Theoretical Study on the Mechanism of Hydrogen Donation and Transfer for Hydrogen-Donor Solvents during Direct Coal Liquefaction

Abstract
:1. Introduction
2. Results
2.1. Stepwise Mechanism
2.2. Concerted Mechanisms
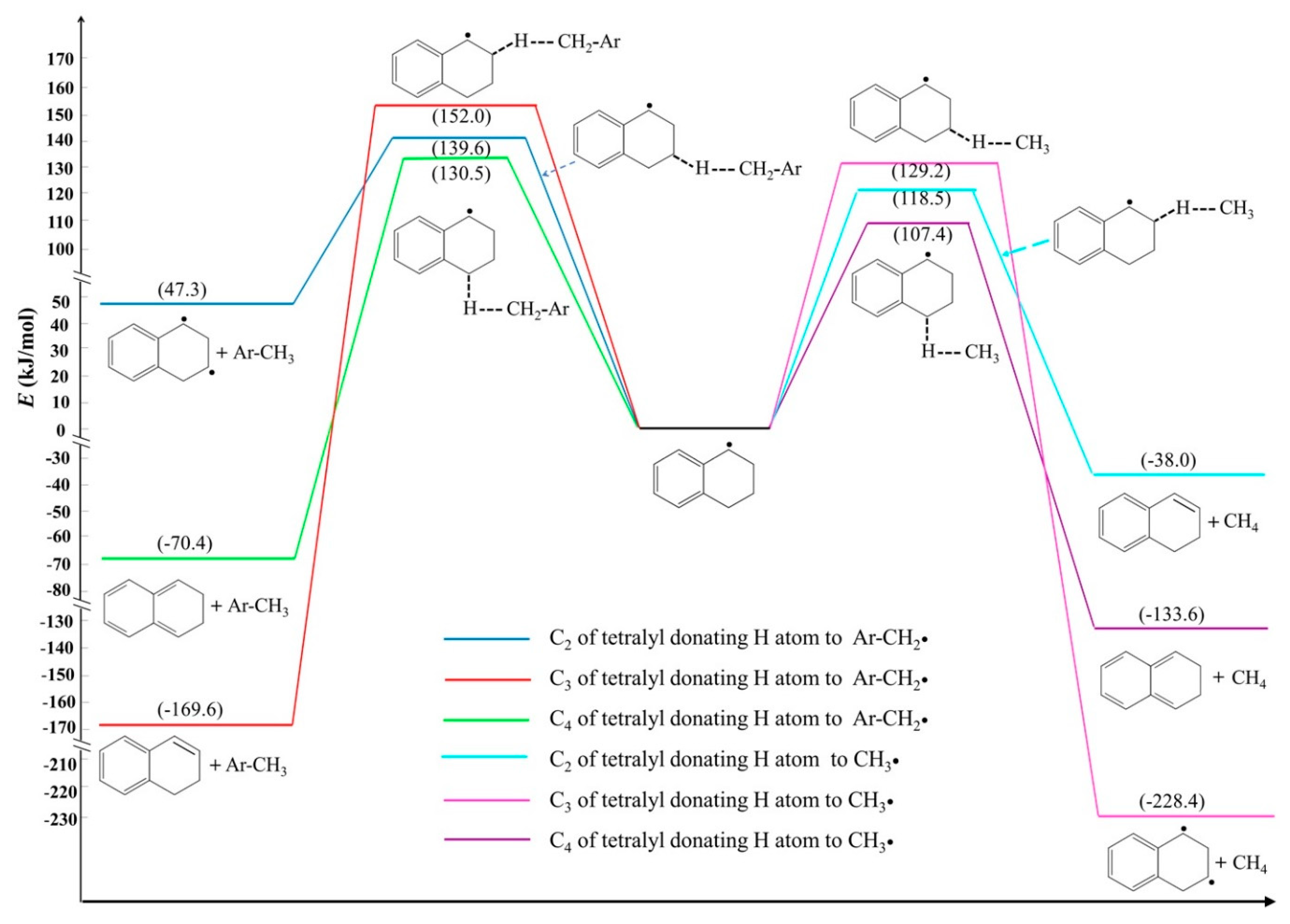
2.3. Donation and Transfer Pathways
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vasireddy, S.; Morreale, B.; Cugini, A.; Song, C.; Spivey, J.J. Clean liquid fuels from direct coal liquefaction: Chemistry, catalysis, technological status and challenges. Energy Environ. Sci. 2011, 4, 311–345. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, S.; Li, Y. Coal liquefaction technologies-Development in China and challenges in chemical reaction engineering. Chem. Eng. Sci. 2010, 65, 12–17. [Google Scholar] [CrossRef]
- Shui, H.; Cai, Z.; Xu, C. Recent advances in direct coal liquefaction. Energies 2010, 3, 155–170. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, D.; Li, K. Effect of recycle solvent hydrotreatment on oil yield of direct coal liquefaction. Energies 2015, 8, 6795–6805. [Google Scholar] [CrossRef]
- Ouchi, K.; Makabe, M. Hydrogen transfer in the hydrogenation of model compounds. Fuel 1988, 67, 1536–1541. [Google Scholar] [CrossRef]
- Kabe, T.; Nitoh, O.; Funatsu, E.; Yamamoto, K. Studies on hydrogen transfer mechanisms in coal liquefaction by means of 3H and 14C trancer techniques. Fuel Process. Technol. 1986, 14, 91–101. [Google Scholar] [CrossRef]
- Bate, K.; Harrison, G. Fate of hydrogen-donor molecules in two-stage liquefaction using model solvents. Fuel 1992, 71, 289–305. [Google Scholar] [CrossRef]
- Vernon, L.W. Free radical chemistry of coal liquefaction: Role of molecular hydrogen. Fuel 1980, 59, 102–106. [Google Scholar] [CrossRef]
- Skowronski, R.P.; Ratto, J.J.; Goldberg, I.B.; Heredy, L.A. Hydrogen incorporation during coal liquefaction. Fuel 1984, 63, 440–448. [Google Scholar] [CrossRef]
- Robinson, K.K. Reaction engineering of direct coal liquefaction. Energies 2009, 2, 976–1006. [Google Scholar] [CrossRef]
- Miller, R.L.; Silver, H.F. Solvent Effects on the Hydro-liquefaction of Wyodak Coal. Energy Sources 1980, 5, 211–222. [Google Scholar] [CrossRef]
- Franck, H.G.; Stadelhofer, J.W.; Biermann, D. Solubilization of bituminous coal in aromatic and hydroaromatic solvents. Fuel 1983, 62, 78–80. [Google Scholar] [CrossRef]
- Joseph, J.T. Liquefaction behaviour of solvent-swollen coals. Fuel 1991, 70, 139–144. [Google Scholar] [CrossRef]
- Nishioka, M. Role of solvation for coal swelling in slurry. Energy Fuels 2002, 16, 1109–1115. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. Transport of penetrants in the macromolecular structure of coals 4. Models for analysis of dynamic penetrant transport. Fuel 1987, 66, 815–826. [Google Scholar] [CrossRef]
- Malhotra, R.; McMillen, D.F. Relevance of cleavage of strong bonds in coal liquefaction. Energy Fuels 1993, 7, 227–233. [Google Scholar] [CrossRef]
- Autrey, T.; Alborn, E.A.; Franz, J.A.; Camaioni, D.M. Solvent-induced scission of diarylmethanes in dihydroarene donor solvents. An experimental and mechanistic modeling study of hydrogen-transfer pathways. Energy Fuels 1995, 9, 420–428. [Google Scholar] [CrossRef]
- McMillen, D.F.; Malhotra, R.; Chang, S.J.; Ogier, W.C.; Nigenda, S.E.; Flemingt, R.H. Mechanisms of hydrogen transfer and bond scission of strongly bonded coal structures in donor-solvent systems. Fuel 1987, 66, 1611–1620. [Google Scholar] [CrossRef]
- Malhotra, R.; McMillen, D.F. A mechanistic numerical model for coal liquefaction involving hydrogenolysis of strong bonds. Rationlization of interactive effect of solvent aromaticity and hydrogen pressure. Energy Fuels 1990, 4, 184–193. [Google Scholar] [CrossRef]
- Wei, X.; Ogata, E.; Zong, Z.; Zhou, S.; Qin, Z.; Liu, J.; Shen, K.; Li, H. Advances in the study of hydrogen transfer to model compounds for coal liquefaction. Fuel Process. Technol. 2000, 62, 103–107. [Google Scholar] [CrossRef]
- Li, X.; Hu, S.; Jin, L.; Hu, H. Role of iron-based catalyst and hydrogen transfer in direct coal liquefaction. Energy Fuels 2008, 22, 1126–1129. [Google Scholar] [CrossRef]
- Mochida, I.; Iwamoto, K.; Tahara, T.; Korai, Y.; Fujitsu, H.; Takeshita, K. Liquefaction of subbituminous coals under apparently non-hydrogenative conditions. Fuel 1982, 61, 603–609. [Google Scholar] [CrossRef]
- Kuhlmann, E.J.; Jung, D.Y.; Guptill, R.P.; Dyke, C.A.; Zang, H.K. Coal liquefaction using a hydrogenated creosote oil solvent: H-atom transfer from hydrogen donor components in the solvent. Fuel 1985, 64, 1552–1557. [Google Scholar] [CrossRef]
- Godo, M.; Saito, M.; Sasahara, J.; Ishihara, A.; Kabe, T. Elucidation of coal liquefaction mechanism using a tritium tracer method. Effect of H2S and H2O on hydrogen exchange reaction of tetralin with tritiated molecular hydrogen. Energy Fuels 1997, 11, 470–476. [Google Scholar] [CrossRef]
- Khorasheh, F.; Gray, M.R. High-pressure thermal cracking of n-hexadecane in tetralin. Energy Fuels 1993, 7, 960–967. [Google Scholar] [CrossRef]
- Khorasheh, F.; Gray, M.R. High-pressure thermal cracking of n-hexadecane in aromatic solvents. Ind. Eng. Chem. Res. 1993, 32, 1864–1876. [Google Scholar] [CrossRef]
- Johnson, E.R.; Clarkin, O.J.; DiLabio, G.A. Density Functional Theory Based Model Calculations for Accurate Bond Dissociation Enthalpies. 3. A Single Approach for X−H, X−X, and X−Y (X, Y = C, N, O, S, Halogen) Bonds. J. Phys. Chem. A 2003, 107, 9953–9963. [Google Scholar] [CrossRef]
- Hou, P.; Zhou, Y.; Guo, W.; Ren, P.; Guo, Q.; Xiang, H.; Yang, Y. Rational Design of Hydrogen-Donor Solvents for Direct Coal Liquefaction. Energy Fuel 2018, 32, 4715–4723. [Google Scholar] [CrossRef]
- Curtis, C.W.; Guin, J.A.; Hale, M.A.; Smith, N.L. Contribution of transferable hydrogen to coal conversion. Fuel 1985, 64, 461–469. [Google Scholar] [CrossRef]
- Kamiya, Y.; Futamura, S.; Mizuki, T.; Kajioka, M.; Koshi, K. Solvent effect on coal liquefaction. Fuel Process. Technol. 1986, 14, 79–90. [Google Scholar] [CrossRef]
- Shi, L.; Liu, Q.; Guo, X.; Wu, W.; Liu, Z. Pyrolysis behavior and bonding information of coal-a TGA study. Fuel Process. Technol. 2013, 108, 125–132. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B. Gaussian 09, Revision, A. 02, Gaussian, Inc.: Wallingford, CT, USA, 2009; 200.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
 : coal).
: coal).
: coal).
: coal).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | C6=C7 | C7=C8 | C8=C9 | C1–C2 | C1–C9 | C2–C3 | C1–H | C2–H | C8–H | C7–H |
|---|---|---|---|---|---|---|---|---|---|---|
| Gas | 570.4 | 587.1 | 571.2 | 267.3 | 377.4 | 318.6 | 303.2 | 361.3 | 418.2 | 428.0 |
| Liquid | 569.5 | 585.2 | 569.3 | 266.7 | 377.0 | 318.0 | 304.3 | 361.0 | 422.7 | 427.4 |
| Chemical Bond Type | BDE (kJ/mol) | Temperature of Bond Cleavage (°C) | |
|---|---|---|---|
| 1 | Release of bonded water and decomposition of carboxylic acid | <150 | <300 |
| 2 | Breakage of bonds between Cal and O, S, and N, and S–S | 150–230 | 300–400 |
| 3 | Breakage of bonds between Cal and Cal, H, O, and Car-N | 210–320 | 400–500 |
| 4 | Breakage of bonds between Cal and Cal and O and S | 300–430 | 500–600 |
| 5 | Decomposition of carbonate in coals to generate CO2 | - | ~700 |
| 6 | Condensation of aromatics rings to release H2 | >400 | 740–800 |
| Model Compound | Tetralin | H2 | |||
|---|---|---|---|---|---|
| C1–H (or C4–H) | C4–H (or C1–H) | C2–H (or C3–H) | C3–H (or C2–H) | ||
| CH3• | 96.4 | 99.8 | 192.2 | 125.0 | 107.4 |
| Ar-CH2• | 111.3 | 72.7 | 104.6 | 129.8 | 142.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, H.; Chang, T.; Cui, L.; Sun, R.; Gao, R. Theoretical Study on the Mechanism of Hydrogen Donation and Transfer for Hydrogen-Donor Solvents during Direct Coal Liquefaction. Catalysts 2018, 8, 648. https://doi.org/10.3390/catal8120648
Hao H, Chang T, Cui L, Sun R, Gao R. Theoretical Study on the Mechanism of Hydrogen Donation and Transfer for Hydrogen-Donor Solvents during Direct Coal Liquefaction. Catalysts. 2018; 8(12):648. https://doi.org/10.3390/catal8120648
Chicago/Turabian StyleHao, Haigang, Tong Chang, Linxia Cui, Ruiqing Sun, and Rui Gao. 2018. "Theoretical Study on the Mechanism of Hydrogen Donation and Transfer for Hydrogen-Donor Solvents during Direct Coal Liquefaction" Catalysts 8, no. 12: 648. https://doi.org/10.3390/catal8120648
APA StyleHao, H., Chang, T., Cui, L., Sun, R., & Gao, R. (2018). Theoretical Study on the Mechanism of Hydrogen Donation and Transfer for Hydrogen-Donor Solvents during Direct Coal Liquefaction. Catalysts, 8(12), 648. https://doi.org/10.3390/catal8120648