Gas-Phase Phosphorous Poisoning of a Pt/Ba/Al2O3 NOx Storage Catalyst

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
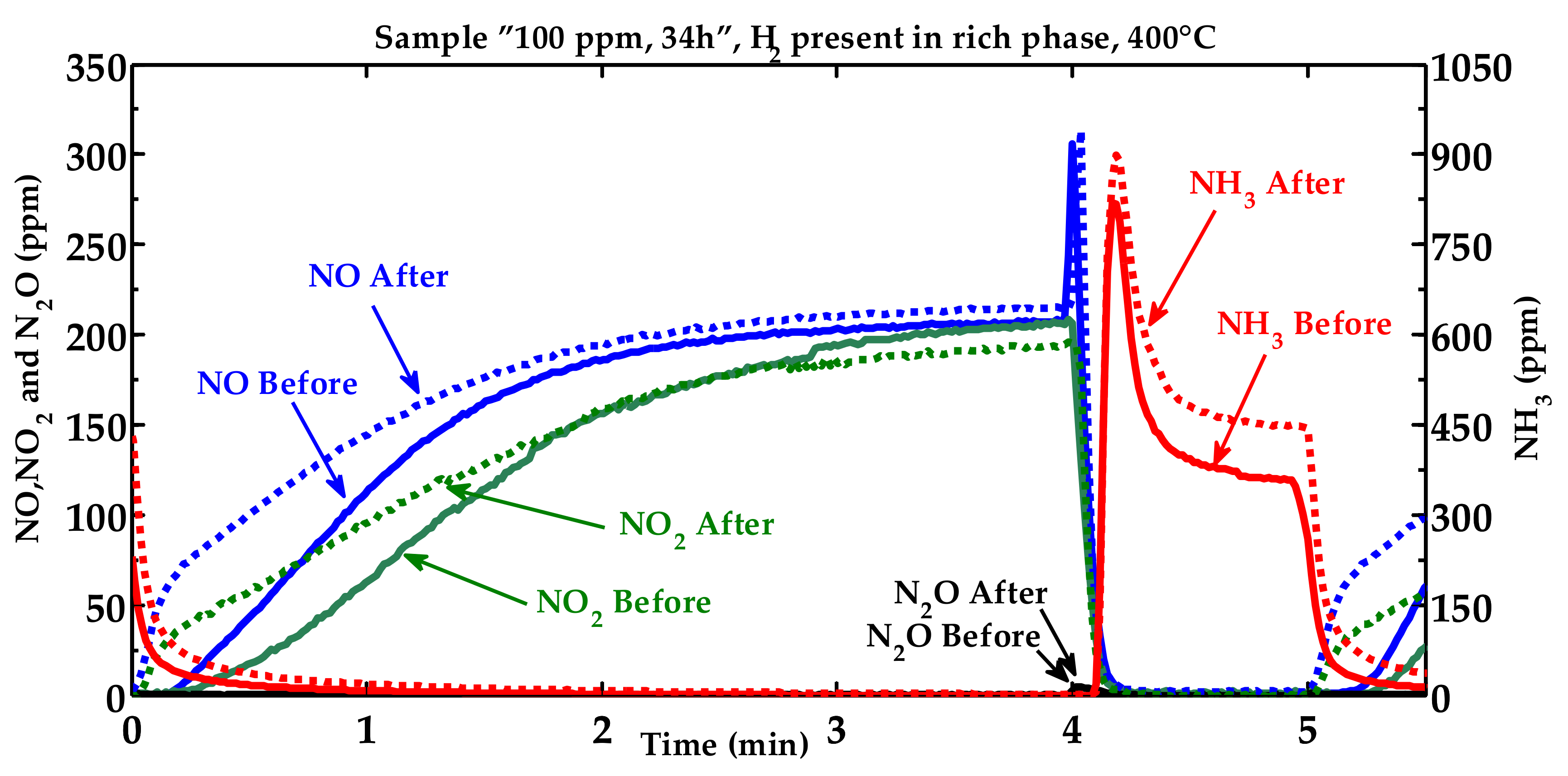
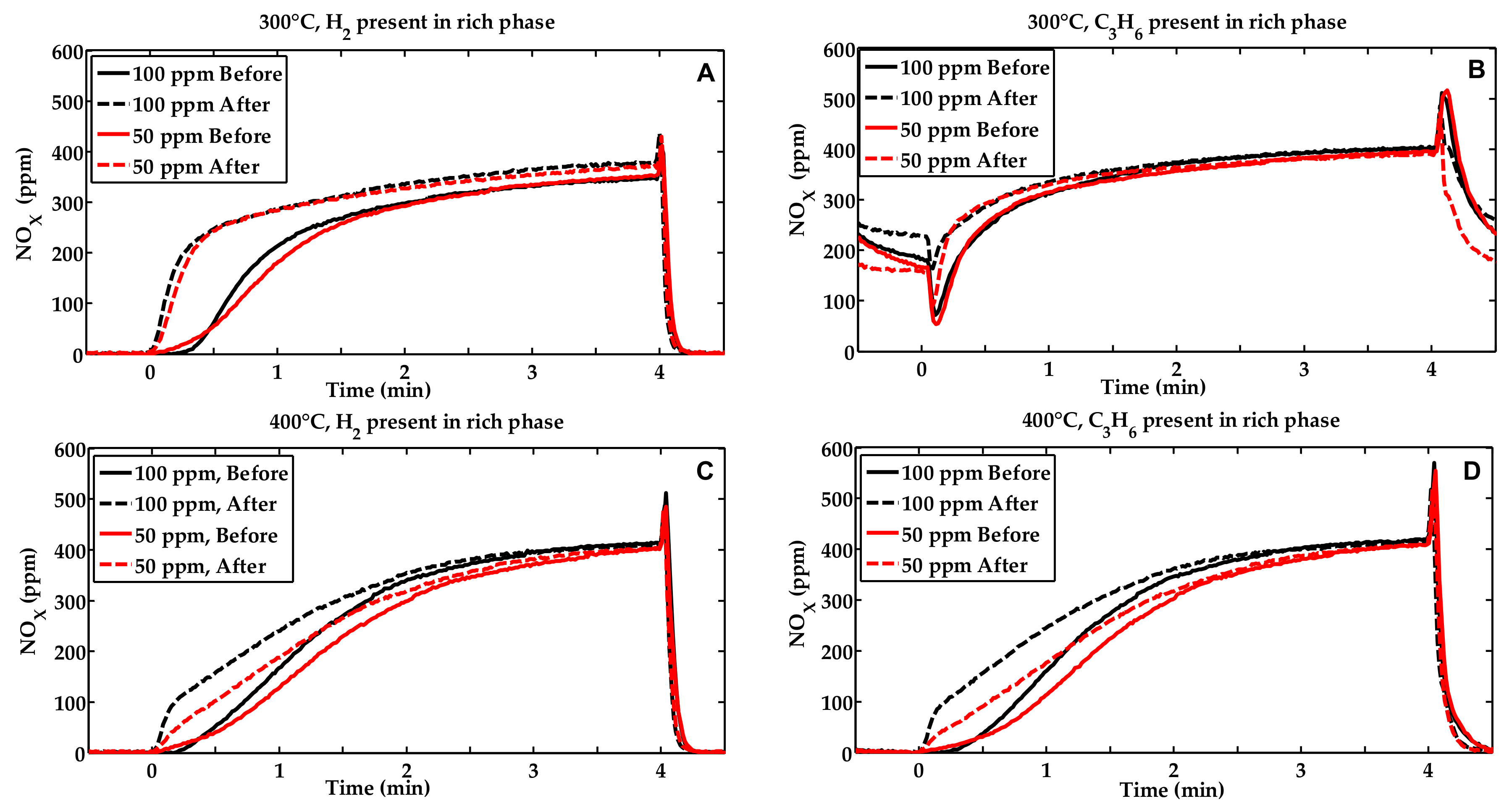
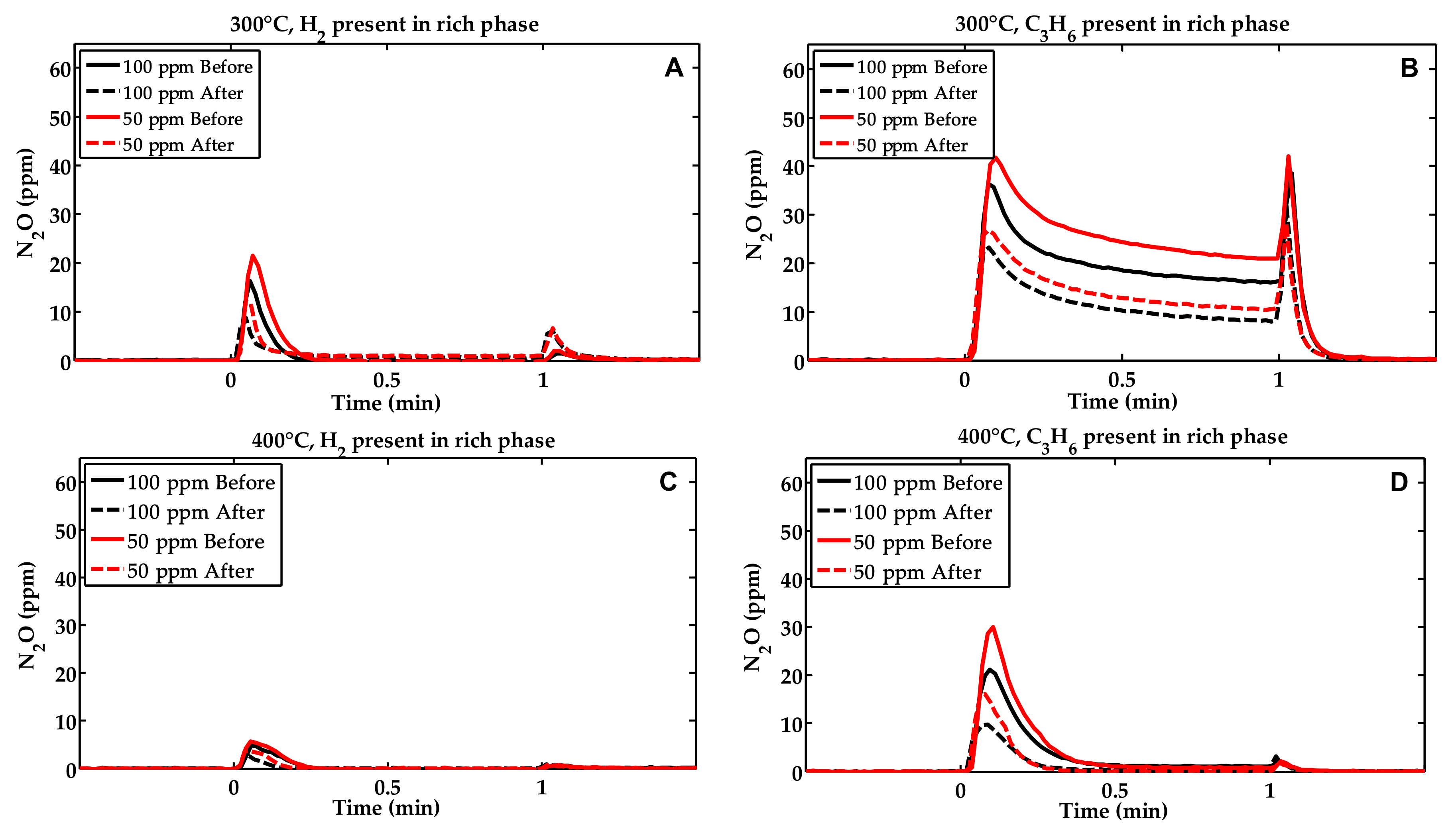
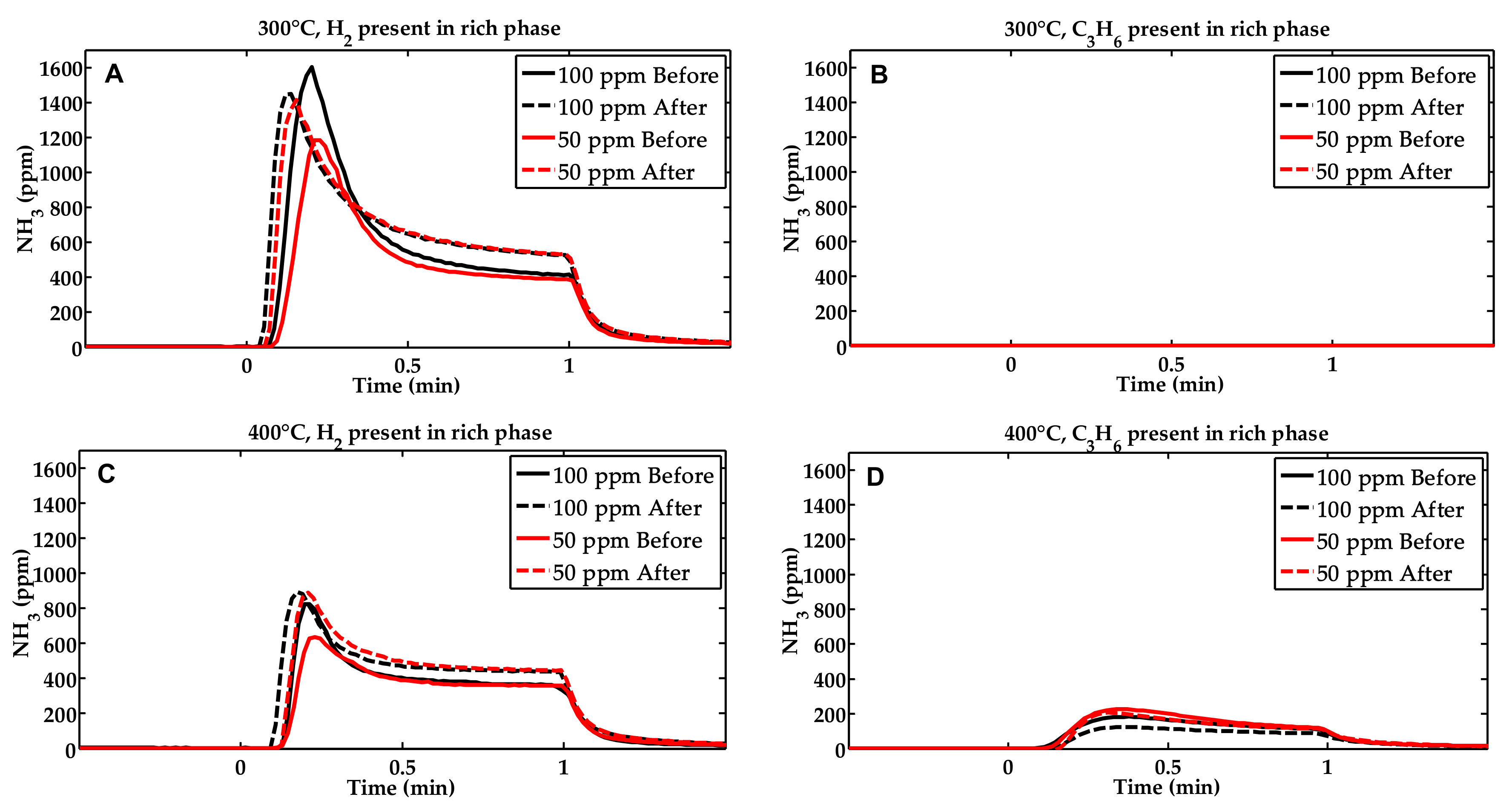
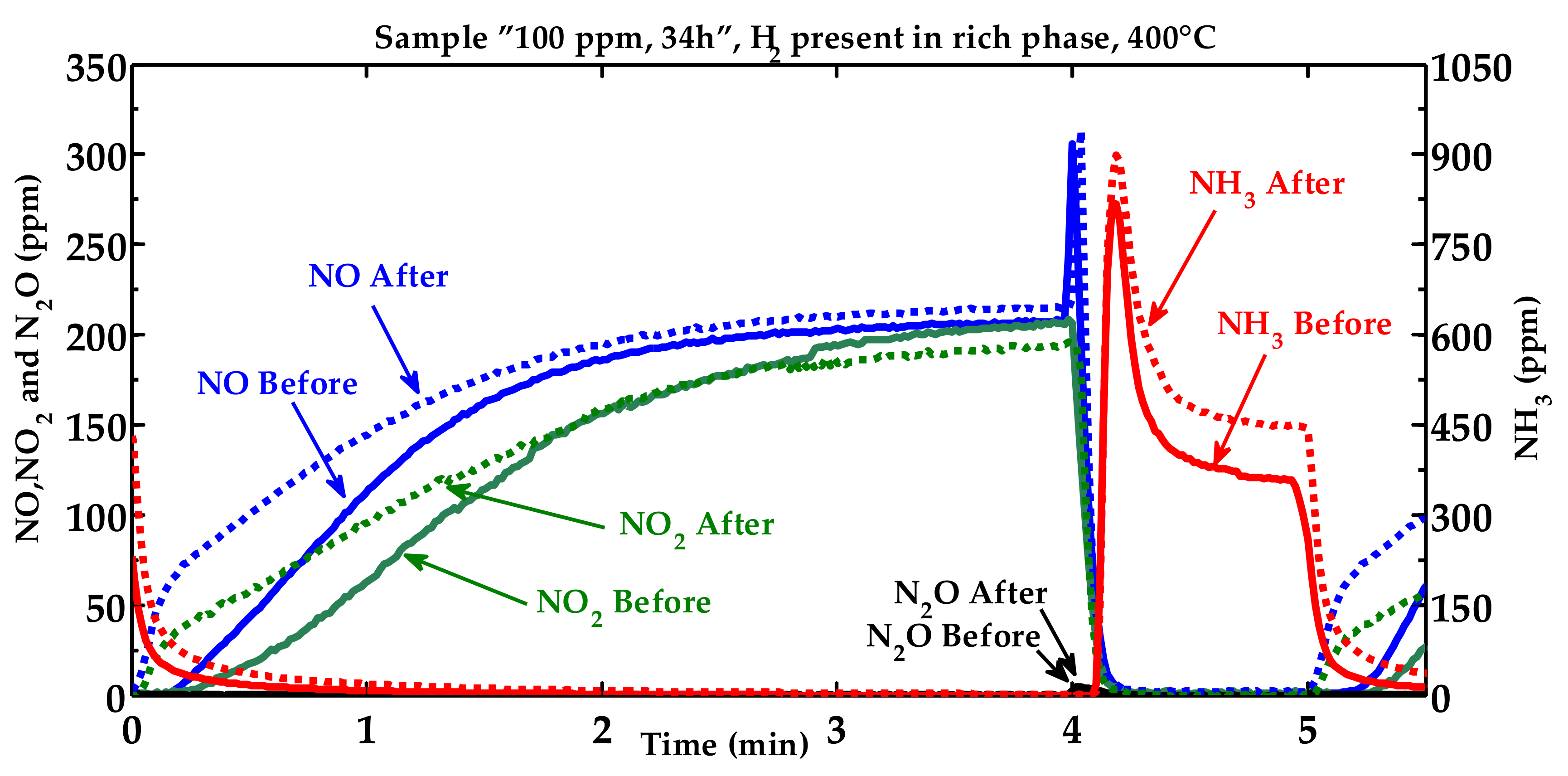
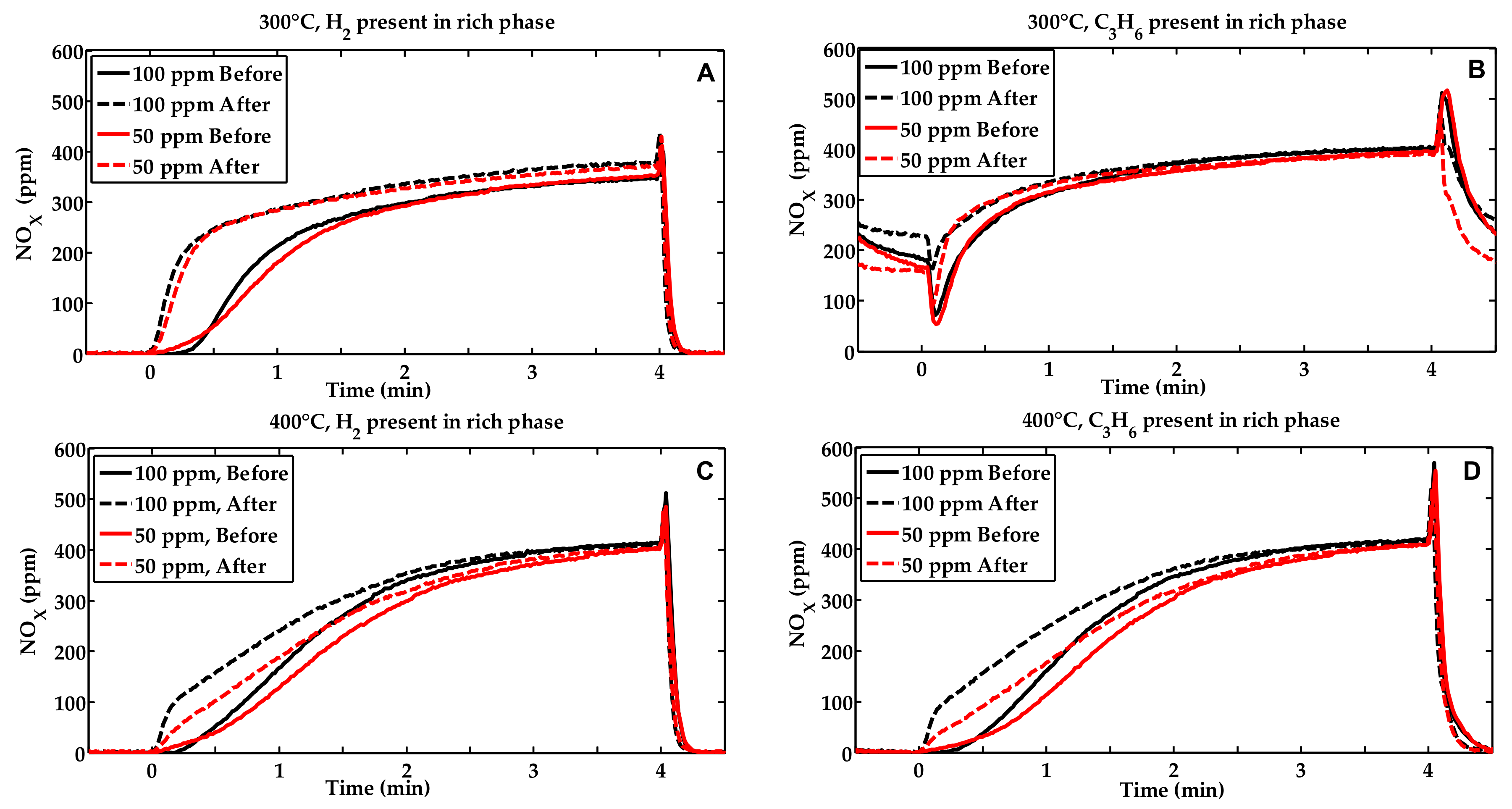
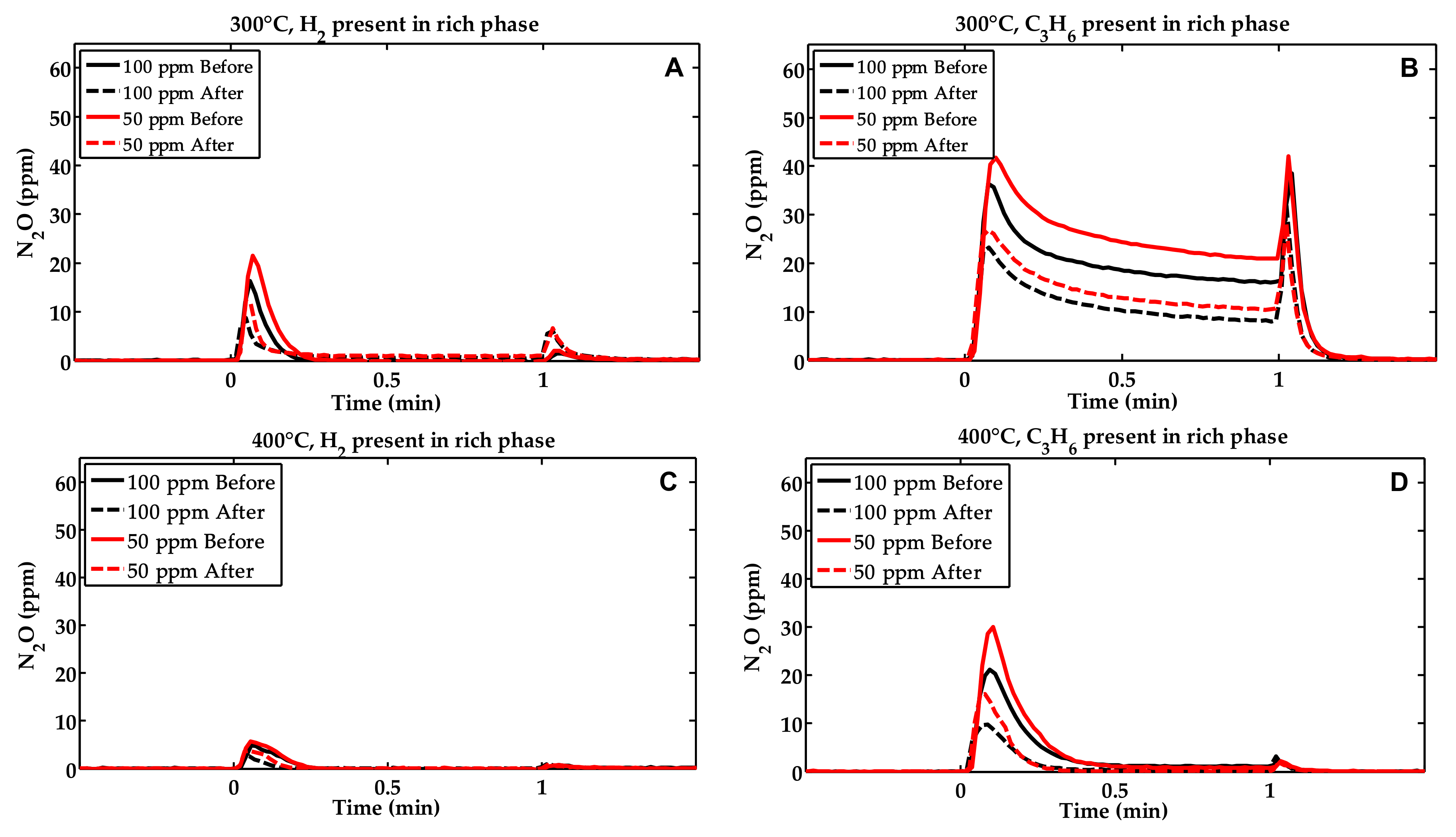
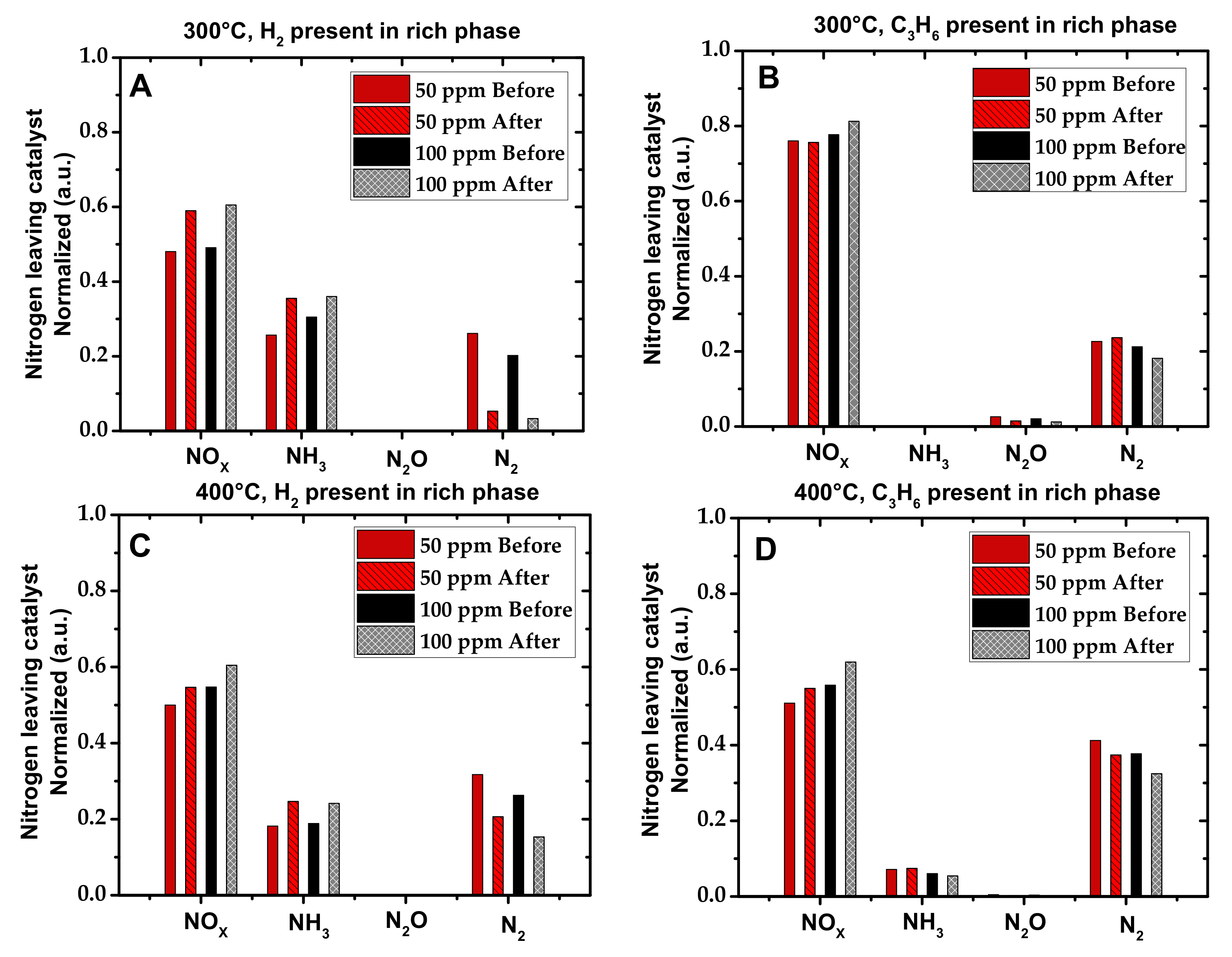
2.1. Activity Measurements
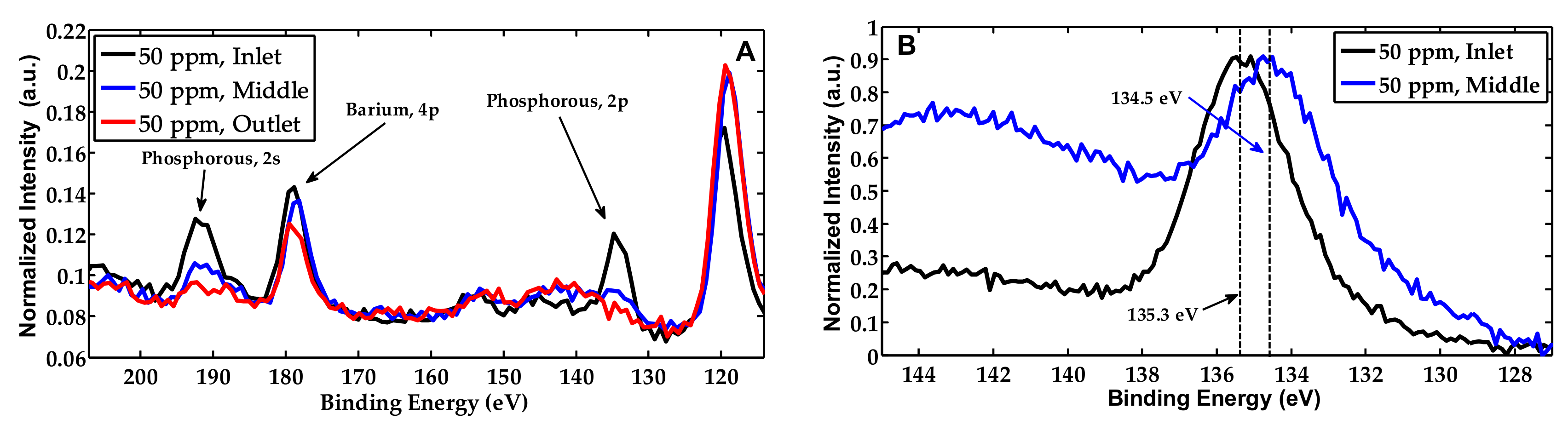
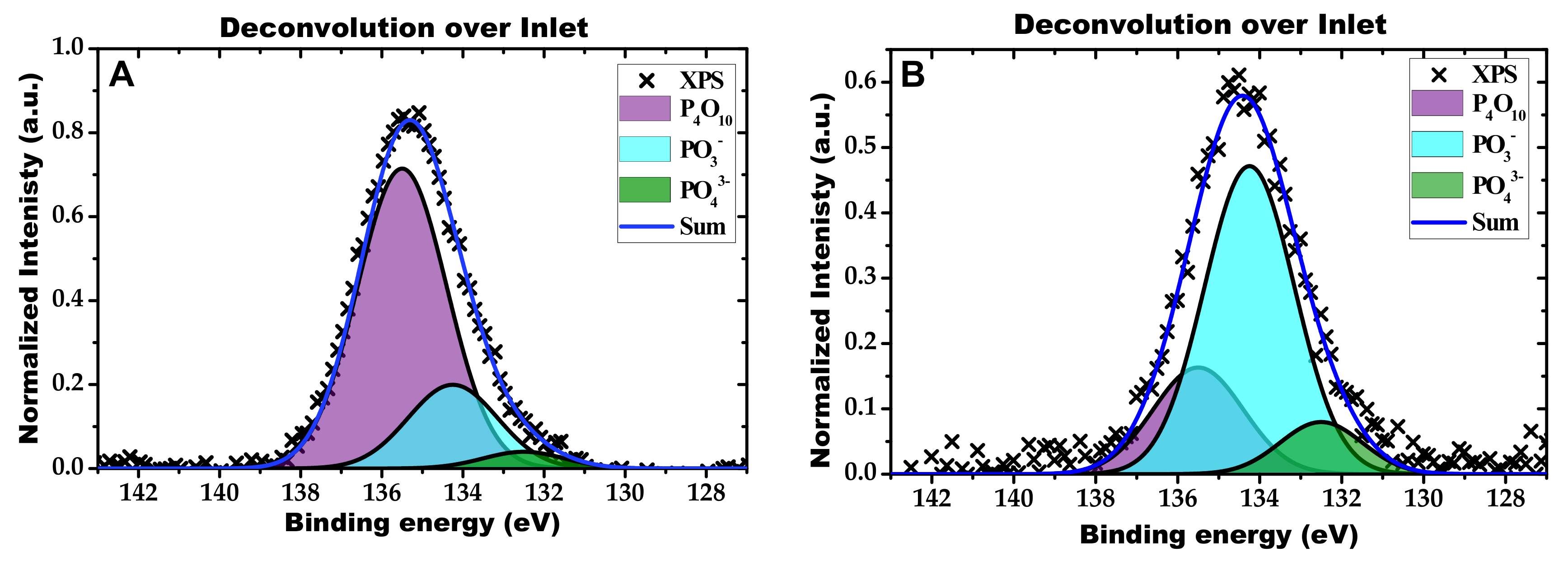
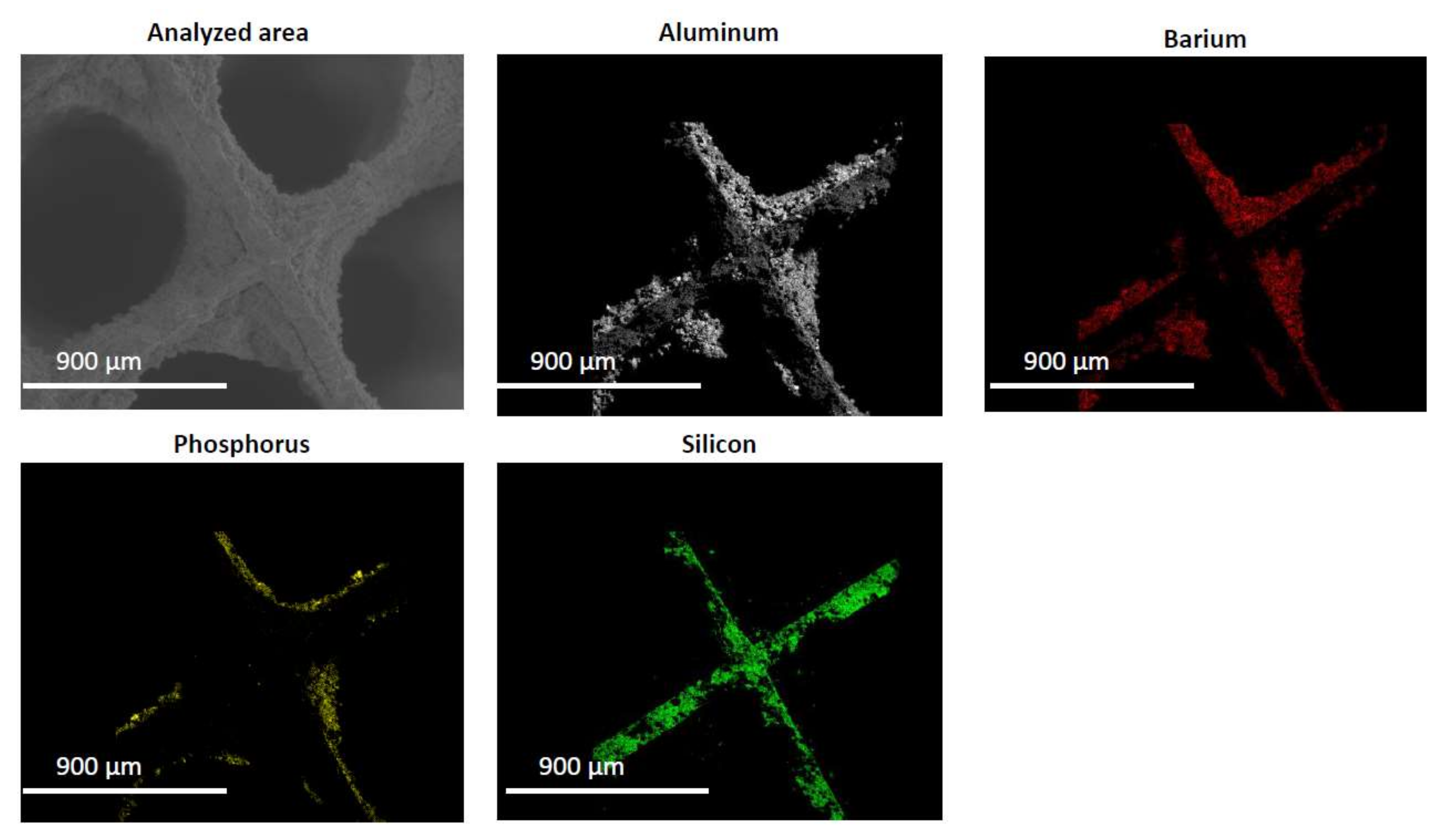
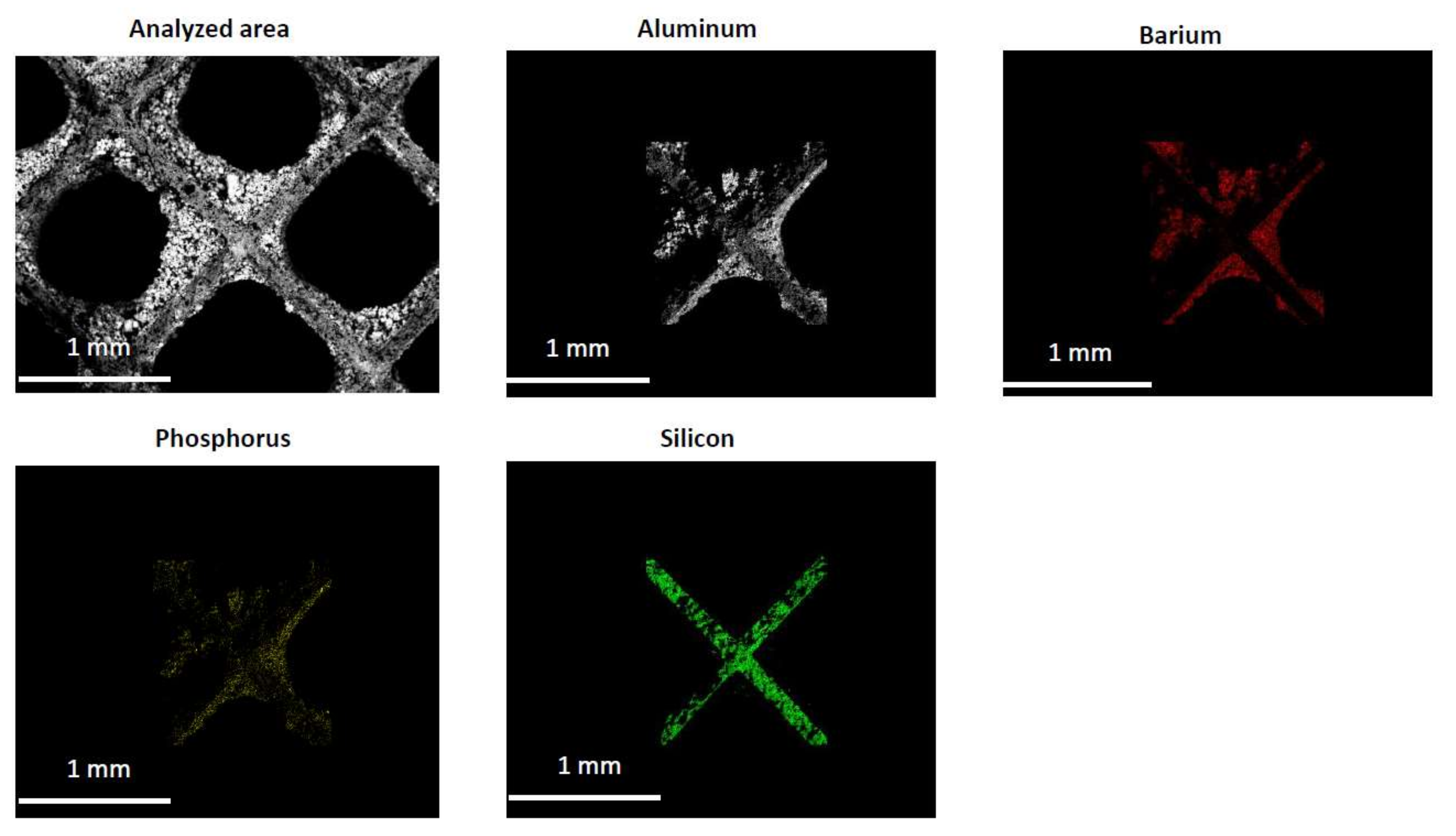
2.2. Characterization of the Phosphorous Exposed Samples
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Monolith Preparation
3.3. Flow-Reactor Experiments
3.4. Phosphorous Exposure
3.5. XPS
3.6. N2-Physisorption and ICP-AES
3.7. ESEM
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guerreiro, C.; Gonzalez Ortiz, A.; de Leeuw, F.; Viana, M.; Horalek, J. Air Quality in Europe—2016 Report; European Environment Agency (EEA): Copenhagen, Denmark, 2016; ISBN 9789292138240. [Google Scholar]
- Takahashi, N.; Shinjoh, H.; Iijima, T.; Suzuki, T.; Yamazaki, K.; Yokota, K.; Suzuki, H.; Miyoshi, N.; Matsumoto, S.; Tanizawa, T.; et al. The new concept 3-way catalyst for automotive lean-burn engine: NOx storage and reduction catalyst. Catal. Today 1996, 27, 63–69. [Google Scholar] [CrossRef]
- Epling, W.S.; Campbell, L.E.; Yezerets, A.; Currier, N.W.; Parks, J.E. Overview of the fundamental reactions and degradation mechanisms of NOx storage/reduction catalysts. Catal. Rev. Sci. Eng. 2004, 46, 163–245. [Google Scholar] [CrossRef]
- Matsumoto, S. Recent advances in automobile exhaust catalysts. Catal. Today 2004, 90, 183–190. [Google Scholar] [CrossRef]
- Fridell, E.; Skoglundh, M.; Westerberg, B.; Johansson, S.; Smedler, G. NOx Storage in Barium-Containing Catalysts. J. Catal. 1999, 183, 196–209. [Google Scholar] [CrossRef]
- Lindholm, A.; Currier, N.W.; Li, J.; Yezerets, A.; Olsson, L. Detailed kinetic modeling of NOx storage and reduction with hydrogen as the reducing agent and in the presence of CO2 and H2O over a Pt/Ba/Al catalyst. J. Catal. 2008, 258, 273–288. [Google Scholar] [CrossRef]
- Lindholm, A.; Currier, N.W.; Fridell, E.; Yezerets, A.; Olsson, L. NOx storage and reduction over Pt based catalysts with hydrogen as the reducing agent. Influence of H2O and CO2. Appl. Catal. B Environ. 2007, 75, 78–87. [Google Scholar] [CrossRef]
- Lindholm, A.; Sjövall, H.; Olsson, L. Reduction of NOx over a combined NSR and SCR system. Appl. Catal. B Environ. 2010, 98, 112–121. [Google Scholar] [CrossRef]
- Nova, I.; Castoldi, L.; Lietti, L.; Tronconi, E.; Forzatti, P.; Prinetto, F.; Ghiotti, G. NOx adsorption study over Pt-Ba/alumina catalysts: FT-IR and pulse experiments. J. Catal. 2004, 222, 377–388. [Google Scholar] [CrossRef]
- Partridge, W.P.; Choi, J.S. NH3 formation and utilization in regeneration of Pt/Ba/Al2O3 NOx storage-reduction catalyst with H2. Appl. Catal. B Environ. 2009, 91, 144–151. [Google Scholar] [CrossRef]
- De Abreu Goes, J.E.; Olsson, L.; Berggrund, M.; Kristoffersson, A.; Gustafson, L.; Hicks, M. Performance Studies and Correlation between Vehicle- and Rapid-Aged Commercial Lean NOx Trap Catalysts. SAE Int. J. Engines 2017, 10. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanism of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Bunting, B.G.; More, K.; Lewis, S.; Toops, T. Phosphorous Poisoning and Phosphorous Exhaust Chemistry with Diesel Oxidation Catalysts. SAE Tech. Pap. 2005. [Google Scholar] [CrossRef]
- Sedlmair, C.; Seshan, K.; Jentys, A.; Lercher, J.A. Studies on the deactivation of NOx storage-reduction catalysts by sulfur dioxide. Catal. Today 2002, 75, 413–419. [Google Scholar] [CrossRef]
- Huang, H.Y.; Long, R.Q.; Yang, R.T. A highly sulfur resistant Pt-Rh/TiO2/Al2O3 storage catalyst for NOx reduction under lean-rich cycles. Appl. Catal. B Environ. 2001, 33, 127–136. [Google Scholar] [CrossRef]
- Umeno, T.; Hanzawa, M.; Hayashi, Y.; Hori, M. Development of New Lean NOx Trap Technology with High Sulfur Resistance. SAE Tech. Pap. 2014. [Google Scholar] [CrossRef]
- Le Phuc, N.; Corbos, E.C.; Courtois, X.; Can, F.; Marecot, P.; Duprez, D. NOx storage and reduction properties of Pt/CexZr1−xO2 mixed oxides: Sulfur resistance and regeneration, and ammonia formation. Appl. Catal. B Environ. 2009, 93, 12–21. [Google Scholar] [CrossRef]
- Darr, S.T.; Choksi, R.A.; Hubbard, C.P.; Johnson, M.D.; Mccabe, R.W.; Co, F.M. Effects of Oil-Derived Contaminants on Emissions from TWC-Equipped Vehicles. SAE Tech. Pap. 2000. [Google Scholar] [CrossRef]
- Sumida, H.; Koda, Y.; Sadai, A.; Ichikawa, S.; Kyogoku, M.; Takato, M.; Miwa, Y. Analysis of Phosphorus Poisoning on Exhaust Catalysts from Compact-Class Vehicle. SAE Tech. Pap. 2004. [Google Scholar] [CrossRef]
- Rokosz, M.J.; Chen, A.E.; Lowe-Ma, C.K.; Kucherov, A.V.; Benson, D.; Paputa Peck, M.C.; McCabe, R.W. Characterization of phosphorus-poisoned automotive exhaust catalysts. Appl. Catal. B Environ. 2001, 33, 205–215. [Google Scholar] [CrossRef]
- Angove, D.E.; Cant, N.W. Position dependent phenomena during deactivation of three-way catalytic converters on vehicles. Catal. Today 2000, 63, 371–378. [Google Scholar] [CrossRef]
- Ball, D.J.; Mohammed, A.G.; Schmidt, W.A. Application of Accelerated Rapid Aging Test (RAT) Schedules with Poisons: The Effects of Oil Derived Poisons, Thermal Degradation and Catalyst Volume on FTP Emissions. SAE Tech. Pap. 1997. [Google Scholar] [CrossRef]
- Guevremont, J.M.; Guinther, G.; Jao, T.; Herlihy, T.; White, R.; Howe, J. Total Phosphorus Detection and Mapping in Catalytic Converters. Powertrain Fluid Syst. Conf. Exhib. 2007. [Google Scholar] [CrossRef]
- Cabello Galisteo, F.; López Granados, M.; Martín Alonso, D.; Mariscal, R.; Fierro, J.L.G. Loss of NOx storage capacity of Pt-Ba/Al2O3 catalysts due to incorporation of phosphorous. Catal. Commun. 2008, 9, 327–332. [Google Scholar] [CrossRef]
- Lindholm, A.; Currier, N.W.; Dawody, J.; Hidayat, A.; Li, J.; Yezerets, A.; Olsson, L. The influence of the preparation procedure on the storage and regeneration behavior of Pt and Ba based NOx storage and reduction catalysts. Appl. Catal. B Environ. 2009, 88, 240–248. [Google Scholar] [CrossRef]
- Sharma, M.; Harold, M.P.; Balakotaiah, V. Analysis of periodic storage and reduction of NOx in catalytic monoliths. Ind. Eng. Chem. Res. 2005, 44, 6264–6277. [Google Scholar] [CrossRef]
- Lietti, L.; Nova, I.; Forzatti, P. Role of ammonia in the reduction by hydrogen of NOx stored over Pt-Ba/Al2O3 lean NOx trap catalysts. J. Catal. 2008, 257, 270–282. [Google Scholar] [CrossRef]
- Nova, I.; Castoldi, L.; Lietti, L.; Tronconi, E.; Forzatti, P. How to control the selectivity in the reduction of NOx with H2 over Pt-Ba/Al2O3 Lean NOx Trap catalysts. Top. Catal. 2007, 42–43, 21–25. [Google Scholar] [CrossRef]
- Tonkyn, R.G.; Disselkamp, R.S.; Peden, C.H.F. Nitrogen release from a NOx storage and reduction catalyst. Catal. Today 2006, 114, 94–101. [Google Scholar] [CrossRef]
- Olsson, L.; Fridell, E.; Skoglundh, M.; Andersson, B. Mean field modelling of NOx storage on Pt/BaO/Al2O3. Catal. Today 2002, 73, 263–270. [Google Scholar] [CrossRef]
- Clayton, R.D.; Harold, M.P.; Balakotaiah, V. Selective catalytic reduction of NO by H2 in O2 on Pt/BaO/Al2O3 monolith NOx storage catalysts. Appl. Catal. B Environ. 2008, 81, 161–181. [Google Scholar] [CrossRef]
- Choi, J.-S.; Partridge, W.P.; Pihl, J.A.; Kim, M.-Y.; Kočí, P.; Daw, C.S. Spatiotemporal distribution of NOx storage and impact on NH3 and N2O selectivities during lean/rich cycling of a Ba-based lean NOx trap catalyst. Catal. Today 2012, 184, 20–26. [Google Scholar] [CrossRef]
- Bamwenda, G. The role of the metal during NO2 reduction by C3H6 over alumina and silica-supported catalysts. J. Mol. Catal. A Chem. 1997, 126, 151–159. [Google Scholar] [CrossRef]
- Ji, Y.; Easterling, V.; Graham, U.; Fisk, C.; Crocker, M.; Choi, J.S. Effect of aging on the NOx storage and regeneration characteristics of fully formulated lean NOx trap catalysts. Appl. Catal. B Environ. 2011, 103, 413–427. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Perkin Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Andonova, S.; Vovk, E.; Sjöblom, J.; Ozensoy, E.; Olsson, L. Chemical deactivation by phosphorous under lean hydrothermal conditions over Cu/BEA NH3-SCR catalysts. Appl. Catal. B Environ. 2014, 147, 251–263. [Google Scholar] [CrossRef]
- Shwan, S.; Jansson, J.; Olsson, L.; Skoglundh, M. Chemical deactivation of Fe-BEA as NH3-SCR catalyst-Effect of phosphorous. Appl. Catal. B Environ. 2014, 147, 111–123. [Google Scholar] [CrossRef]
- Andersson, J.; Antonsson, M.; Eurenius, L.; Olsson, E.; Skoglundh, M. Deactivation of diesel oxidation catalysts: Vehicle- and synthetic aging correlations. Appl. Catal. B Environ. 2007, 72, 71–81. [Google Scholar] [CrossRef]
- Beck, D.; Monroe, D.; Di Maggio, C.; Sommers, J. Impact of Oil-Derived Catalyst Poisons on FTP Performance of LEV Catalyst Systems. SAE Trans. 1997. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
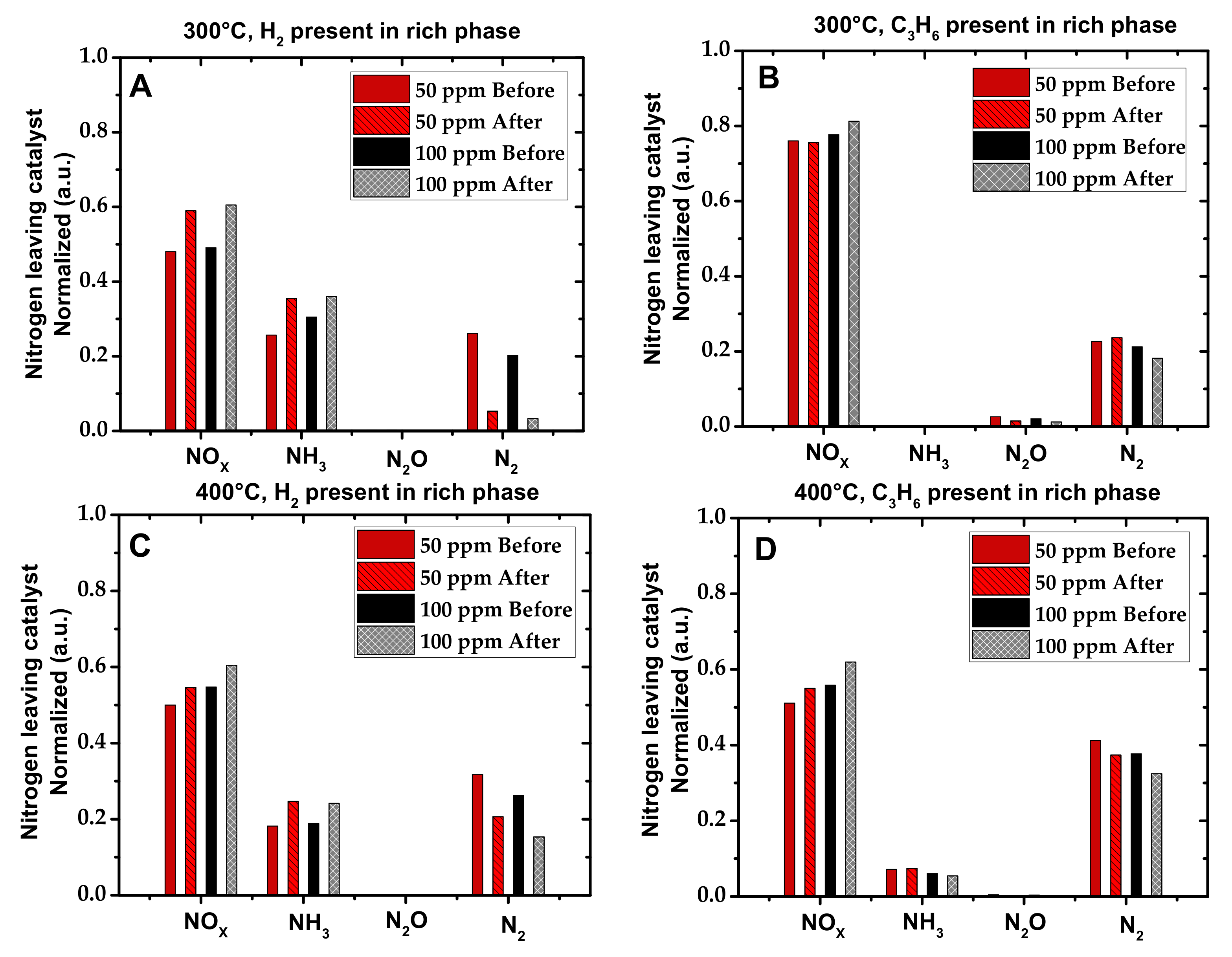

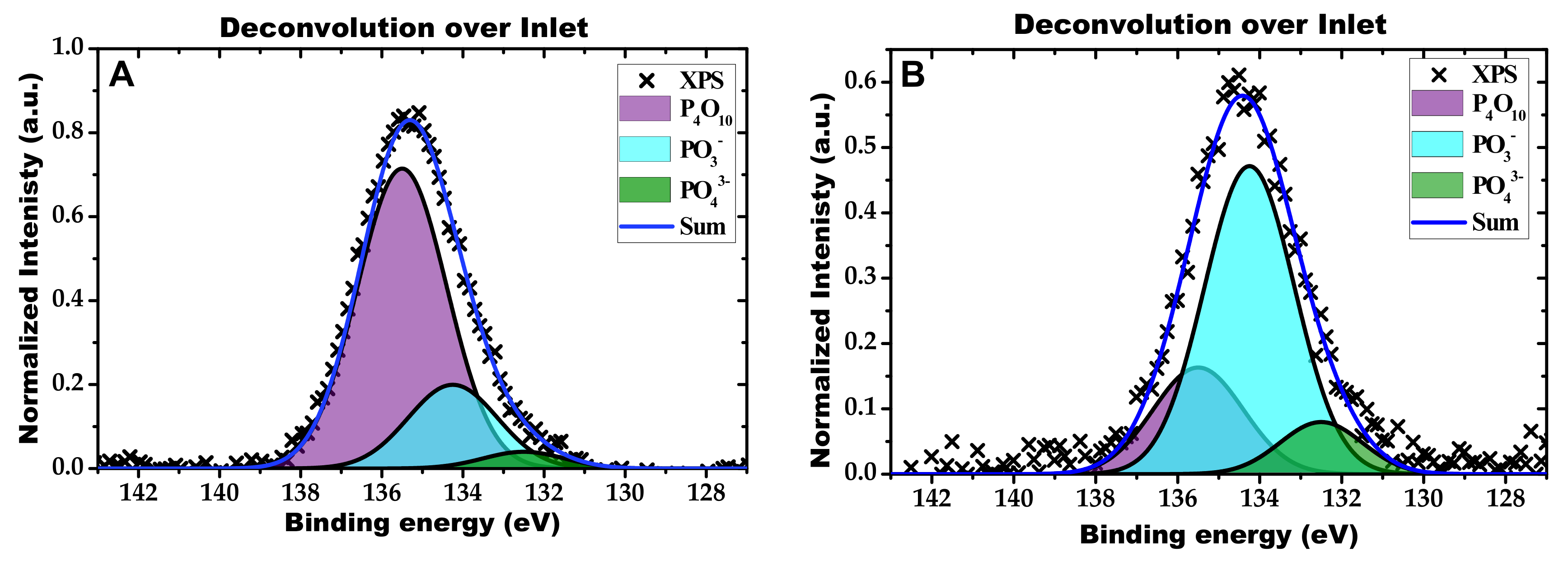
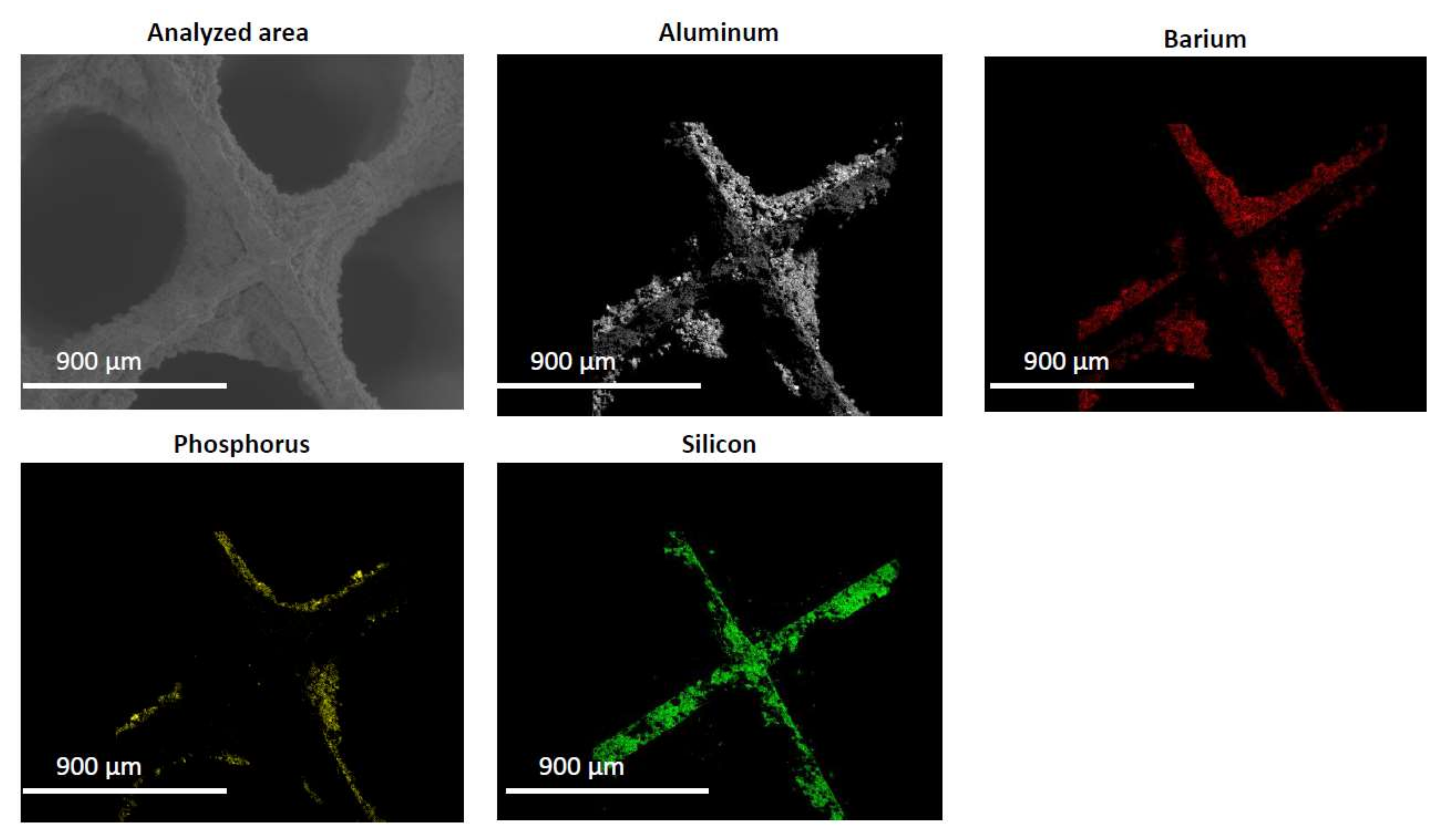
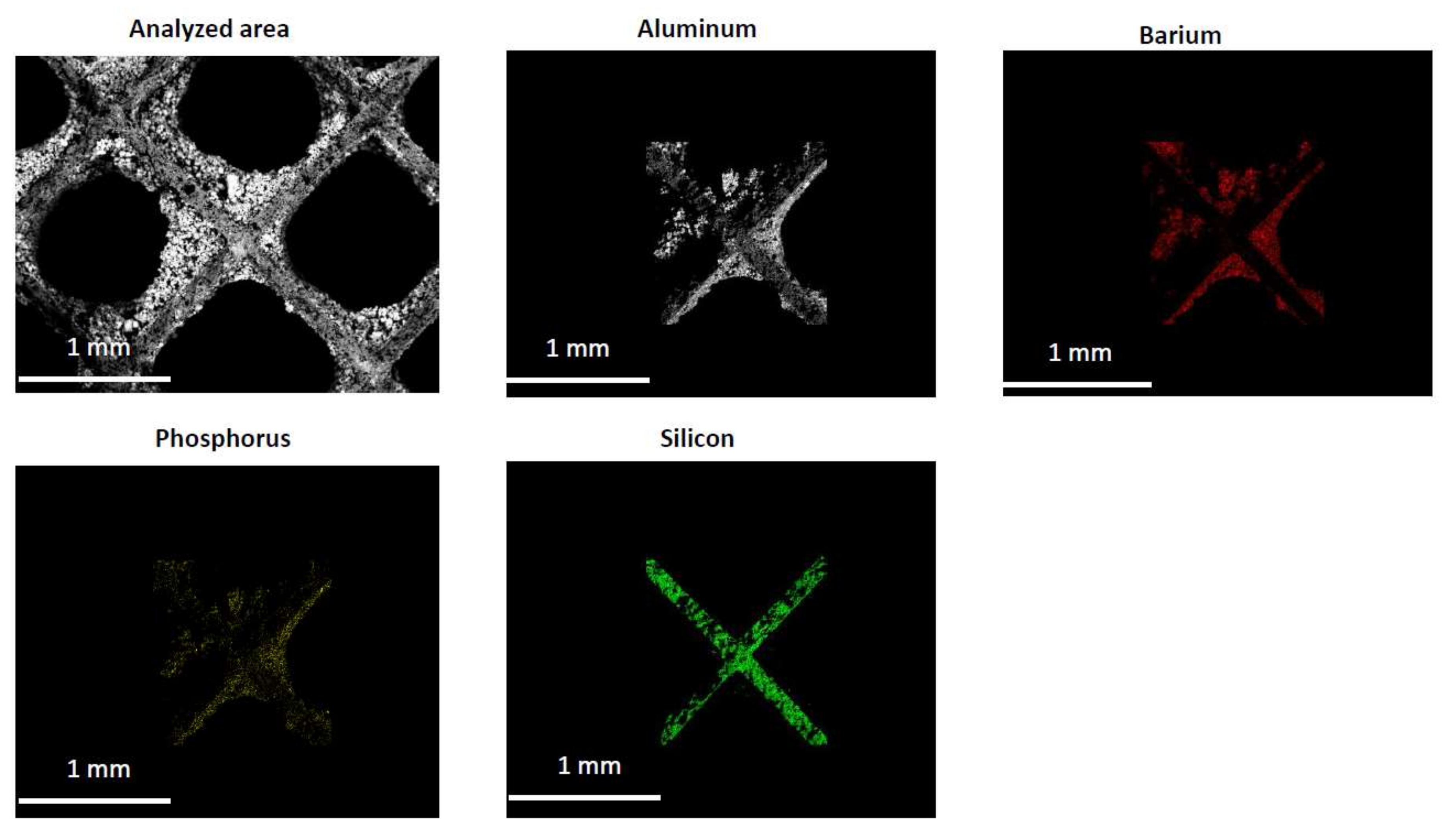

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | NOx Storage Capacity (μmol) | Loss in NOx Storage Capacity (μmol) |
|---|---|---|
| NO+H2, 300 °C, fresh | 92.4 | - |
| NO+H2, 300 °C, 50 ppm P | 56.2 | 36.2 |
| NO+H2, 300 °C, fresh | 88.8 | - |
| NO+H2, 300 °C, 100 ppm P | 51.4 | 37.4 |
| NO+H2, 400 °C, fresh | 85.9 | - |
| NO+H2, 400 °C, 50 ppm P | 70.6 | 15.3 |
| NO+H2, 400 °C, fresh | 70.3 | - |
| NO+H2, 400 °C, 100 ppm P | 51.5 | 18.8 |
| P-content, (wt.-%) | SBET (m2/(g washcoat)) 1 | VP (cm3/(g washcoat)) 1 | |
|---|---|---|---|
| Fresh | - | 134 | 0.46 |
| 50 ppm, 34 h, inlet | 2.2 | 98 | 0.36 |
| 50 ppm, 34 h, middle | 0.40 | 130 | 0.45 |
| 50 ppm, 34 h, outlet | 0.11 | 134 | 0.45 |
| 100 ppm, 34 h, inlet | 2.0 | 113 | 0.35 |
| 100 ppm, 34 h, middle | 0.38 | 123 | 0.39 |
| 100 ppm, 34 h, outlet | 0.07 | 132 | 0.39 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonsson, R.; Mihai, O.; Woo, J.; Skoglundh, M.; Olsson, E.; Berggrund, M.; Olsson, L. Gas-Phase Phosphorous Poisoning of a Pt/Ba/Al2O3 NOx Storage Catalyst. Catalysts 2018, 8, 155. https://doi.org/10.3390/catal8040155
Jonsson R, Mihai O, Woo J, Skoglundh M, Olsson E, Berggrund M, Olsson L. Gas-Phase Phosphorous Poisoning of a Pt/Ba/Al2O3 NOx Storage Catalyst. Catalysts. 2018; 8(4):155. https://doi.org/10.3390/catal8040155
Chicago/Turabian StyleJonsson, Rasmus, Oana Mihai, Jungwon Woo, Magnus Skoglundh, Eva Olsson, Malin Berggrund, and Louise Olsson. 2018. "Gas-Phase Phosphorous Poisoning of a Pt/Ba/Al2O3 NOx Storage Catalyst" Catalysts 8, no. 4: 155. https://doi.org/10.3390/catal8040155
APA StyleJonsson, R., Mihai, O., Woo, J., Skoglundh, M., Olsson, E., Berggrund, M., & Olsson, L. (2018). Gas-Phase Phosphorous Poisoning of a Pt/Ba/Al2O3 NOx Storage Catalyst. Catalysts, 8(4), 155. https://doi.org/10.3390/catal8040155