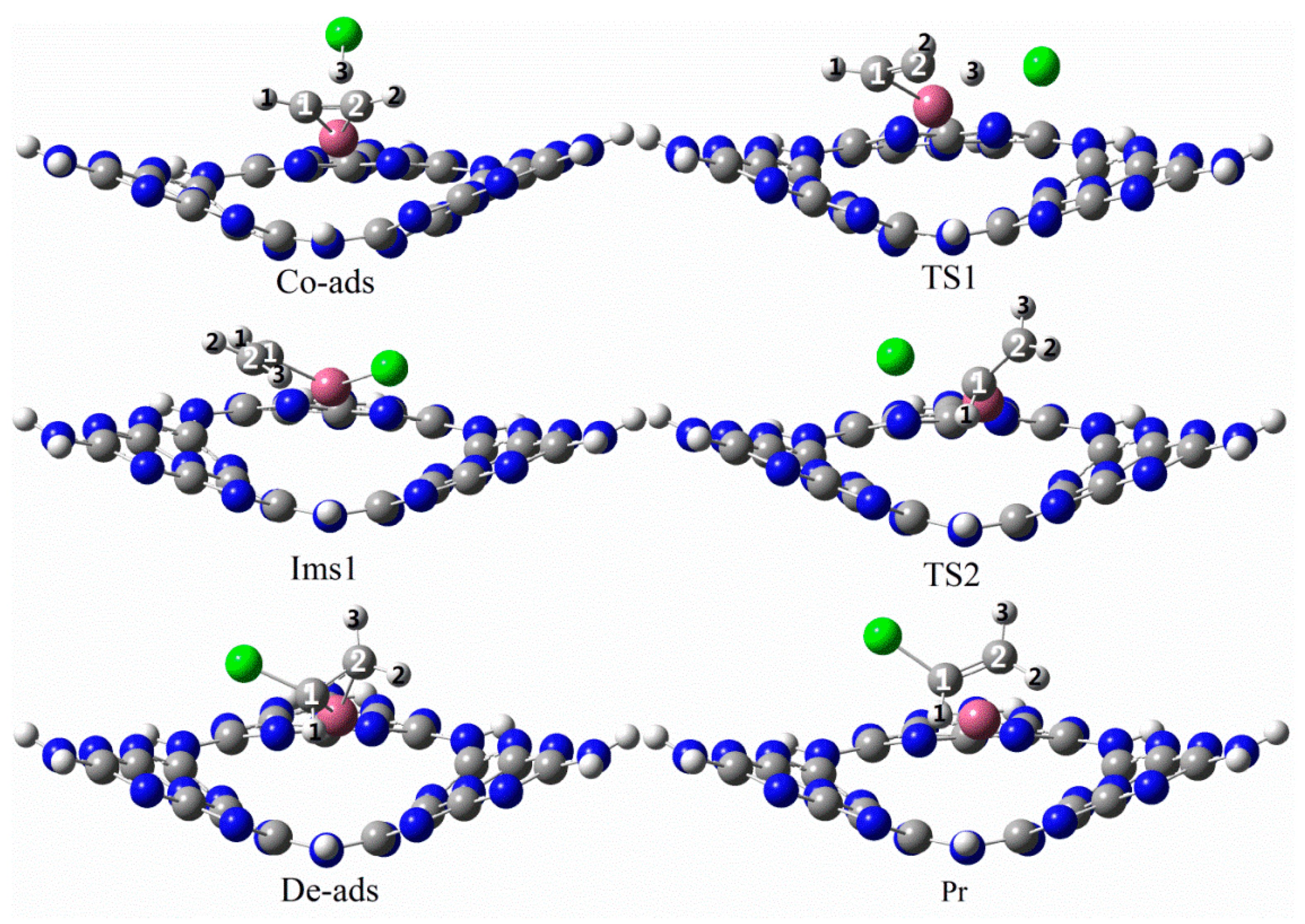
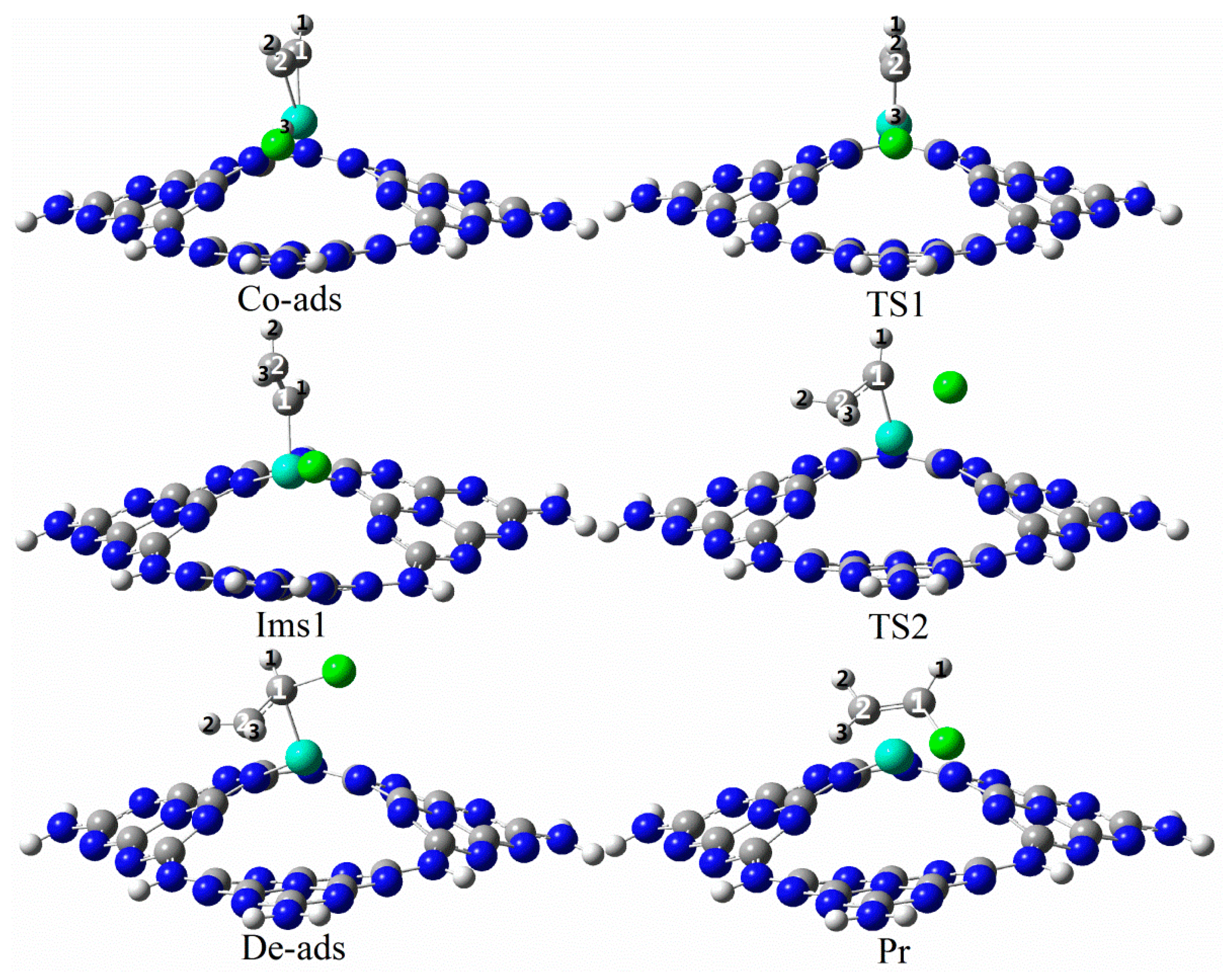
3.3. The Possible Reaction Mechanisms for Acetylene Hydrogenation on SACs
3.3.1. Reaction Mechanism Utilizing Au1/g-C3N4
3.3.2. Reaction Mechanism Utilizing Pd1/g-C3N4
3.3.3. Reaction Mechanism Utilizing Ru1/g-C3N4
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liang, S.; Shi, Y.; Hao, C. The Power of Single-Atom Catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Accounts Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gu, X.; Cao, Q.; Hu, P.; Hao, J.; Li, J.; Tang, X. Catalytically Active Single-Atom Sites Fabricated from Silver Particles. Angew. Chem. Int. Ed. 2012, 51, 4198–4203. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Allard, L.F.; Flytzani-Stephanopoulos, M. Atomically Dispersed Au–(OH)x Species Bound on Titania Catalyze the Low-Temperature Water-Gas Shift Reaction. J. Am. Chem. Soc. 2013, 135, 3768–3771. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.R.; Du, A. Single Atom (Pd/Pt) Supported on Graphitic Carbon Nitride as an Efficient Photocatalyst for Visible-Light Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef]
- Gong, W.; Zhao, F.; Kang, L. Novel nitrogen-doped Au-embedded graphene single-atom catalysts for acetylene hydrochlorination: A density functional theory study. Comput. Theor. Chem. 2018, 1130, 83–89. [Google Scholar] [CrossRef]
- Johnston, P.; Carthey, N.; Hutchings, G.J. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar] [CrossRef]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Lu, L.; Dawson, S.; Thetford, A.; Gibson, E.K.; Morgan, D.J.; Jones, W.; et al. Identification of single-site gold catalysis in acetylene hydrochlorination. Science 2017, 355, 1399–1403. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.; Heirene, C.; Willock, D.; Johnston, P.; Herzing, A.; Kiely, C.; Hutchings, G. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, J.; Xu, J.; Zhang, T.; Di, X.; Ni, J.; Li, X. Enhancement of Au/AC acetylene hydrochlorination catalyst activity and stability via nitrogen-modified activated carbon support. Chem. Eng. J. 2015, 262, 1152–1160. [Google Scholar] [CrossRef]
- Malta, G.; Freakley, S.J.; Kondrat, S.A.; Hutchings, G.J. Acetylene hydrochlorination using Au/carbon: A journey towards single site catalysis. Chem. Commun. 2017, 53, 11733–11746. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shi, H.; Qian, W.; Luo, G.; Jin, Y.; Wei, F. Gas-Phase Catalytic Hydrochlorination of Acetylene in a Two-Stage Fluidized-Bed Reactor. Ind. Eng. Chem. Res. 2009, 48, 128–133. [Google Scholar] [CrossRef]
- Zhou, K.; Wang, W.; Zhao, Z.; Luo, G.; Miller, J.T.; Wong, M.S.; Wei, F. Synergistic Gold–Bismuth Catalysis for Non-Mercury Hydrochlorination of Acetylene to Vinyl Chloride Monomer. ACS Catal. 2014, 4, 3112–3116. [Google Scholar] [CrossRef]
- Zhao, W.; Zhu, M.; Dai, B. Cobalt-nitrogen-activated carbon as catalyst in acetylene hydrochlorination. Catal. Commun. 2017, 98, 22–25. [Google Scholar] [CrossRef]
- Hutchings, G. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Nkosi, B.; Coville, N.J.; Hutchings, G.J. Reactivation of a supported gold catalyst for acetylene hydrochlorination. J. Chem. Soc. Chem. Commun. 1988, 1, 71–72. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, M.; Kang, L.; Su, Y.; Dai, B. MClx (M = Hg, Au, Ru; x = 2, 3) catalyzed hydrochlorination of acetylene—A density functional theory study. Can. J. Chem. 2013, 91, 120–125. [Google Scholar]
- Zhu, M.; Wang, Q.; Chen, K.; Wang, Y.; Huang, C.; Dai, H.; Yu, F.; Kang, L.; Dai, B. Development of a Heterogeneous Non-Mercury Catalyst for Acetylene Hydrochlorination. ACS Catal. 2015, 5, 5306–5316. [Google Scholar] [CrossRef]
- Zhang, J.; Sheng, W.; Guo, C.; Li, W. Acetylene hydrochlorination over bimetallic Ru-based catalysts. RSC Adv. 2013, 3, 21062. [Google Scholar] [CrossRef]
- Pu, Y.; Zhang, J.; Yu, L.; Jin, Y.; Li, W. Active ruthenium species in acetylene hydrochlorination. Appl. Catal. A Gen. 2014, 488, 28–36. [Google Scholar] [CrossRef]
- Li, G.; Pu, Y.; Sun, M.; Zhang, H. Non-mercury catalytic acetylene hydrochlorination over Ru catalysts enhanced by carbon nanotubes. RSC Adv. 2015, 5, 9002–9008. [Google Scholar] [CrossRef]
- Li, G.; Pu, Y.; Jin, Y.; Zhang, J. Effects of potassium additive on the activity of Ru catalyst for acetylene hydrochlorination. RSC Adv. 2015, 5, 37774–37779. [Google Scholar]
- Sun, M.; Han, Y.; Li, W.; Zhang, J. Influence of chlorine coordination number on the catalytic mechanism of ruthenium chloride catalysts in the acetylene hydrochlorination reaction: A DFT study. Phys. Chem. Chem. Phys. 2015, 17, 7720–7730. [Google Scholar]
- Xu, J.; Zhang, T.; Di, X.; Gu, S.; Zhao, J.; Ni, J.; Li, X. Ultra-low Ru-promoted CuCl2 as highly active catalyst for the hydrochlorination of acetylene. RSC Adv. 2015, 5, 38159–38163. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.; Wang, J.; Tang, X.; Zhao, Y.; Yang, D.; Jia, F.; Hao, T. Hydrochlorination of acetylene to vinyl chloride over Pd supported on zeolite Y. React. Kinet. Mech. Catal. 2013, 110, 187–194. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.; Wang, J. Catalytic properties of Pd/HY catalysts modified with NH4F for acetylene hydrochlorination. Catal. Commun. 2015, 65, 41–45. [Google Scholar] [CrossRef]
- Hu, J.; Yang, Q.; Yang, L.; Zhang, Z.; Su, B.; Bao, Z.; Ren, Q.; Xing, H.; Dai, S. Confining Noble Metal (Pd, Au, Pt) Nanoparticles in Surfactant Ionic Liquids: Active Non-Mercury Catalysts for Hydrochlorination of Acetylene. ACS Catal. 2015, 5, 6724–6731. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, M.; Kang, L.; Dai, B. Neutral Aun (n = 3–10) clusters catalyze acetylene hydrochlorination: A density functional theory study. RSC Adv. 2014, 4, 38466–38473. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, F.; Kang, L. Catalysis of the acetylene hydrochlorination reaction by Si-doped Au clusters: A DFT study. J. Mol. Model. 2018, 24, 61. [Google Scholar] [CrossRef] [PubMed]
- Fei, Z.; Yang, W.; Lihua, K. A density functional theory study on the performance of graphene and N-doped graphene supported Au 3 cluster catalyst for acetylene hydrochlorination. Can. J. Chem. 2016, 94, 842–847. [Google Scholar]
- Ma, X.; Lv, Y.; Xu, J.; Liu, Y.; Zhang, R.; Zhu, Y. A Strategy of Enhancing the Photoactivity of g-C3N4 via Doping of Nonmetal Elements: A First-Principles Study. J. Phys. Chem. C 2012, 116, 23485–23493. [Google Scholar] [CrossRef]
- Wang, X.; Blechert, S.; Antonietti, M. Polymeric Graphitic Carbon Nitride for Heterogeneous Photocatalysis. ACS Catal. 2012, 2, 1596–1606. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Chen, J.; Liu, J.; Liang, J.; Du, A.; Zhang, W.; Zhu, Z.; Smith, S.C.; Jaroniec, M.; et al. Nanoporous Graphitic-C3N4@Carbon Metal-Free Electrocatalysts for Highly Efficient Oxygen Reduction. J. Am. Chem. Soc. 2011, 133, 20116–20119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Kang, L.; Zhu, M.; Dai, B. A novel, non-metallic graphitic carbon nitride catalyst for acetylene hydrochlorination. J. Catal. 2014, 311, 288–294. [Google Scholar] [CrossRef]
- Zhao, W.; Zhu, M.; Dai, B. The Preparation of Cu-g-C3N4/AC Catalyst for Acetylene Hydrochlorination. Catal. 2016, 6, 193. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Zhu, Y.; Cai, Q.; Vasileff, A.; Li, L.H.; Han, Y.; Chen, Y.; Qiao, S.-Z. Molecule-Level g-C3N4 Coordinated Transition Metals as a New Class of Electrocatalysts for Oxygen Electrode Reactions. J. Am. Chem. Soc. 2017, 139, 3336–3339. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-L.; Yin, H.; Kan, X.; Gan, L.-Y.; Schwingenschlögl, U.; Zhao, Y. Potential of transition metal atoms embedded in buckled monolayer g-C 3 N 4 as single-atom catalysts. Phys. Chem. Chem. Phys. 2017, 19, 30069–30077. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09. Revision C.01, Gaussian Inc.: Wallingford, CT, USA, 2010.
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Krieg, H.; Grimme, S.; Ehrlich, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- González, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Kroke, E.; Schwarz, M.; Horath-Bordon, E.; Kroll, P.; Noll, B.; Norman, A.D. Tri-s-triazine derivatives. Part I. From trichloro-tri-s-triazine to graphitic C3N4 structures. New J. Chem. 2002, 26, 508–512. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, M.; Kang, L. The DFT Study of Single-Atom Pd1/g-C3N4 Catalyst for Selective Acetylene Hydrogenation Reaction. Catal. Lett. 2018, 148, 2992–3002. [Google Scholar] [CrossRef]
- Gracia, J.; Kroll, P. Corrugated layered heptazine-based carbon nitride: The lowest energy modifications of C3N4 ground state. J. Mater. Chem. 2009, 19, 3013. [Google Scholar] [CrossRef]
- Wang, X.; Dai, B.; Wang, Y.; Yu, F. Nitrogen-Doped Pitch-Based Spherical Active Carbon as a Nonmetal Catalyst for Acetylene Hydrochlorination. ChemCatChem 2014, 6, 2339–2344. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C2H2 | HCl | Co-Adsorption | |
|---|---|---|---|
| Au1/g-C3N4 | −14.07 | −10.64 | −26.38 |
| Pd1/g-C3N4 | −28.08 | −12.70 | −43.32 |
| Ru1/g-C3N4 | −35.81 | −3.38 | −51.38 |
| Catalyst | Energy Barrier | Reference |
|---|---|---|
| Au1/g-C3N4 | 13.66 | This work |
| Pd1/g-C3N4 | 30.89 | This work |
| Ru1/g-C3N4 | 30.37 | This work |
| g-C3N4/AC | 77.94 | [36] |
| AuG-SAC | 30.59 | [8] |
| PSAC-N | 28.83 | [48] |
| AuCl3 | 11.86 | [19] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Zhu, M.; Kang, L. Single-Atom X/g-C3N4(X = Au1, Pd1, and Ru1) Catalysts for Acetylene Hydrochlorination: A Density Functional Theory Study. Catalysts 2019, 9, 808. https://doi.org/10.3390/catal9100808
Zhou X, Zhu M, Kang L. Single-Atom X/g-C3N4(X = Au1, Pd1, and Ru1) Catalysts for Acetylene Hydrochlorination: A Density Functional Theory Study. Catalysts. 2019; 9(10):808. https://doi.org/10.3390/catal9100808
Chicago/Turabian StyleZhou, Xuening, Mingyuan Zhu, and Lihua Kang. 2019. "Single-Atom X/g-C3N4(X = Au1, Pd1, and Ru1) Catalysts for Acetylene Hydrochlorination: A Density Functional Theory Study" Catalysts 9, no. 10: 808. https://doi.org/10.3390/catal9100808
APA StyleZhou, X., Zhu, M., & Kang, L. (2019). Single-Atom X/g-C3N4(X = Au1, Pd1, and Ru1) Catalysts for Acetylene Hydrochlorination: A Density Functional Theory Study. Catalysts, 9(10), 808. https://doi.org/10.3390/catal9100808