Straightforward Design for Phenoxy-Imine Catalytic Activity in Ethylene Polymerization: Theoretical Prediction


Abstract
1. Introduction
2. Results and Discussion
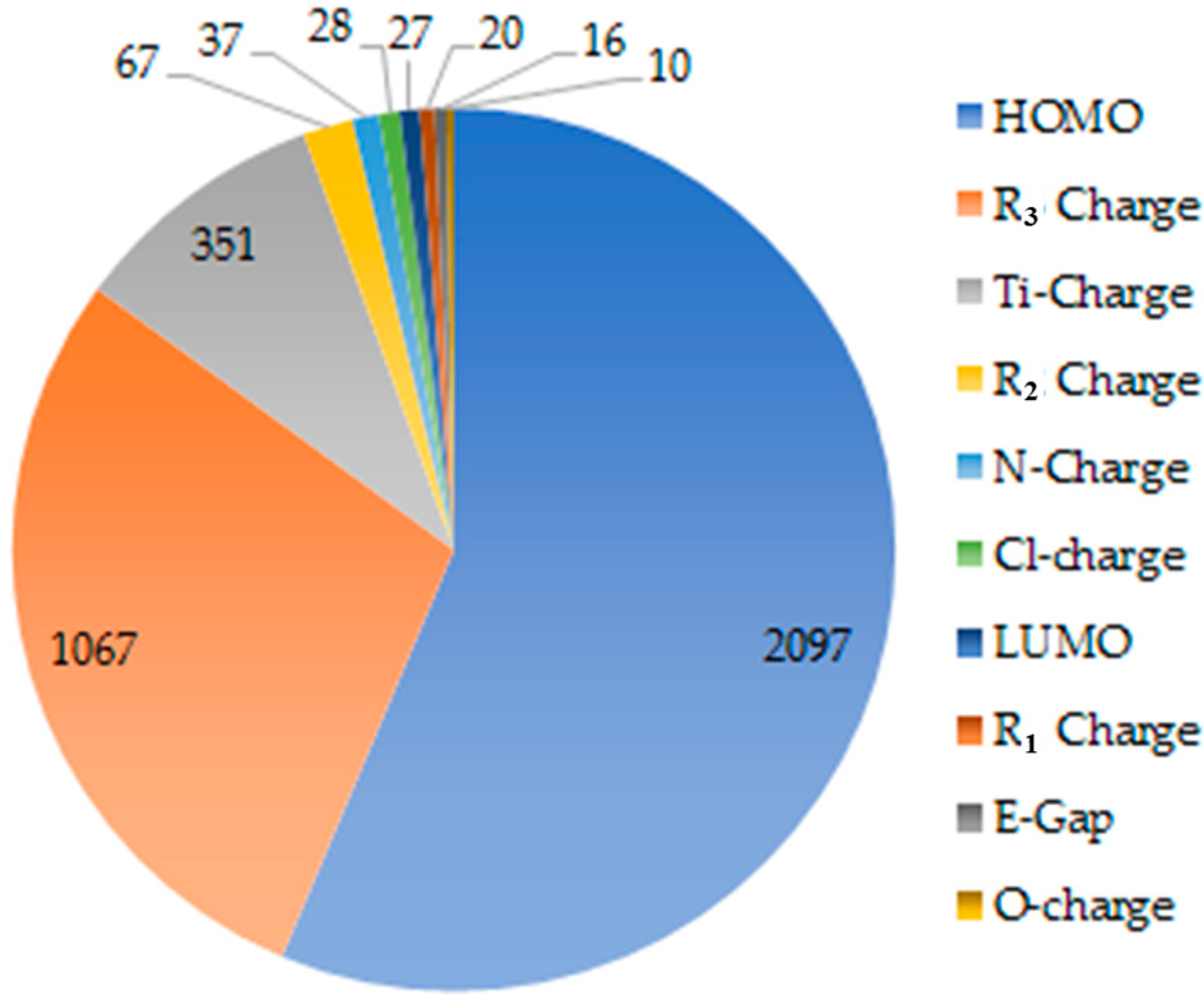
2.1. Electronic Properties
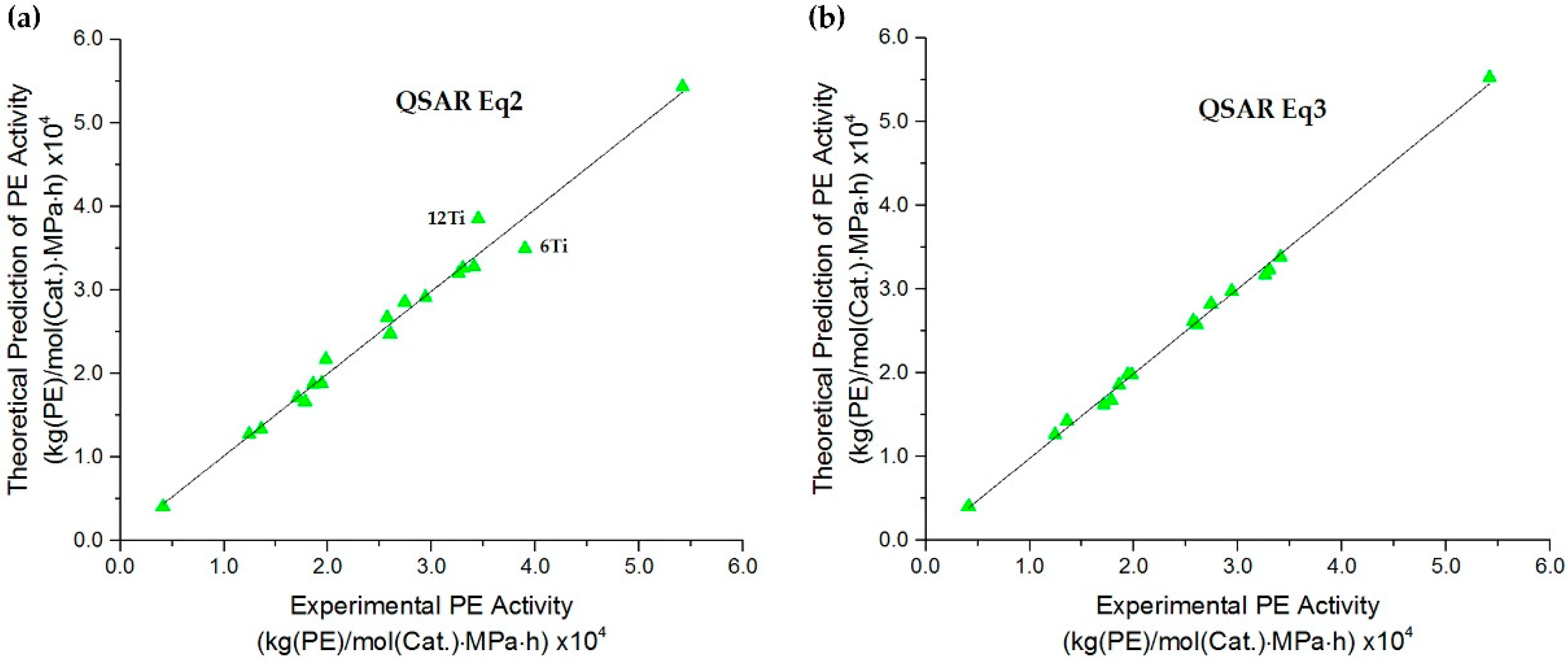
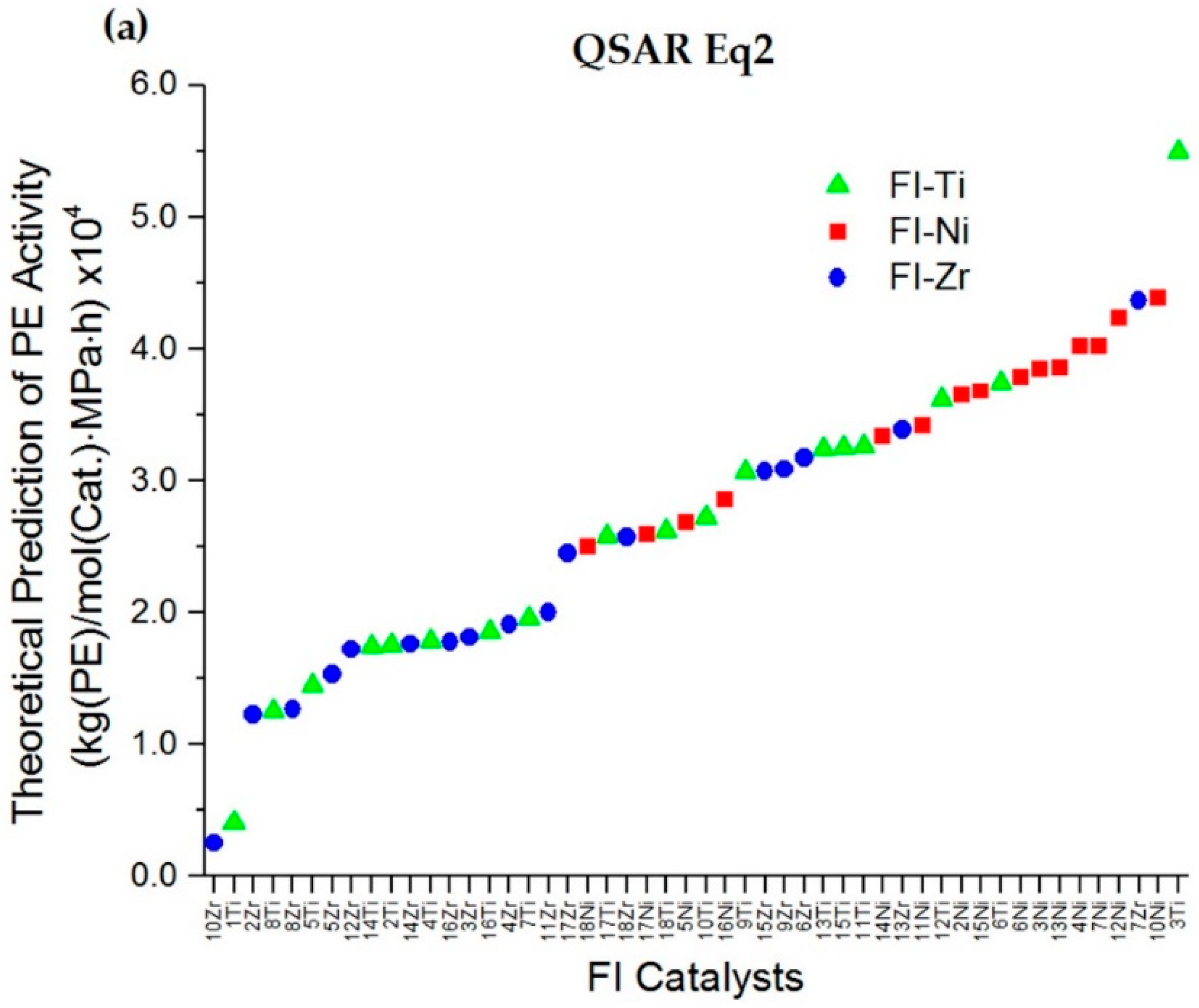
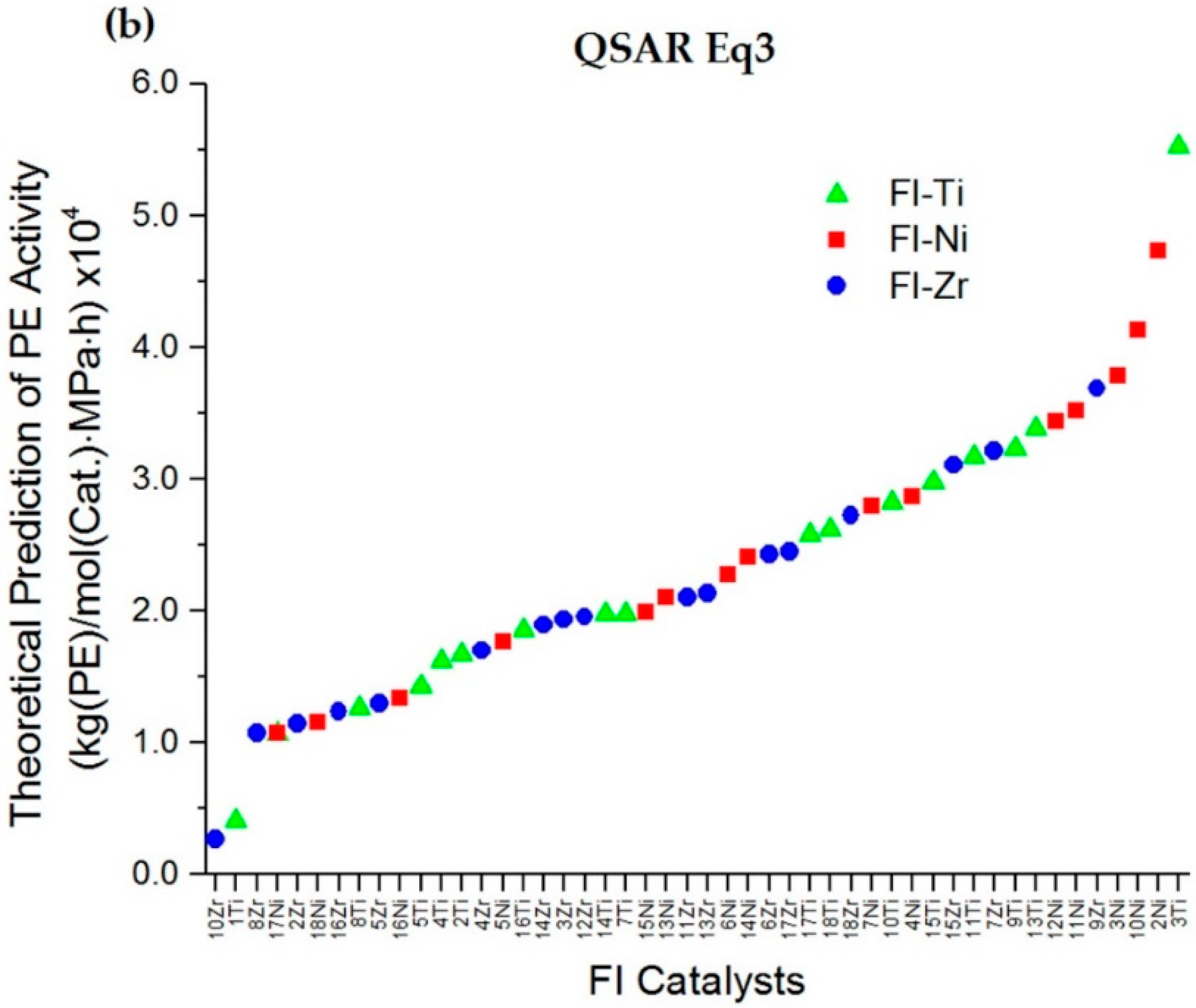
2.2. QSAR Model Relationship
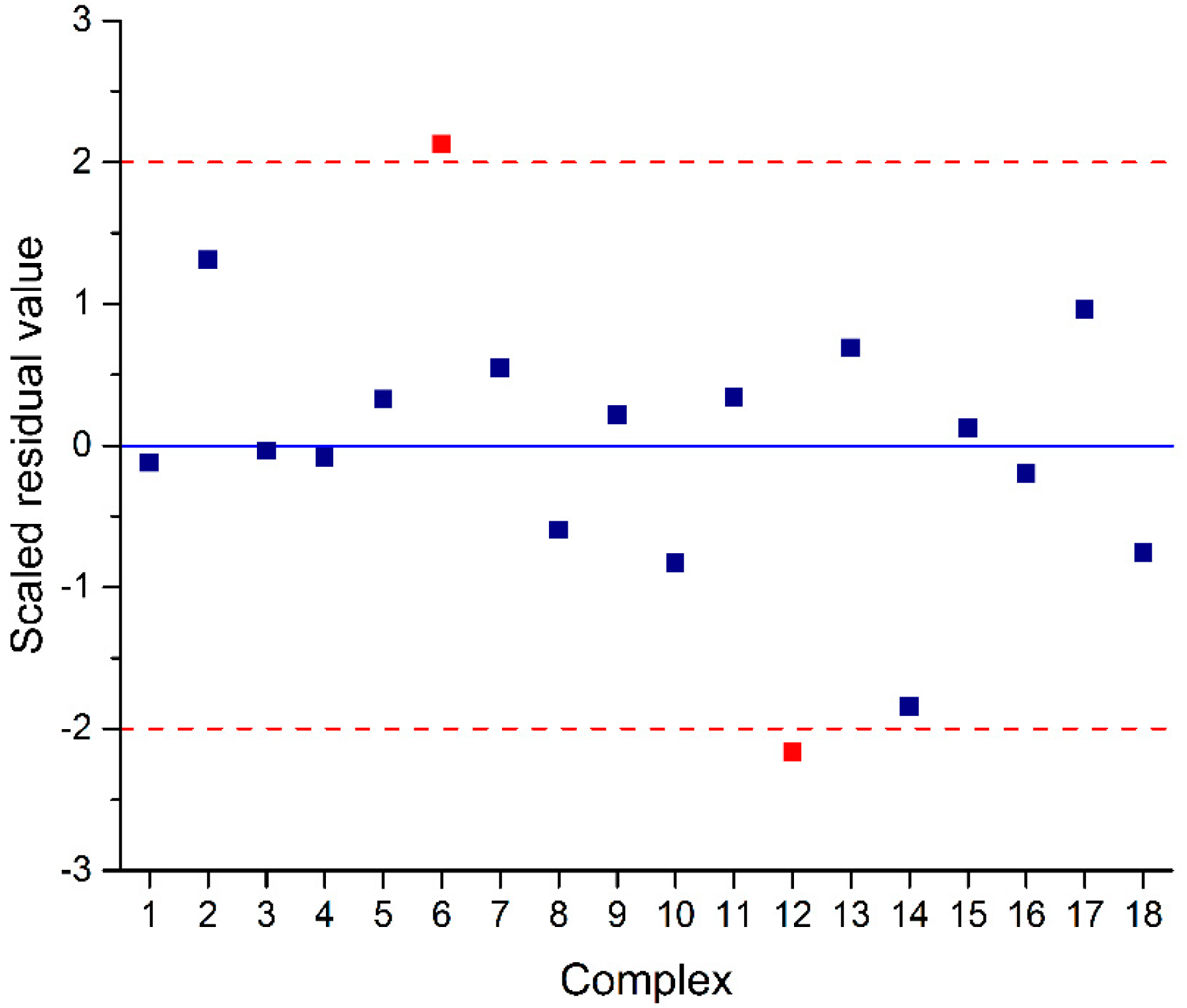
2.3. QSAR Model Improvement via the Elimination of Insignificant Descriptors and Outliers
2.4. Prediction of PE Polymerization Activity for Newly Designed Zr and Ni Phenoxyimine Catalysts
3. Materials and Methods
3.1. Source of Experimental Datasets
3.2. Structural Optimization
3.3. QSAR Model
3.4. Statistical Terms
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xie, H.; Hua, X.; Liu, B.; Wu, C.; Cui, D. Phosphinimino-amino supported complex: Synthesis, polymerization of ethylene and dearomatisation of pyridine. J. Organomet. Chem. 2015, 798, 335–340. [Google Scholar] [CrossRef]
- Nyamato, G.S.; Ojwach, S.O.; Akerman, M.P. Ethylene oligomerization studies by nickel(II) complexes chelated by (amino)pyridine ligands: Experimental and density functional theory studies. Dalton Trans. 2016, 45, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Do, L.H. Customizing polyolefin morphology by selective pairing of alkali ions with nickel phenoxyimine-polyethylene glycol catalysts. Organometallics 2017, 36, 4691–4698. [Google Scholar] [CrossRef]
- Yu, F.; Yang, Y.; He, D.; Gong, D.; Chen, Z.-R. Pressure-sensitive supported FI catalyst for the precise synthesis of uni- and bimodal polyethylene. Ind. Eng. Chem. Res. 2017, 56, 4684–4689. [Google Scholar] [CrossRef]
- Goerl, C.; Betthausen, E.; Alt, H.G. Di- and trinuclear iron/titanium and iron/zirconium complexes with heterocyclic ligands as catalysts for ethylene polymerization. Polyhedron 2016, 118, 37–51. [Google Scholar] [CrossRef]
- Woo, J.O.; Kang, S.K.; Park, J.-E.; Son, K.-S. Synthesis, characterization, and ethylene polymerization behavior of cr(III) catalysts based on bis(pyrazolylmethyl)pyridine and its derivatives. J. Mol. Catal. A Chem. 2015, 404–405, 204–210. [Google Scholar] [CrossRef]
- Steelman, D.K.; Xiong, S.; Pletcher, P.D.; Smith, E.; Switzer, J.M.; Medvedev, G.A.; Delgass, W.N.; Caruthers, J.M.; Abu-Omar, M.M. Effects of pendant ligand binding affinity on chain transfer for 1-hexene polymerization catalyzed by single-site zirconium amine bis-phenolate complexes. J. Am. Chem. Soc. 2013, 135, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Martínez, S.; Cruz, V.L.; Ramos, J.; Martínez-Salazar, J. Polymerization activity prediction of zirconocene single-site catalysts using 3d quantitative structure–activity relationship modeling. Organometallics 2012, 31, 1673–1679. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Song, B.; Yoon, S.-W.; Go, M.; Lee, J.; Lee, B. Preparation of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl titanium complexes for ethylene/α-olefin copolymerization. Catalysts 2013, 3, 104. [Google Scholar] [CrossRef]
- Shamiri, A.; Chakrabarti, M.; Jahan, S.; Hussain, M.; Kaminsky, W.; Aravind, P.; Yehye, W. The influence of ziegler-natta and metallocene catalysts on polyolefin structure, properties, and processing ability. Materials 2014, 7, 5069. [Google Scholar] [CrossRef] [PubMed]
- Alt, H.G.; Köppl, A. Effect of the nature of metallocene complexes of group iv metals on their performance in catalytic ethylene and propylene polymerization. Chem. Rev. 2000, 100, 1205–1222. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, A.; Liu, S.; Ye, H.; Li, Z. 2-benzimidazol-6-pyrazol-pyridine chromium(III) trichlorides: Synthesis, characterization, and application for ethylene oligomerization and polymerization. Organometallics 2016, 35, 3045–3050. [Google Scholar] [CrossRef]
- Bariashir, C.; Wang, Z.; Du, S.; Solan, G.A.; Huang, C.; Liang, T.; Sun, W.-H. Cycloheptyl-fused nno-ligands as electronically modifiable supports for M(II) (M = Co, Fe) chloride precatalysts; probing performance in ethylene oligo-/polymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3980–3989. [Google Scholar] [CrossRef]
- Du, S.; Wang, X.; Zhang, W.; Flisak, Z.; Sun, Y.; Sun, W.-H. A practical ethylene polymerization for vinyl-polyethylenes: Synthesis, characterization and catalytic behavior of α,α′-bisimino-2,3:5,6-bis(pentamethylene)pyridyliron chlorides. Polym. Chem. 2016, 7, 4188–4197. [Google Scholar] [CrossRef]
- Park, J.-E.; Kang, S.K.; Woo, J.O.; Son, K.-S. Highly active chromium(III) complexes based on tridentate pyrazolyl pyridyl ligands for ethylene polymerization and oligomerization. Dalton Trans. 2015, 44, 9964–9969. [Google Scholar] [CrossRef] [PubMed]
- Nyamato, G.S.; Alam, M.G.; Ojwach, S.O.; Akerman, M.P. (pyrazolyl)-(phosphinoyl)pyridine iron(II), cobalt(II) and nickel(II) complexes: Synthesis, characterization and ethylene oligomerization studies. J. Organomet. Chem. 2015, 783, 64–72. [Google Scholar] [CrossRef]
- Falivene, L.; Cavallo, L.; Talarico, G. Buried volume analysis for propene polymerization catalysis promoted by group 4 metals: A tool for molecular mass prediction. ACS Catal. 2015, 5, 6815–6822. [Google Scholar] [CrossRef]
- Nomura, K.; Mitsudome, T.; Igarashi, A.; Nagai, G.; Tsutsumi, K.; Ina, T.; Omiya, T.; Takaya, H.; Yamazoe, S. Synthesis of (adamantylimido)vanadium(V) dimethyl complex containing (2-anilidomethyl)pyridine ligand and selected reactions: Exploring the oxidation state of the catalytically active species in ethylene dimerization. Organometallics 2017, 36, 530–542. [Google Scholar] [CrossRef]
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. Fi catalysts for olefin polymerization—A comprehensive treatment. Chem. Rev. 2011, 111, 2363–2449. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Furuyama, R.; Mohri, J.-I.; Saito, J.; Ishii, S.; Terao, H.; Nakano, T.; Tanaka, H.; Fujita, T. Syndiospecific living propylene polymerization catalyzed by titanium complexes having fluorine-containing phenoxy−imine chelate ligands. J. Am. Chem. Soc. 2003, 125, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Mohri, J.-I.; Yoshida, Y.; Saito, J.; Ishii, S.; Tsuru, K.; Matsui, S.; Furuyama, R.; Nakano, T.; Tanaka, H.; et al. Living polymerization of ethylene catalyzed by titanium complexes having fluorine-containing phenoxy-imine chelate ligands. J. Am. Chem. Soc. 2002, 124, 3327–3336. [Google Scholar] [CrossRef] [PubMed]
- Saito, J.; Mitani, M.; Matsui, S.; Tohi, Y.; Makio, H.; Nakano, T.; Tanaka, H.; Kashiwa, N.; Fujita, T. A new titanium complex having two phenoxy-imine chelate ligands for ethylene polymerization. Macromol. Chem. Phys. 2002, 203, 59–65. [Google Scholar] [CrossRef]
- Terao, H.; Ishii, S.; Mitani, M.; Tanaka, H.; Fujita, T. Ethylene/polar monomer copolymerization behavior of bis(phenoxy-imine)ti complexes: Formation of polar monomer copolymers. J. Am. Chem. Soc. 2008, 130, 17636–17637. [Google Scholar] [CrossRef] [PubMed]
- Makio, H.; Kashiwa, N.; Fujita, T. Fi catalysts: A new family of high performance catalysts for olefin polymerization. Adv. Synth. Catal. 2002, 344, 477–493. [Google Scholar] [CrossRef]
- Wang, Z.; Solan, G.A.; Mahmood, Q.; Liu, Q.; Ma, Y.; Hao, X.; Sun, W.-H. Bis(imino)pyridines incorporating doubly fused eight-membered rings as conformationally flexible supports for cobalt ethylene polymerization catalysts. Organometallics 2018, 37, 380–389. [Google Scholar] [CrossRef]
- Ishii, S.; Nakano, T.; Kawamura, K.; Kinoshita, S.; Ichikawa, S.; Fujita, T. Development of new selective ethylene trimerization catalysts based on highly active ethylene polymerization catalysts. Catal. Today 2018, 303, 263–270. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Solan, G.A.; Sun, W.-H. Recent advances in ni-mediated ethylene chain growth: Nimine-donor ligand effects on catalytic activity, thermal stability and oligo-/polymer structure. Coord. Chem. Rev. 2017, 350, 68–83. [Google Scholar] [CrossRef]
- Preston, A.Z.; Kim, J.; Medvedev, G.A.; Delgass, W.N.; Caruthers, J.M.; Abu-Omar, M.M. Steric and solvation effects on polymerization kinetics, dormancy, and tacticity of Zr-salan catalysts. Organometallics 2017, 36, 2237–2244. [Google Scholar] [CrossRef]
- Mitchell, N.E.; Anderson, W.C., Jr.; Long, B.K. Mitigating chain-transfer and enhancing the thermal stability of co-based olefin polymerization catalysts through sterically demanding ligands. J. Polym. Sci. Part A. Polym. Chem. 2017, 55, 3990–3995. [Google Scholar] [CrossRef]
- D’Auria, I.; Milione, S.; Caruso, T.; Balducci, G.; Pellecchia, C. Synthesis of hyperbranched low molecular weight polyethylene oils by an iminopyridine nickel(II) catalyst. Polym. Chem. 2017, 8, 6443–6454. [Google Scholar] [CrossRef]
- Tuskaev, V.A.; Gagieva, S.C.; Kurmaev, D.A.; Kolosov, N.A.; Fedyanin, I.V.; Bulychev, B.M. Titanium (IV) complexes stabilized by 2,6-bis-(α,α′-diphenyl(hydroxy)methyl)-pyridine: Catalytic activity in olefin polymerization and impact of lithium and magnesium chlorides. Inorg. Chim. Acta 2015, 425, 275–281. [Google Scholar] [CrossRef]
- Ratanasak, M.; Rungrotmongkol, T.; Saengsawang, O.; Hannongbua, S.; Parasuk, V. Towards the design of new electron donors for ziegler–natta catalyzed propylene polymerization using qspr modeling. Polymer 2015, 56, 340–345. [Google Scholar] [CrossRef]
- Ratanasak, M.; Parasuk, V. Roles of malonate donor on activity and stereoselectivity of ziegler-natta catalyzed propylene polymerization. J. Organomet. Chem. 2015, 775, 6–11. [Google Scholar] [CrossRef]
- Nyamato, G.S.; Ojwach, S.O.; Akerman, M.P. Potential hemilabile (imino)pyridine palladium(II) complexes as selective ethylene dimerization catalysts: An experimental and theoretical approach. Organometallics 2015, 34, 5647–5657. [Google Scholar] [CrossRef]
- Nikitin, S.V.; Nikitin, V.V.; Oleynik, I.I.; Oleynik, I.V.; Bagryanskaya, E.G. Activity of phenoxy-imine titanium catalysts in ethylene polymerization: A quantum chemical approach. J. Mol. Catal. A Chem. 2016, 423, 285–292. [Google Scholar] [CrossRef]
- Yang, W.; Yi, J.; Ma, Z.; Sun, W.-H. 2d-qsar modeling on the catalytic activities of 2-azacyclyl-6-aryliminopyridylmetal precatalysts in ethylene oligomerization. Catal. Commun. 2017, 101, 40–43. [Google Scholar] [CrossRef]
- Manz, T.A. Deactivation of Ti and Zr half-metallocene complexes activated with B(C6F5)3: A case study in constructing DFT-based qsars to predict unimolecular rate constants. RSC Adv. 2015, 5, 48246–48254. [Google Scholar] [CrossRef]
- Cruz, V.L.; Martinez, S.; Ramos, J.; Martinez-Salazar, J. 3D-QSAR as a tool for understanding and improving single-site polymerization catalysts. Organometallics 2014, 33, 2944–2959. [Google Scholar] [CrossRef]
- Cruz, V.L.; Martinez, S.; Martinez-Salazar, J.; Polo-Cerón, D.; Gómez-Ruiz, S.; Fajardo, M.; Prashar, S. 3D-QSAR study of ANSA-metallocene catalytic behavior in ethylene polymerization. Polymer 2007, 48, 4663–4674. [Google Scholar] [CrossRef]
- Melanie, M. Genetic algorithms: An overview. Complexity 1995, 1, 31–39. [Google Scholar]
- McCall, J. Genetic algorithms for modelling and optimisation. J. Comput. Appl. Math. 2005, 184, 205–222. [Google Scholar] [CrossRef]
- Fisz, J.J. Combined genetic algorithm and multiple linear regression (GA-MLR) optimizer: Application to multi-exponential fluorescence decay surface. J. Phys. Chem. A 2006, 110, 12977–12985. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Comp. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, F.; Fabiano, E.; Constantin, L.A. Kinetic-energy-density dependent semilocal exchange-correlation functionals. Int. J. Quantum Chem. 2016, 116, 1641–1694. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four m06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Remya, K.; Suresh, C.H. Which density functional is close to ccsd accuracy to describe geometry and interaction energy of small noncovalent dimers? A benchmark study using gaussian09. J. Comput. Chem. 2013, 34, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Rakhi, R.; Suresh, C.H. A dft study on dihydropyrazine annulated linear polyacenes: Aromaticity, stability and homo-lumo energy modulation. PCCP 2016, 18, 24631–24641. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.M. Design and training of a neural network for predicting the solvent accessibility of proteins. J. Comput. Chem. 2003, 24, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Hunger, J.; Huttner, G. Optimization and analysis of force field parameters by combination of genetic algorithms and neural networks. J. Comput. Chem. 1999, 20, 455–471. [Google Scholar] [CrossRef]
- Waller, C.L.; Bradley, M.P. Development and validation of a novel variable selection technique with application to multidimensional quantitative structure-activity relationship studies. J. Chem. Inf. Comp. Sci. 1999, 39, 345–355. [Google Scholar] [CrossRef]
- Aires-de-Sousa, J.; Hemmer, M.C.; Gasteiger, J. Prediction of 1H NMR chemical shifts using neural networks. Anal. Chem. 2002, 74, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Srivastava, D.C.; Gupta, P.K. The genetic algorithm: A robust method for stress inversion. J. Struct. Geol. 2017, 94, 227–239. [Google Scholar] [CrossRef]
- Douguet, D.; Thoreau, E.; Grassy, G. A genetic algorithm for the automated generation of small organic molecules: Drug design using an evolutionary algorithm. J. Comput. Aided Mol. Des. 2000, 14, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Kubiny, H. Variable selection in QSAR studies. I. An evolutionary algorithm. Quant. Struct. Act. Relat. 1994, 13, 285–294. [Google Scholar] [CrossRef]
- Leardi, R. 3-genetic algorithms in feature selection. In Genetic Algorithms in Molecular Modeling; Devillers, J., Ed.; Academic Press: Cambridge, MA, USA, 1996; pp. 67–86. [Google Scholar]
- Wold, S.; Eriksson, L.; Clementi, S. Statistical validation of QSAR results. In Chemometric Methods in Molecular Design; VCH, P.O. Box: Weinheim, Germany, 2008; pp. 309–338. [Google Scholar]
- Balakrishnan, N.; Colton, T.; Everitt, B.; Piegorsch, W.; Ruggeri, F.; Teugels, J.L. Ramp function. Wiley StatsRef Stat. Ref. Online 2014. [Google Scholar] [CrossRef]
- Mudelsee, M. Ramp function regression: A tool for quantifying climate transitions. Comput. Geosci. 2000, 26, 293–307. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, revision B.01; Gaussian Inc.: Wallingford UK, 2009. [Google Scholar]
- Pourbasheer, E.; Aalizadeh, R.; Ganjali, M.R.; Norouzi, P.; Shadmanesh, J. QSAR study of ACK1 inhibitors by genetic algorithm-multiple linear regression (GA-MLR). J. Saudi Chem. Soc. 2014, 18, 681–688. [Google Scholar] [CrossRef]
- Materials Studio Modeling; release 7.0; Accelrys Software Inc.: San Diego, CA, USA, 2013.
- Smith, H.J.; Williams, H. Introduction to the Principles of Drug Design, 2nd ed.; The stonebrige press: Butterworth, Malaysian, 1988; pp. 240–264. [Google Scholar]
- Leardi, R. Genetic algorithms in chemometrics and chemistry: A review. J. Chemom. 2001, 15, 559–569. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Complex | R1 | R2 | R3 | Activity (A) (kg(PE)/mol(Cat.)·MPa·h) | ln(A) |
|---|---|---|---|---|---|
| 1Ti | t-Bu | H | H | 4100 | 3.613 |
| 2Ti | t-Bu | H | m-OAll | 17,800 | 4.250 |
| 3Ti | t-Bu | H | p-OAll | 54,200 | 4.734 |
| 4Ti | t-Bu | Me | H | 17,100 | 4.233 |
| 5Ti | t-Bu | Me | m-OAll | 13,600 | 4.134 |
| 6Ti | t-Bu | Me | p-OAll | 39,000 | 4.591 |
| 7Ti | t-Bu | t-Bu | H | 19,400 | 4.288 |
| 8Ti | t-Bu | t-Bu | m-OAll | 12,400 | 4.093 |
| 9Ti | t-Bu | t-Bu | p-OAll | 33,000 | 4.519 |
| 10Ti | Cumyl | H | H | 27,400 | 4.438 |
| 11Ti | Cumyl | H | m-OAll | 32,600 | 4.513 |
| 12Ti | Cumyl | H | p-OAll | 34,500 | 4.538 |
| 13Ti | Cumyl | Me | H | 34,050 | 4.532 |
| 14Ti | Cumyl | Me | m-OAll | 19,800 | 4.297 |
| 15Ti | Cumyl | Me | p-OAll | 29,400 | 4.468 |
| 16Ti | Cumyl | Cumyl | H | 18,550 | 4.268 |
| 17Ti | Cumyl | Cumyl | m-OAll | 26,000 | 4.415 |
| 18Ti | Cumyl | Cumyl | p-OAll | 25,700 | 4.410 |

| Complex | Ti a | O b | N b | Cl b | R1 c | R2 c | R3 c | HOMO d | LUMO d | E-Gap d |
|---|---|---|---|---|---|---|---|---|---|---|
| 1Ti | 1.235 | −0.704 | −0.586 | −0.374 | −1.062 | 0.136 | 0.136 | −0.199 | −0.131 | 1.840 |
| 2Ti | 1.232 | −0.703 | −0.585 | −0.374 | −1.062 | 0.345 | −1.117 | −0.197 | −0.129 | 1.858 |
| 3Ti | 1.247 | −0.706 | −0.588 | −0.376 | −1.057 | 0.130 | −1.230 | −0.194 | −0.127 | 1.843 |
| 4Ti | 1.234 | −0.707 | −0.589 | −0.377 | −1.058 | −0.450 | 0.134 | −0.193 | −0.129 | 1.736 |
| 5Ti | 1.227 | −0.705 | −0.589 | −0.378 | −1.062 | −0.446 | −1.109 | −0.191 | −0.126 | 1.770 |
| 6Ti | 1.250 | −0.709 | −0.592 | −0.380 | −1.055 | −0.442 | −1.134 | −0.191 | −0.124 | 1.812 |
| 7Ti | 1.252 | −0.713 | −0.610 | −0.372 | −1.056 | −1.083 | 0.134 | −0.197 | −0.129 | 1.863 |
| 8Ti | 1.235 | −0.707 | −0.588 | −0.380 | −1.033 | −1.054 | −1.097 | −0.191 | −0.124 | 1.817 |
| 9Ti | 1.236 | −0.707 | −0.586 | −0.381 | −1.030 | −1.054 | −1.116 | −0.187 | −0.124 | 1.720 |
| 10Ti | 1.243 | −0.708 | −0.607 | −0.362 | −1.147 | 0.139 | 0.136 | −0.198 | −0.133 | 1.765 |
| 11Ti | 1.273 | −0.706 | −0.603 | −0.372 | −1.303 | 0.137 | −1.129 | −0.199 | −0.130 | 1.867 |
| 12Ti | 1.232 | −0.694 | −0.578 | −0.374 | −1.254 | 0.122 | −1.131 | −0.189 | −0.124 | 1.765 |
| 13Ti | 1.242 | −0.696 | −0.584 | −0.376 | −1.248 | −0.026 | 0.133 | −0.189 | −0.125 | 1.739 |
| 14Ti | 1.269 | −0.707 | −0.606 | −0.376 | −1.302 | −0.445 | −1.125 | −0.191 | −0.127 | 1.764 |
| 15Ti | 1.224 | −0.700 | −0.579 | −0.376 | −1.257 | −0.432 | −1.121 | −0.185 | −0.123 | 1.688 |
| 16Ti | 1.286 | −0.703 | −0.538 | −0.385 | −1.222 | −1.353 | 0.120 | −0.189 | −0.121 | 1.856 |
| 17Ti | 1.236 | −0.705 | −0.602 | −0.370 | −1.216 | −1.350 | −1.101 | −0.189 | −0.123 | 1.794 |
| 18Ti | 1.270 | −0.709 | −0.598 | −0.384 | −1.262 | −1.305 | −1.102 | −0.190 | −0.125 | 1.750 |
| Des. | ln(A) | Ti a | O b | N b | Cl b | R1 c | R2 c | R3 c | HOMO | LUMO | E-Gap |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ln(A) | 1.000 | 0.155 | 0.119 | −0.084 | 0.033 | −0.298 | 0.041 | −0.380 | 0.355 | 0.327 | −0.225 |
| Tia | 0.155 | 1.000 | −0.312 | 0.119 | −0.294 | −0.468 | −0.290 | 0.114 | −0.156 | 0.040 | 0.391 |
| Ob | 0.119 | −0.312 | 1.000 | 0.175 | −0.003 | −0.268 | 0.027 | −0.169 | 0.449 | 0.296 | −0.472 |
| Nb | -0.084 | 0.119 | 0.175 | 1.000 | −0.568 | −0.032 | −0.128 | 0.159 | 0.397 | 0.550 | 0.043 |
| Clb | 0.033 | −0.294 | −0.003 | −0.568 | 1.000 | −0.065 | 0.501 | 0.219 | −0.524 | −0.636 | 0.087 |
| R1c | -0.298 | −0.468 | −0.268 | −0.032 | −0.065 | 1.000 | 0.002 | 0.112 | −0.280 | −0.225 | 0.230 |
| R2c | 0.041 | −0.290 | 0.027 | −0.128 | 0.501 | 0.002 | 1.000 | 0.017 | −0.480 | −0.567 | 0.103 |
| R3c | -0.380 | 0.114 | −0.169 | 0.159 | 0.219 | 0.112 | 0.017 | 1.000 | −0.347 | −0.396 | 0.096 |
| HOMO | 0.355 | −0.156 | 0.449 | 0.397 | −0.524 | −0.280 | −0.480 | −0.347 | 1.000 | 0.890 | −0.684 |
| LUMO | 0.327 | 0.040 | 0.296 | 0.550 | −0.636 | −0.225 | −0.567 | −0.396 | 0.890 | 1.000 | −0.275 |
| E-Gap | -0.225 | 0.391 | −0.472 | 0.043 | 0.087 | 0.230 | 0.103 | 0.096 | −0.684 | −0.275 | 1.000 |
| No | Equation c | R2 | R2cv | F Value |
|---|---|---|---|---|
| Equation (1) | ln(Activity) = 26.09 × (R3 Charge + 0.22) − 7.47 × (R3 Charge + 1.18) + 497.87 × (HOMO + 0.19) − 494.72 × (HOMO + 0.19) − 7.81 × ( 1.24 − Ti-Charge) + 1943.36 × (−0.20 - HOMO) − 3345.07 × (−0.20 − HOMO) + 4.73 | 0.992 | 0.936 | 168.295 |
| Equation (2) a | ln(Activity) = −4.91 × (R3 Charge + 1.22) + 23.54 × (R3 Charge + 0.13) + 33949.84 × (−0.19 − HOMO) − 2361.91 × (−0.20 − HOMO) + 743.80 × (−0.20 − HOMO) − 33941.61 × (−0.19 − HOMO) + 5.00 | 0.992 | 0.984 | 230.917 |
| Equation (3) b | ln(Activity) = −9.15 × (R3 Charge + 1.17) + 30.70 × (R3 Charge + 0.24) − 257.00 × (HOMO + 0.189) + 225.65 × (HOMO + 0.19) −1938.84 × (−0.20 − HOMO) + 533.89 * (−0.20 − HOMO) + 4.74 | 0.997 | 0.927 | 564.305 |
| Complex | Substituents | Descriptors | Predicted PE Activity a | ||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R3-Charge | HOMO | |||
| Equation (2) | Equation (3) | ||||||
| 2-Zr | t-Bu | H | m-OAll | −1.115 | −0.199 | 12,260 | 11,474 |
| 3-Zr | t-Bu | H | p-OAll | −1.124 | −0.194 | 18,134 | 19,368 |
| 4-Zr | t-Bu | Me | H | 0.135 | −0.194 | 19,141 | 17,009 |
| 5-Zr | t-Bu | Me | m-OAll | −1.105 | −0.197 | 15,335 | 12,979 |
| 6-Zr | t-Bu | Me | p-OAll | −1.119 | −0.190 | 31,764 | 24,327 |
| 7-Zr | t-Bu | t-Bu | H | 0.135 | −0.198 | 43,697 | 32,147 |
| 8-Zr | t-Bu | t-Bu | m-OAll | −1.096 | −0.192 | 12,709 | 10,737 |
| 9-Zr | t-Bu | t-Bu | p-OAll | −1.117 | −0.189 | 30,879 | 36,923 |
| 10-Zr | Cumyl | H | H | 0.136 | −0.199 | 25,310 | 2709 |
| 11-Zr | Cumyl | H | m-OAll | −1.128 | −0.197 | 20,015 | 21,072 |
| 12-Zr | Cumyl | H | p-OAll | −1.124 | −0.191 | 17,239 | 19,574 |
| 13-Zr | Cumyl | Me | H | 0.134 | −0.191 | 33,901 | 21,331 |
| 14-Zr | Cumyl | Me | m-OAll | −1.123 | −0.193 | 17,624 | 18,965 |
| 15-Zr | Cumyl | Me | p-OAll | −1.116 | −0.187 | 30,705 | 31,103 |
| 16-Zr | Cumyl | Cumyl | H | 0.119 | −0.190 | 17,817 | 12,411 |
| 17-Zr | Cumyl | Cumyl | m-OAll | −1.096 | −0.189 | 24,490 | 24,515 |
| 18-Zr | Cumyl | Cumyl | p-OAll | −1.101 | −0.189 | 25,768 | 27,286 |
| 2-Ni | t-Bu | H | m-OAll | −1.132 | −0.188 | 36,587 | 47,324 |
| 3-Ni | t-Bu | H | p-OAll | −1.136 | −0.183 | 38,497 | 37,871 |
| 4-Ni | t-Bu | Me | H | 0.138 | −0.183 | 40,245 | 28,649 |
| 5-Ni | t-Bu | Me | m-OAll | −1.104 | −0.182 | 26,809 | 17,645 |
| 6-Ni | t-Bu | Me | p-OAll | −1.135 | −0.177 | 37,849 | 22,786 |
| 7-Ni | t-Bu | t-Bu | H | 0.138 | −0.183 | 40,245 | 27,974 |
| 10-Ni | t-Bu | t-Bu | m-OAll | −0.140 | −0.187 | 43,849 | 41,383 |
| 11-Ni | t-Bu | t-Bu | p-OAll | −1.126 | -0.186 | 34,187 | 35,222 |
| 12-Ni | Cumyl | H | H | −1.145 | −0.180 | 42,380 | 34,431 |
| 13-Ni | Cumyl | H | m-OAll | 0.137 | −0.179 | 38,555 | 21,053 |
| 14-Ni | Cumyl | H | p-OAll | −1.124 | −0.181 | 33,422 | 24,122 |
| 15-Ni | Cumyl | Me | H | −1.132 | −0.176 | 36,794 | 19,909 |
| 16-Ni | Cumyl | Me | m-OAll | 0.130 | −0.178 | 28,557 | 13,369 |
| 17-Ni | Cumyl | Me | p-OAll | −1.101 | −0.176 | 25,915 | 10,772 |
| 18-Ni | Cumyl | Cumyl | H | −1.098 | −0.178 | 25,050 | 11,616 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chasing, P.; Maitarad, P.; Wu, H.; Zhang, D.; Shi, L.; Promarak, V. Straightforward Design for Phenoxy-Imine Catalytic Activity in Ethylene Polymerization: Theoretical Prediction. Catalysts 2018, 8, 422. https://doi.org/10.3390/catal8100422
Chasing P, Maitarad P, Wu H, Zhang D, Shi L, Promarak V. Straightforward Design for Phenoxy-Imine Catalytic Activity in Ethylene Polymerization: Theoretical Prediction. Catalysts. 2018; 8(10):422. https://doi.org/10.3390/catal8100422
Chicago/Turabian StyleChasing, Pongsakorn, Phornphimon Maitarad, Hongmin Wu, Dengsong Zhang, Liyi Shi, and Vinich Promarak. 2018. "Straightforward Design for Phenoxy-Imine Catalytic Activity in Ethylene Polymerization: Theoretical Prediction" Catalysts 8, no. 10: 422. https://doi.org/10.3390/catal8100422
APA StyleChasing, P., Maitarad, P., Wu, H., Zhang, D., Shi, L., & Promarak, V. (2018). Straightforward Design for Phenoxy-Imine Catalytic Activity in Ethylene Polymerization: Theoretical Prediction. Catalysts, 8(10), 422. https://doi.org/10.3390/catal8100422