Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins


 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
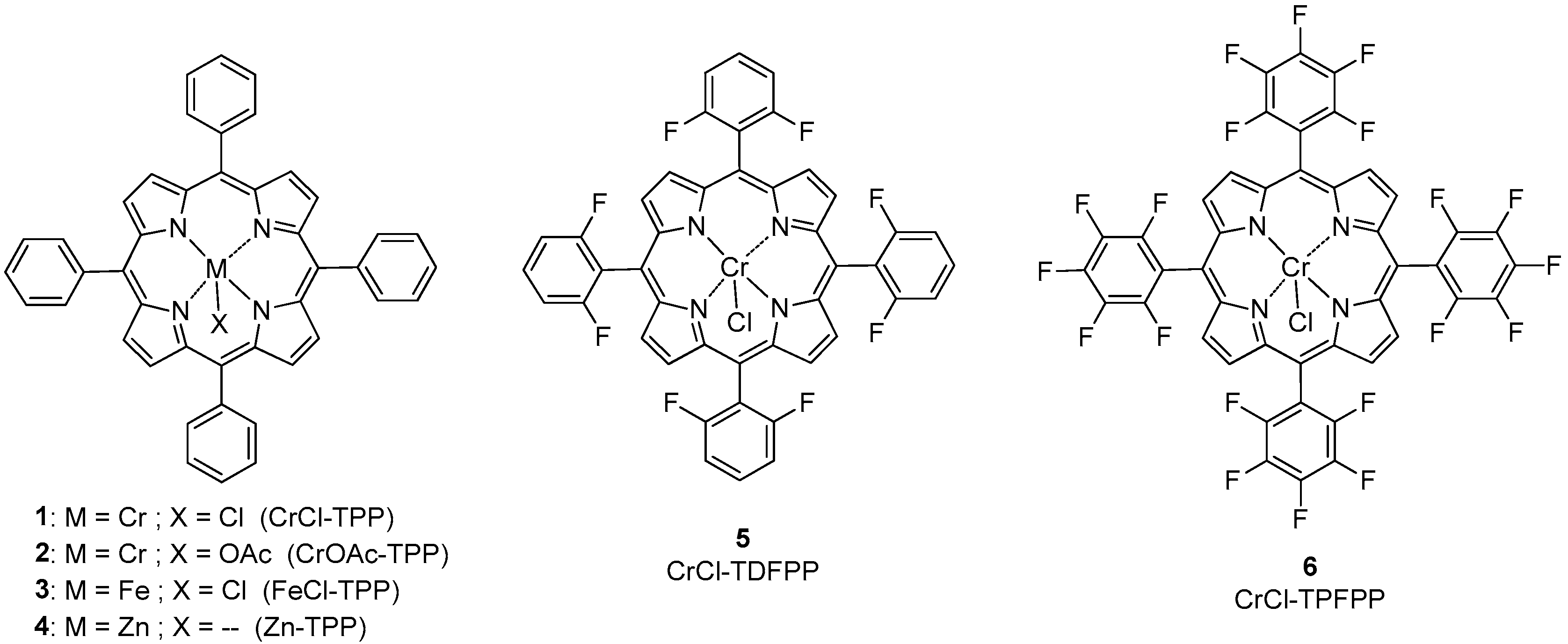
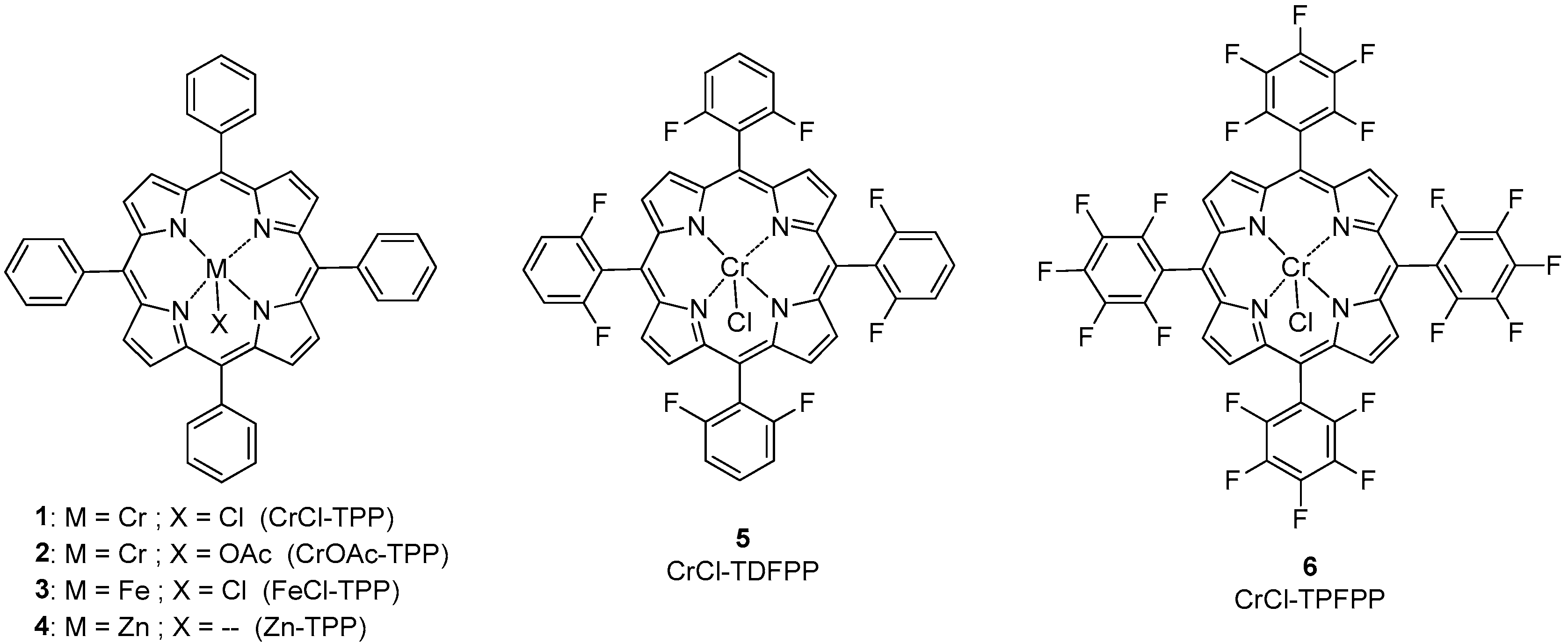
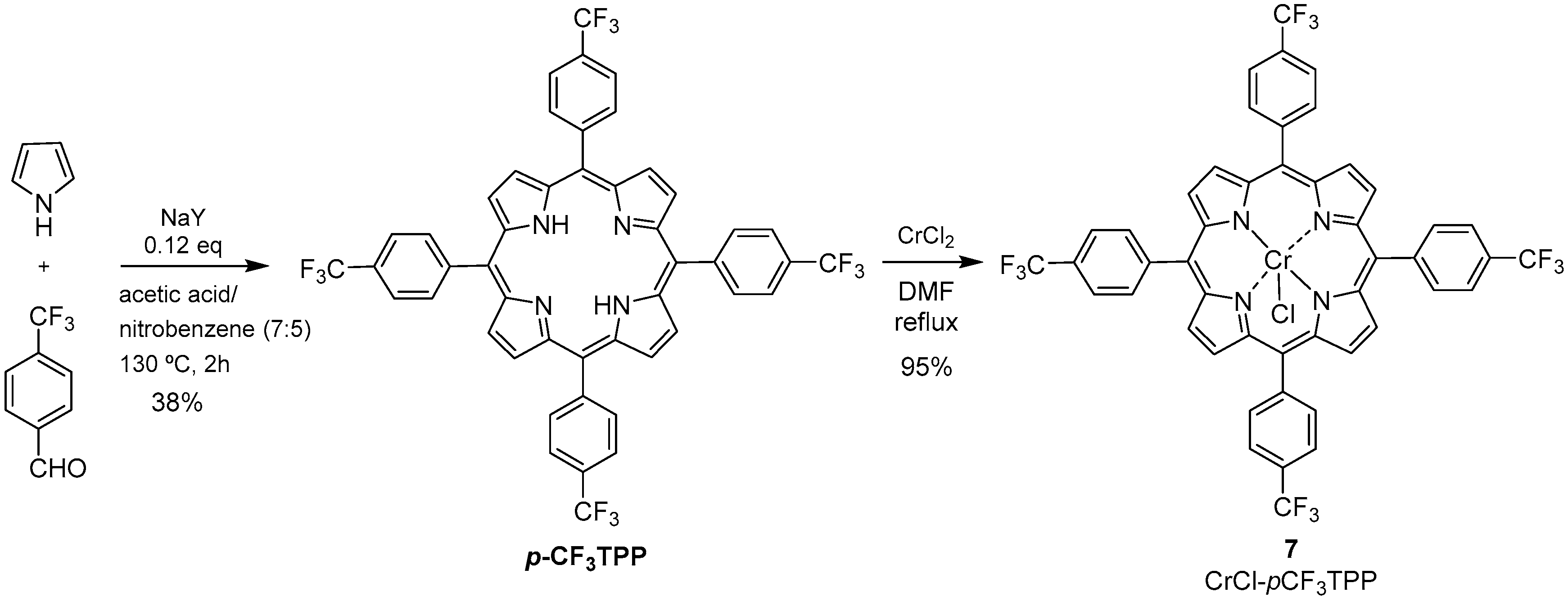
2.1. Synthesis of the Catalysts
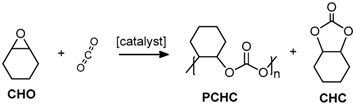
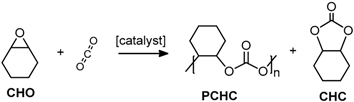
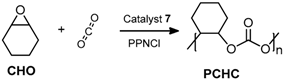
2.2. Coupling Reactions of Cyclohexene Oxide with CO2
2.2.1. Effect of the Metal
2.2.2. Effect of the Fluorinated Groups in Cr(III)-Porphyrin Catalysts
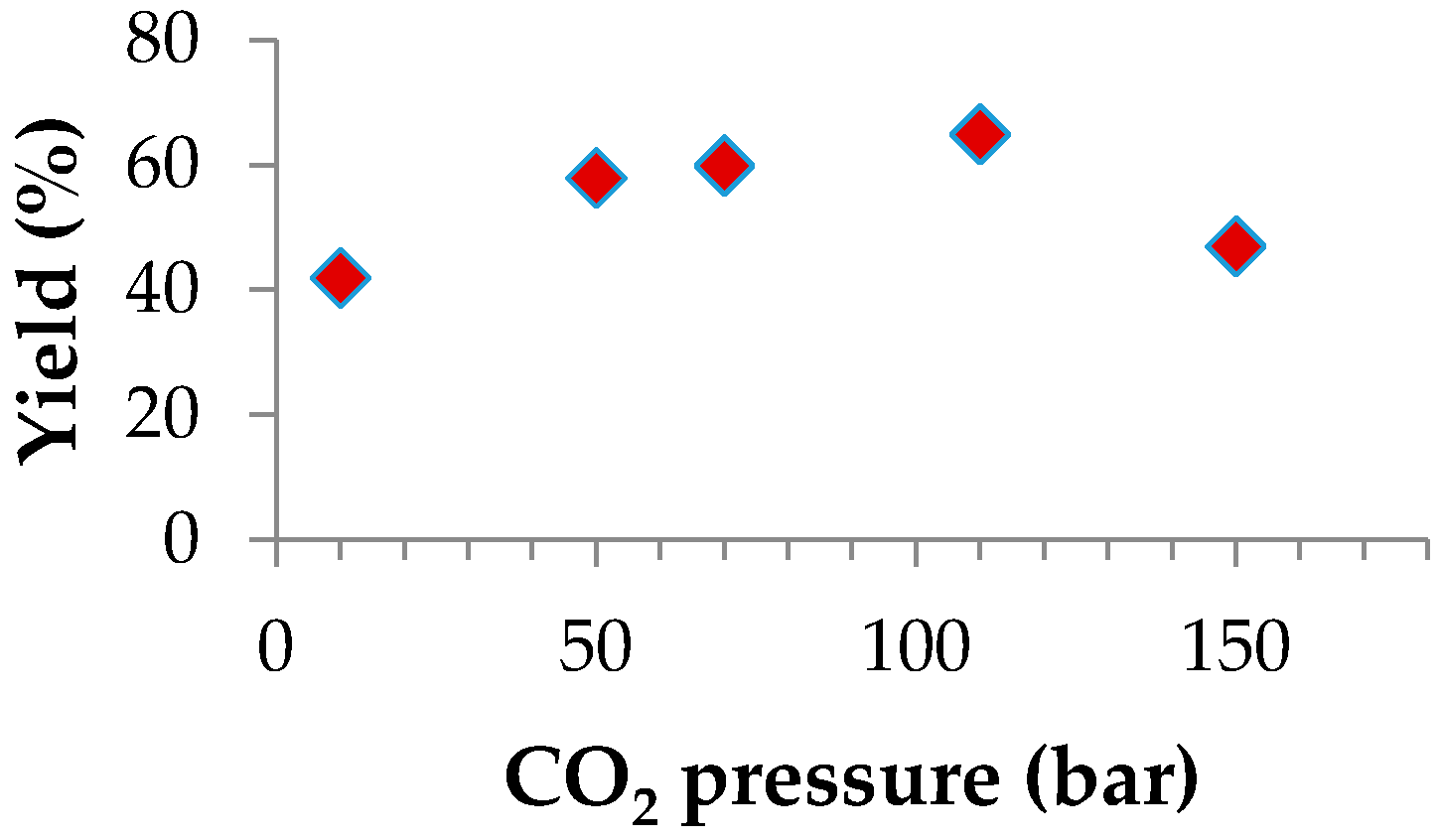
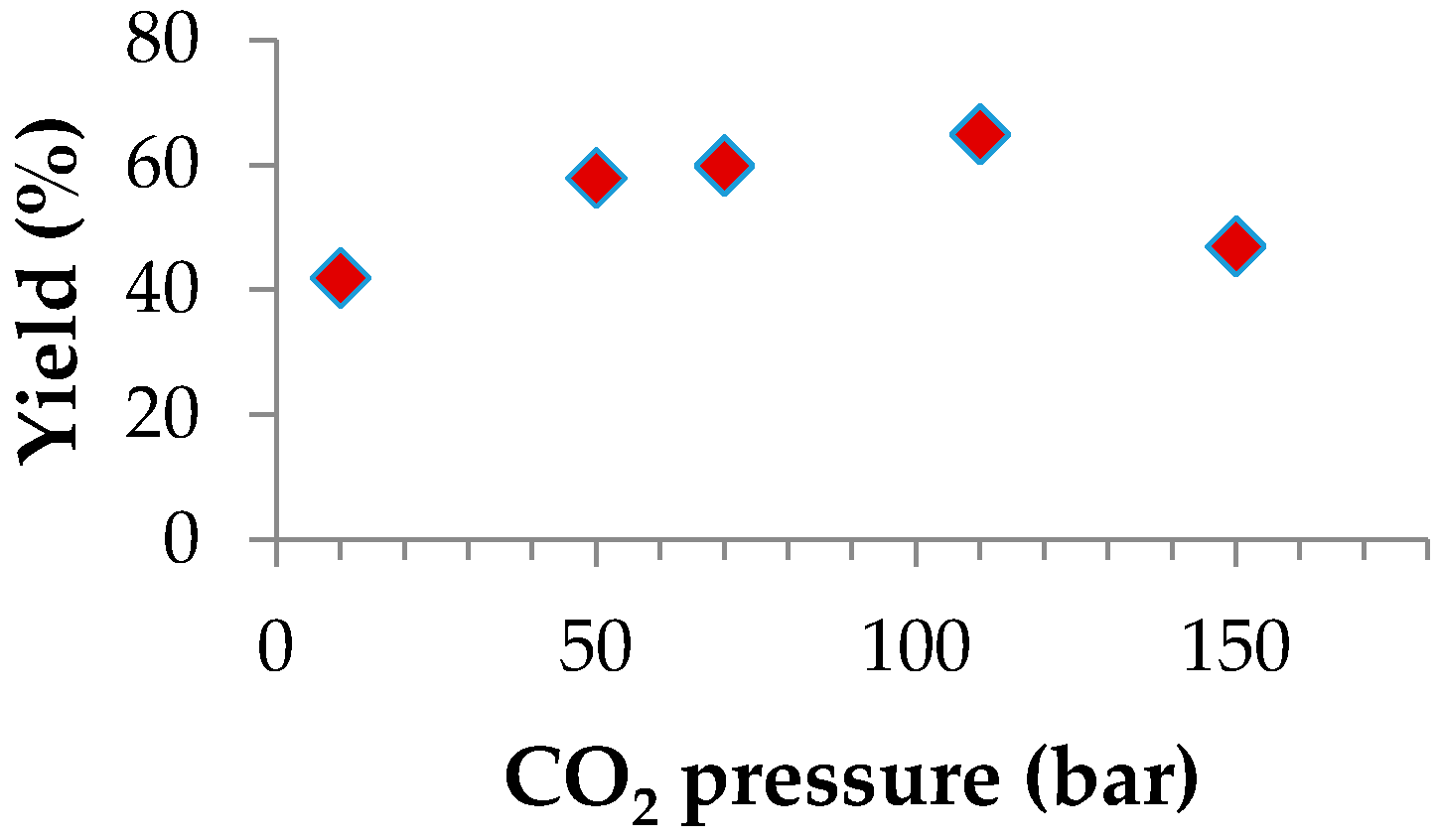
2.2.3. Effect of Temperature and CO2 Pressure
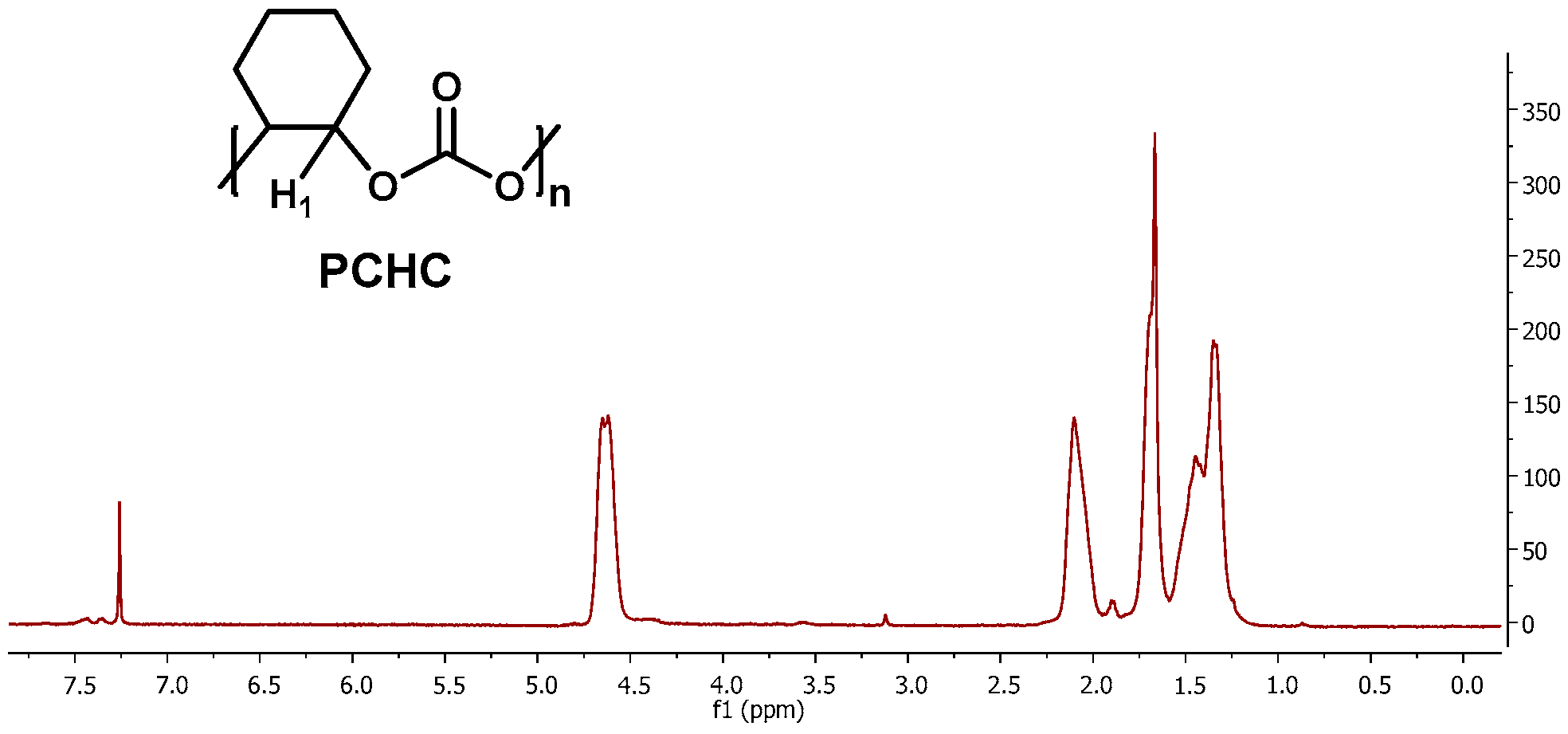
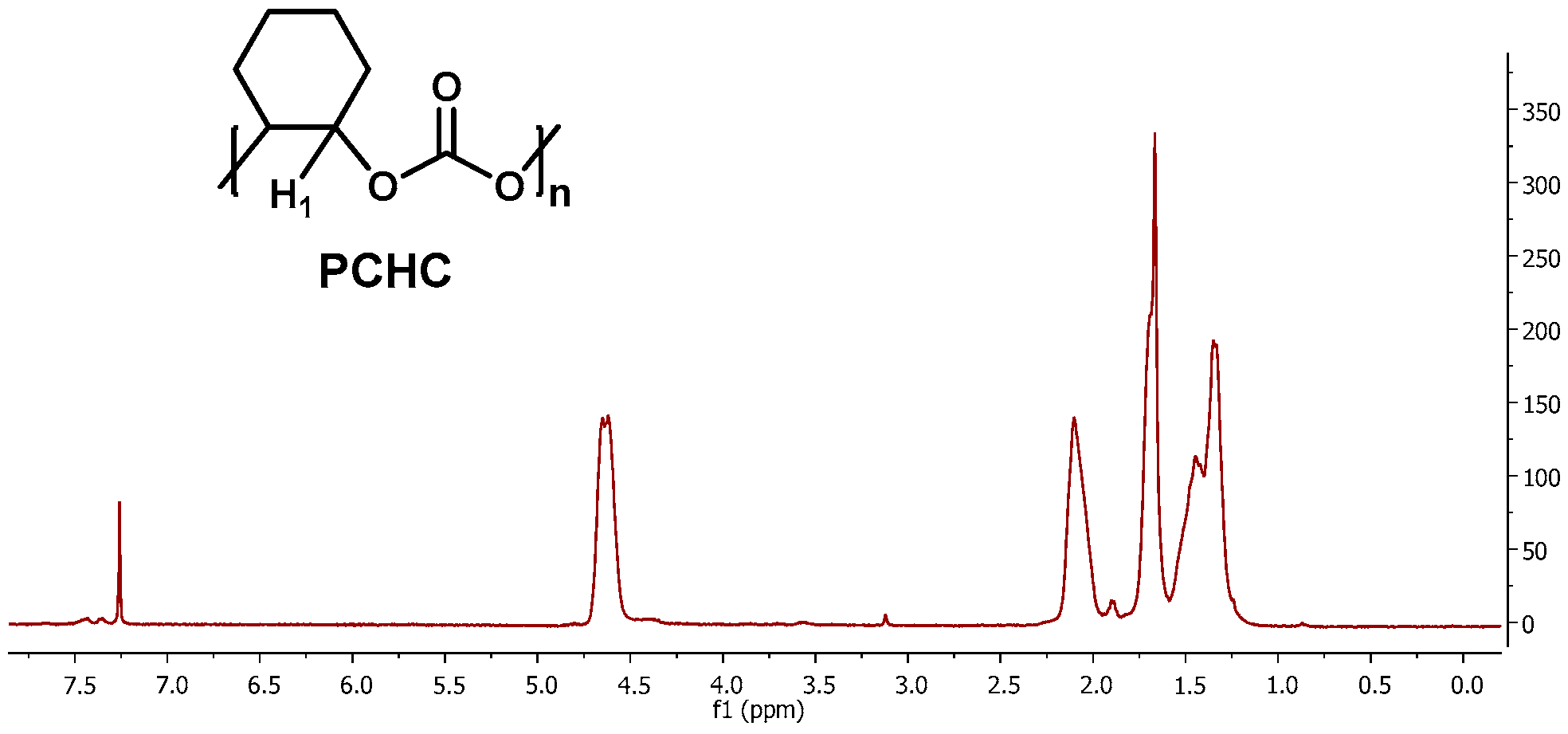
2.2.4. Characterisation of PCHC Copolymers
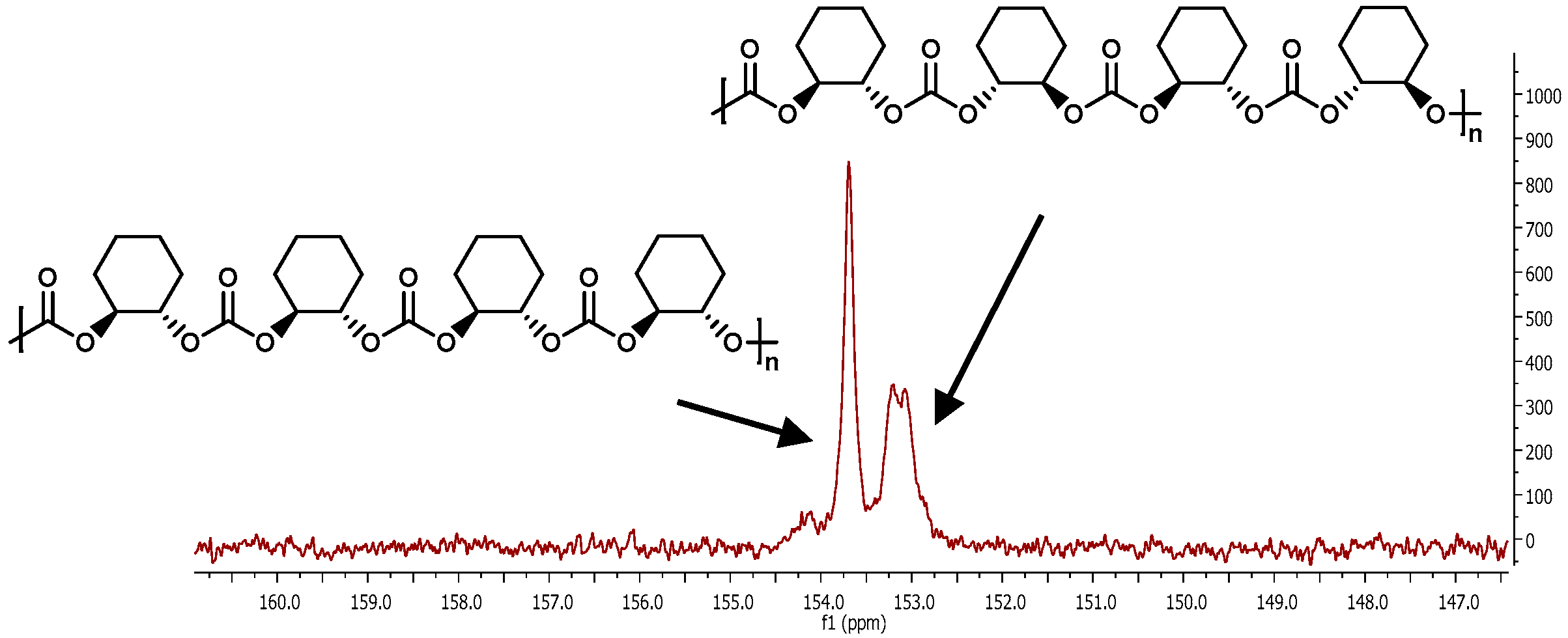
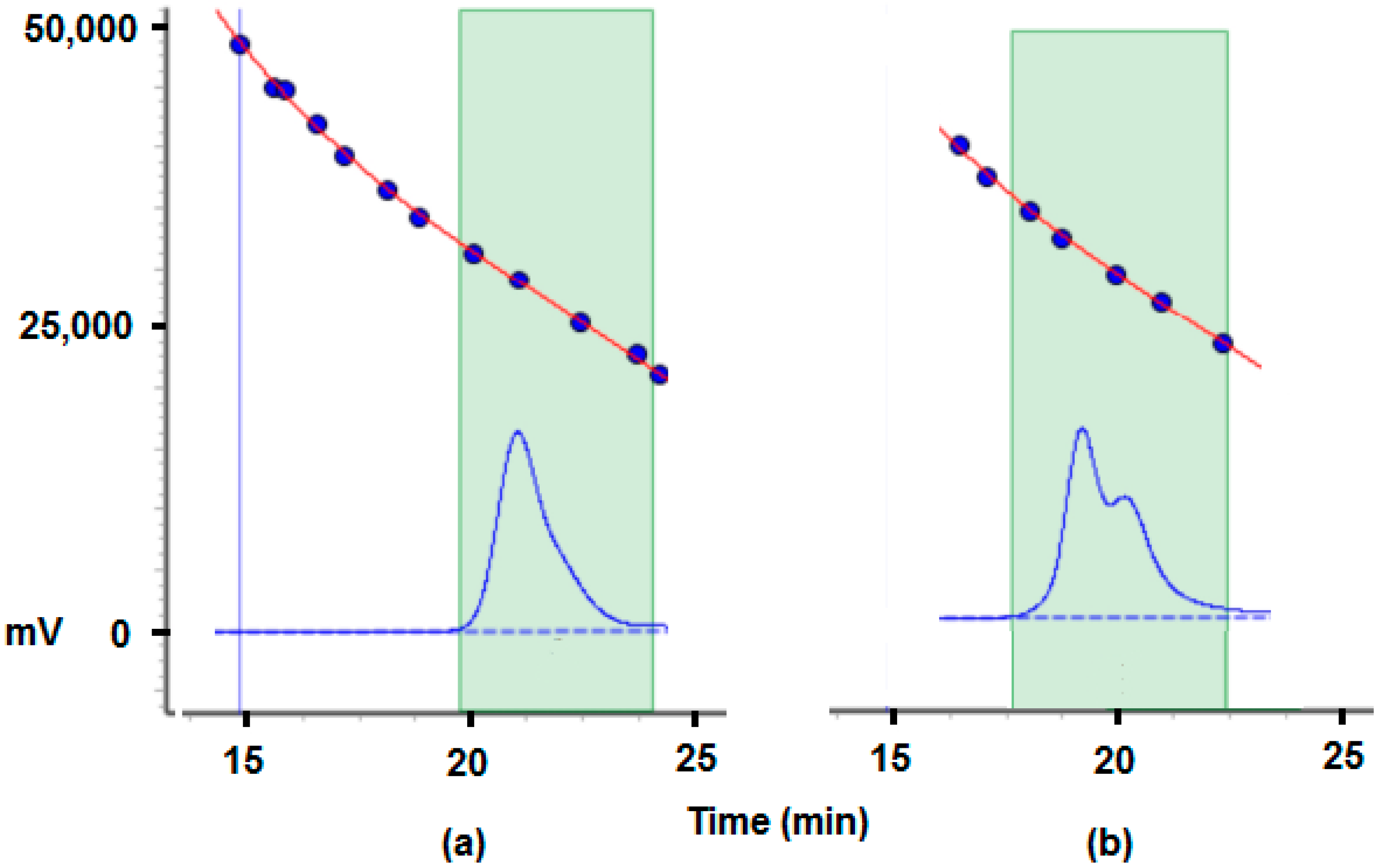
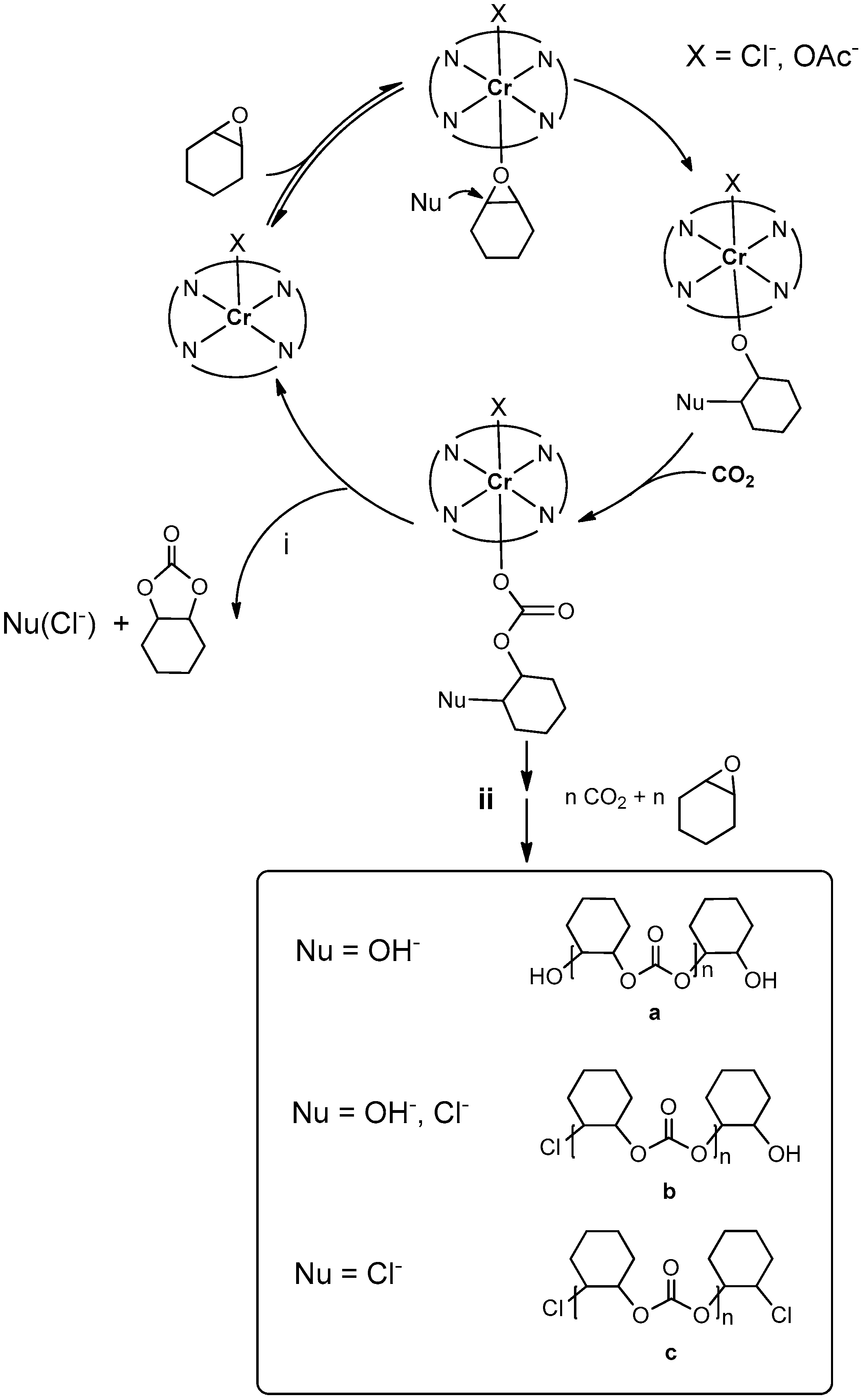
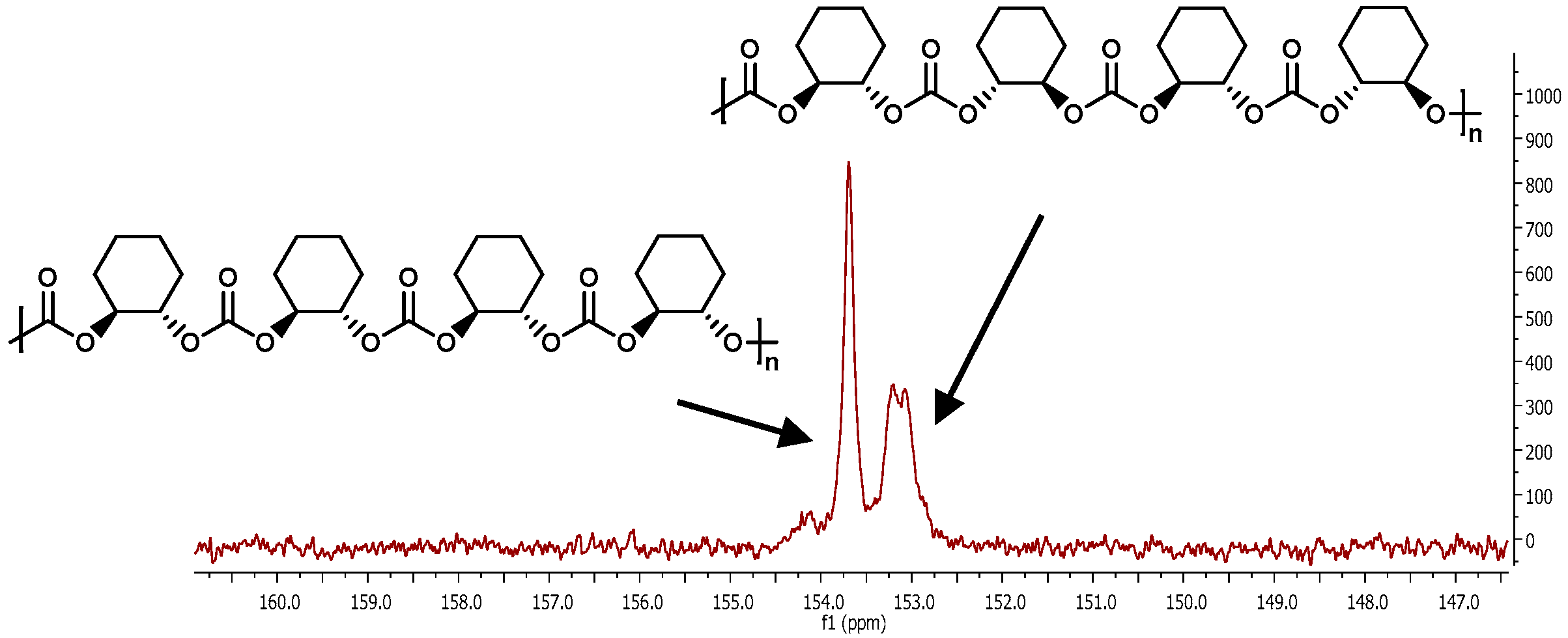
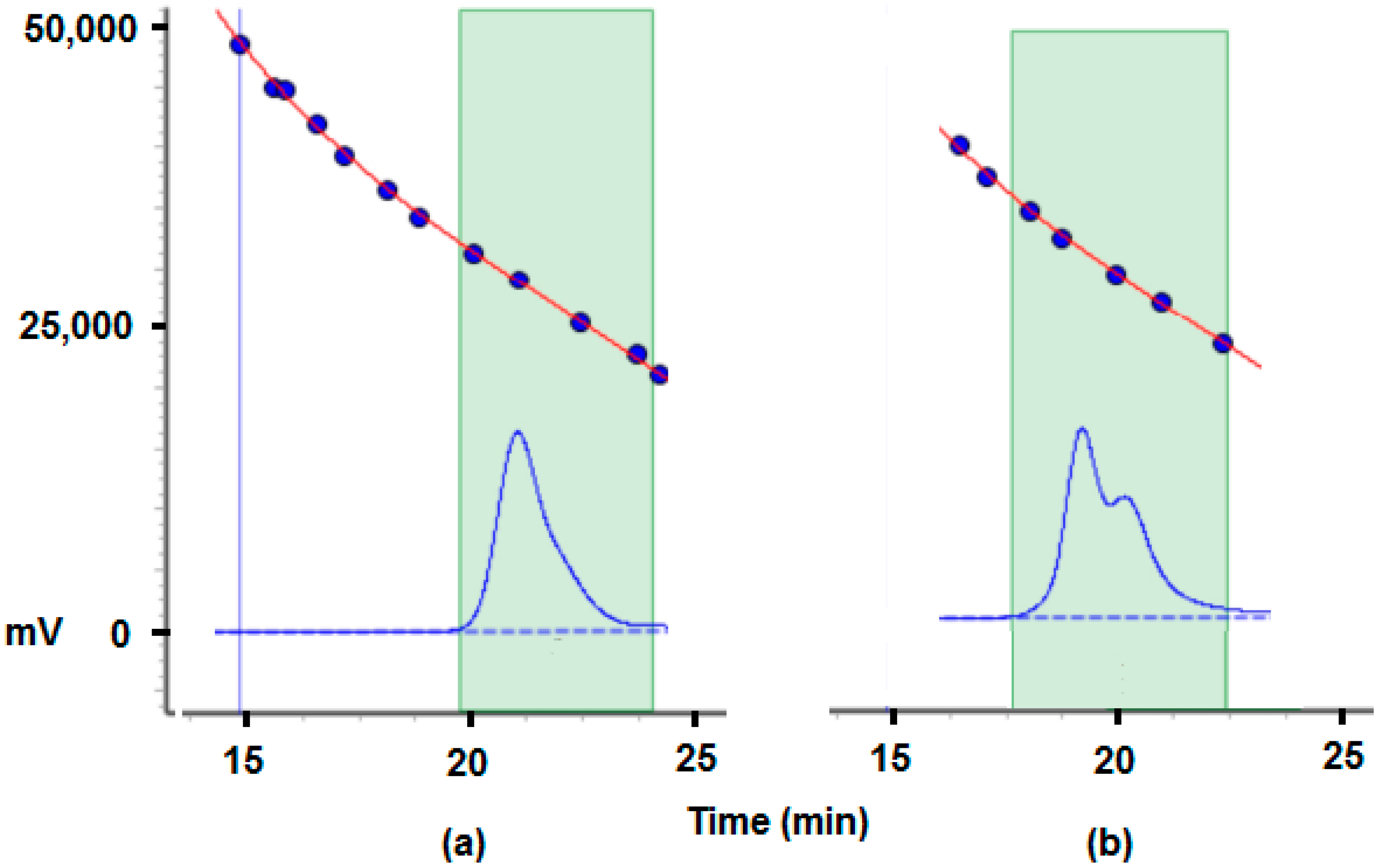
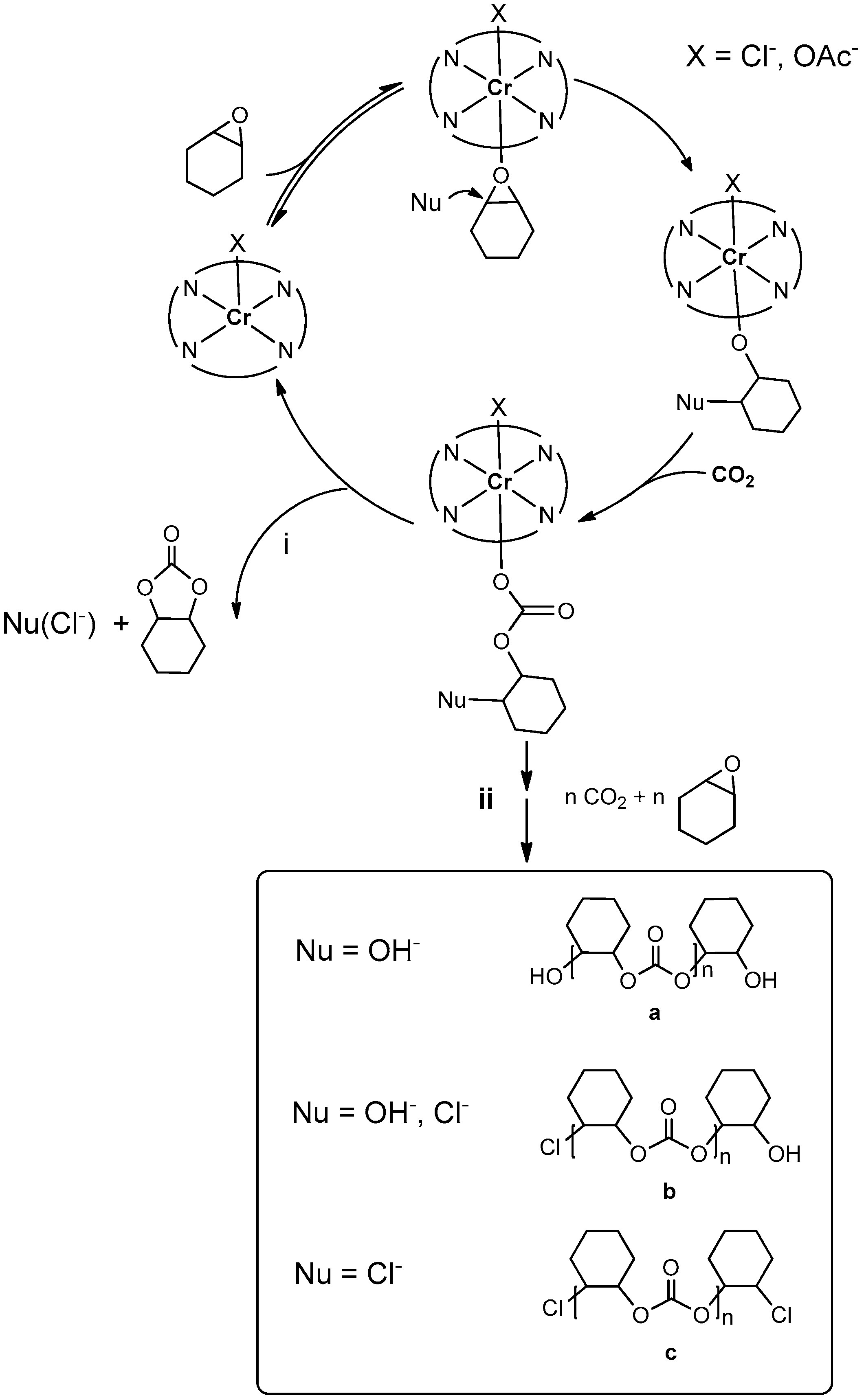
2.2.5. MALDI-TOF Analysis and Mechanistic Considerations
3. Materials and Methods
3.1. Reagents
3.2. Equipment
3.3. Preparation of Metalloporphyrin Catalysts
Synthesis of 5,10,15,20-tetrakis(4-trifluoromethylphenyl)porphyrinatochromium(III) Chloride (7)
3.4. Synthesis and Characterisation of Copolymers
3.4.1. General Procedure of Catalytic Reactions of Epoxides with CO2
3.4.2. Poly(cyclohexenecarbonate) (PCHC)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Whiteoak, C.J.; Kleij, A.W. Catalyst development in the context of ring expansion-addition of carbon dioxide to epoxides to give organic carbonates. Synlett 2013, 24, 1748–1756. [Google Scholar] [CrossRef]
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 21, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-L.; Luo, S.-L.; Yin, S.-F.; Au, C.-T. The direct transformation of carbon dioxide to organic carbonates over heterogeneous catalysts. Appl. Catal A Gen. 2009, 366, 2–12. [Google Scholar] [CrossRef]
- Lu, X.-B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Buchard, A.; Williams, C.K. Catalysts for CO2/epoxide copolymerization. Chem. Commun. 2011, 47, 141–163. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization and mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef] [PubMed]
- Clemens, J.H. Reactive applications of cyclic alkylene carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Martin, C.; Fiorani, G.; Kleij, A.W. Recent advances in the catalytic preparation of cyclic organic carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.; Wu, V.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933–5948. [Google Scholar] [CrossRef] [PubMed]
- Omae, I. Recent developments in carbon dioxide utilization for the production of organic chemicals. Coord. Chem. Rev. 2012, 256, 1384–1405. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Razali, N.A.M.; Lee, K.T.; Bhatia, S.; Mohamed, A.R. Heterogeneous catalysts for production of chemicals using carbon dioxide as raw material: A review. Energy Rev. 2012, 16, 4951–4964. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Holtcamp, M.W. Catalysts for the reaction of epoxides and carbon dioxide. Coord. Chem. Rev. 1996, 153, 155–174. [Google Scholar] [CrossRef]
- North, M. Synthesis of cyclic carbonates from epoxides and carbon dioxide using bimetallic aluminium(salen) complexes. ARKIVOC 2012, 2012, 610–628. [Google Scholar]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic carbonate synthesis catalysed by bimetallic aluminium-salen complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- Harrold, N.D.; Li, Y.; Chisholm, M.H. Studies of ring-opening reactions of styrene oxide by chromium tetraphenylporphyrin initiators. Mechanistic and stereochemical considerations. Macromolecules 2013, 46, 692–698. [Google Scholar] [CrossRef]
- Ohkawara, T.; Suzuki, K.; Nakano, K.; Mori, S.; Nozaki, K. Facile estimation of catalytic activity and selectivities in copolymerization of propylene oxide with carbon dioxide mediated by metal complexes with planar tetradentate ligand. J. Am. Chem. Soc. 2014, 136, 10728–10735. [Google Scholar] [CrossRef] [PubMed]
- Mang, S.; Cooper, A.I.; Colclough, M.E.; Chauhan, N.; Holmes, A.B. Copolymerization of CO2 and 1,2-cyclohexene oxide using a CO2-soluble chromium porphyrin catalyst. Macromolecules 2000, 33, 303–308. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Fitch, S.B. An Exploration of the coupling reactions of epoxides and carbon dioxide catalyzed by tetramethyltetraazaannulene chromium(III) derivatives: Formation of copolymers versus cyclic carbonates. Inorg. Chem. 2008, 47, 11868–11878. [Google Scholar] [CrossRef] [PubMed]
- Dengler, J.E.; Lehenmeier, M.W.; Klaus, S.; Anderson, C.E.; Herdtweck, E.; Rieger, B. A one-component iron catalyst for cyclic propylene carbonate synthesis. Eur. J. Inorg. Chem. 2011, 2011, 336–343. [Google Scholar] [CrossRef]
- Cuesta-Aluja, L.; Djoufak, M.; Aghmiz, A.; Rivas, R.; Christ, L.; Masdeu-Bultó, A.M. Novel chromium(III) complexes with N4-donor ligands as catalysts for the coupling of CO2 and epoxides in supercritical CO2. J. Mol. Catal. A Chem. 2014, 381, 161–170. [Google Scholar] [CrossRef]
- Adolph, M.; Zevaco, T.A.; Altesleben, C.; Walter, O.; Dinjus, E. New cobalt, iron and chromium catalysts based on easy-to-handle N4-chelating ligands for the coupling reaction of epoxides with CO2. Dalton Trans. 2014, 43, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Qiao, L.; Qin, Y.; Wang, X.; Wang, F. Highly efficient and quantitative synthesis of a cyclic carbonate by iron complex catalysts. Polyhedron 2014, 74, 129–133. [Google Scholar] [CrossRef]
- Adolph, M.; Zevaco, T.A.; Altesleben, C.; Staudt, S.; Dinjus, E. New zinc catalysts based on easy-to-handle N4-chelating ligands for the coupling reaction of epoxides with CO2. J. Mol. Catal. A Chem. 2015, 400, 104–110. [Google Scholar] [CrossRef]
- Mercadé, E.; Zangrando, E.; Claver, C.; Godard, C. Robust zinc complexes that contain pyrrolidine-based ligands as recyclable catalysts for the synthesis of cyclic carbonates from carbon dioxide and epoxides. ChemCatChem 2016, 8, 234–243. [Google Scholar] [CrossRef]
- Campos-Carrasco, A.; Tortosa-Estorach, C.; Masdeu-Bultó, A.M. Rh(I) complexes with fluorinated 2,2′-bipyridines. Inorg. Chem. Commun. 2012, 18, 61–64. [Google Scholar] [CrossRef]
- Stamp, L.M.; Mang, S.A.; Holmes, A.B.; Knights, K.A.; de Miguel, Y.R.; McConvey, I.F. Polymer supported chromium porphyrin as catalyst for polycarbonate formation in supercritical carbon dioxide. Chem. Commun. 2001, 2502–2503. [Google Scholar] [CrossRef]
- Chatterjee, C.; Chisholm, M.H.; El-Khaldy, A.; McIntosh, R.D.; Miller, J.T.; Wu, T. Influence of the metal (Al, Cr, and Co) and substituents of the porphyrin in controlling reactions involved in copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 3. Cobalt chemistry. Inorg. Chem. 2013, 52, 4547–4553. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Chisholm, M.H. Influence of the metal (Al, Cr, and Co) and the substituents of the porphyrin in controlling the reactions involved in the copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 2. Chromium chemistry. Inorg. Chem. 2012, 51, 12041–12052. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Chisholm, M.H. The influence of the metal (Al, Cr, and Co) and the substituents of the porphyrin in controlling the reactions involved in the copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 1. Aluminum chemistry. Inorg. Chem. 2011, 50, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Duan, S.; Hai, L.; Jing, H. Carbon dioxide fixation by cycloaddition with epoxides, catalyzed by biomimetic metalloporphyrins. ChemCatChem 2012, 4, 1752–1758. [Google Scholar] [CrossRef]
- Bai, D.; Wang, Q.; Song, Y.; Li, B.; Jing, H. Synthesis of cyclic carbonate from epoxide and CO2 catalyzed by magnetic nanoparticle-supported porphyrin. Catal. Commun. 2011, 12, 684–688. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Koyama, S.; Yano, Y.; Sakai, T. A bifunctional catalyst for carbon dioxide fixation: cooperative double activation of epoxides for the synthesis of cyclic carbonates. Chem. Commun. 2012, 48, 4489–4491. [Google Scholar] [CrossRef] [PubMed]
- Ema, T.; Miyazaki, Y.; Taniguchi, T.; Takada, J. Robust porphyrin catalysts immobilized on biogenous iron oxide for the repetitive conversions of epoxides and CO2 into cyclic carbonates. Green Chem. 2013, 15, 2485–2492. [Google Scholar] [CrossRef]
- Cuesta-Aluja, L.; Castilla, J.; Masdeu-Bultó, A.M.; Henriques, C.A.; Calvete, M.J.F.; Pereira, M.M. Halogenated meso-phenyl Mn(III) porphyrins as highly efficient catalysts for the synthesis of polycarbonates and cyclic carbonates using carbon dioxide and epoxides. J. Mol. Catal. A Chem. 2016, 423, 489–494. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Nunes, M.L.P.J.; Pereira, M.M.; Gonsalves, A.M.A.R.; Serra, A.C. Improved Syntheses of 5,10,15,20-Tetrakisaryl- and Tetrakisalkylporphyrins. Heterocycles 1996, 43, 1423–1437. [Google Scholar] [CrossRef]
- Silva, M.; Fernandes, A.; Bebiano, S.S.; Calvete, M.J.F.; Ribeiro, M.F.; Burrows, H.D.; Pereira, M.M. Size and ability do matter! Influence of acidity and pore size on the synthesis of hindered halogenated meso-phenyl porphyrins catalysed by porous solid oxides. Chem. Commun. 2014, 50, 6571–6573. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chisholm, M.H.; Gallucci, J.C.; Zhang, X.; Zhou, Z. Binding of propylene oxide to porphyrin- and salen-M(III) cations, where M = Al, Ga, Cr, and Co. Inorg. Chem. 2005, 44, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Azenha, E.G.; Serra, A.C.; Pineiro, M.; Pereira, M.M.; Melo, J.S.; Arnaut, L.G.; Formosinho, S.J.; Gonsalves, A.M.A.R. heavy-atom effects on metalloporphyrins and polyhalogenated porphyrins. Chem. Phys. 2002, 280, 177–190. [Google Scholar] [CrossRef]
- Liston, D.J.; West, B.O. Oxochromium compounds. 2. Reaction of oxygen with chromium(II) and chromium(III) porphyrins and synthesis of a p-oxo chromium porphyrin Derivative. Inorg. Chem. 1985, 24, 1568–1576. [Google Scholar] [CrossRef]
- Bottomley, L.A.; Neely, F.L. Stereoelectronic aspects of inter-metal nitrogen atom transfer reactions between nitridomanganese(V) and chromium(III) porphyrins. Inorg. Chem. 1997, 36, 5435–5439. [Google Scholar] [CrossRef]
- Huang, T.; Wu, X.; Weare, W.W.; Sommer, R.D. Mono-oxido-bridged heterobimetallic and heterotrimetallic compounds containing titanium(IV) and chromium(III). Eur. J. Inorg. Chem. 2014, 2014, 5662–5674. [Google Scholar] [CrossRef]
- Huang, T.; Wu, X.; Song, X.; Xu, H.; Smirnova, T.I.; Walter, W.; Weare, W.W.; Sommer, R.D. Ferromagnetic coupling in d1–d3 linear oxidobridged heterometallic complexes: Ground-state models of metal-to-metal charge transfer excited states. Dalton Trans. 2015, 44, 18937–18944. [Google Scholar] [CrossRef] [PubMed]
- Super, M.; Beckman, E.J. Copolymerization of CO2 and cyclohexene oxide. Macromol. Symp. 1998, 127, 89–108. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yarbrough, J.C. Mechanistic aspects of the copolymerization reaction of carbon dioxide and epoxides, using a chiral salen chromium chloride catalyst. J. Am. Chem. Soc. 2002, 124, 6335–6342. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Darling, N.A.; Lobkovsky, E.B.; Coates, G.W. Enantiomerically-enriched organic reagents via polymer synthesis: enantioselective copolymerization of cycloalkene oxides and CO2 using homogeneous, zinc-based catalysts. Chem. Commun. 2000, 0, 2007–2008. [Google Scholar] [CrossRef]
- Nakano, K.; Nozaki, K.; Hiyama, T. Spectral assignment of poly[cyclohexene oxide-alt-carbon dioxide]. Macromolecules 2001, 34, 6325–6332. [Google Scholar] [CrossRef]
- Super, M.; Costello, C.; Berluche, E.; Beckman, E. Copolymerization of 1,2-epoxycyclohexane and carbon dioxide using carbon dioxide as both reactant and solvent. Macromolecules 1997, 30, 368–372. [Google Scholar] [CrossRef]
- Kooriyaden, F.R.; Sujatha, S.; Arunkumar, C. Synthesis, spectral, structural and antimicrobial studies of fluorinated porphyrins. Polyhedron 2015, 97, 66–74. [Google Scholar] [CrossRef]
- Grancho, J.C.P.; Pereira, M.M.; Miguel, M.G.; Gonsalves, A.M.R.; Burrows, H.D. Synthesis, spectra and photophysics of some free base tetrafluoroalkyland tetrafluoroaryl porphyrins with potential applications in imaging. Photochem. Photobiol. 2002, 75, 249–256. [Google Scholar] [CrossRef]
- Sun, Z.-C.; She, Y.-B.; Zhou, Y.; Song, X.-F.; Li, K. Synthesis, Characterization and spectral properties of substituted tetraphenylporphyrin iron chloride complexes. Molecules 2011, 16, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Ohshima, H.; Inoue, S. Alternating copolymerization of carbon dioxide and epoxide by manganese porphyrin: The first example of polycarbonate synthesis from 1-atm carbon dioxide. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3549–3555. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Co-cat | Conversion b (%) | TON c | Selectivity d | |
|---|---|---|---|---|---|---|
| PCHC (%) | CHC (%) | |||||
| 1 | 1 (CrCl-TPP) | - | 0 | 0 | - | - |
| 2 | 1 (CrCl-TPP) | PPNCl | 79 | 1128 | 98 | 2 |
| 3 | 2 (CrOAc-TPP) | PPNCl | 74 | 1056 | 92 | 8 |
| 4 | 3 (FeCl-TPP) | - | 0 | 0 | - | - |
| 5 | 3 (FeCl-TPP) | PPNCl | 14 | 192 | 0 | 100 |
| 6 | 4 (Zn-TPP) | - | 0 | 0 | - | - |
| 7 | 4 (Zn-TPP) | PPNCl | 34 | 480 | 0 | 100 |
| Entry | Catalyst | Conversion b (%) | TON c | Selectivity d | |
|---|---|---|---|---|---|
| PCHC (%) | CHC (%) | ||||
| 1 | 7 (CrCl-p-CF3TPP) | 86 | 1224 | 99 | 1 |
| 2 | 5 (CrCl-TDFPP) | 79 | 1128 | 92 | 8 |
| 3 | 6 (CrCl-TPFPP) | 50 | 720 | 91 | 9 |
| Entry | T (°C) | PCO2 (bar) | Conv. b (%) | TON c | Polymers (%) | Isolated Yield (%) d | % CO2 e | Mn·103 f | Mw/Mn f |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 50 | 50 | 77 | 1080 | 99 | 30 | 98 | 6.8 | 1.43 |
| 2 | 80 | 50 | 86 | 1224 | 99 | 58 | 98 | 4.8 | 1.25 |
| 3 | 80 | 10 | 70 | 1008 | 99 | 42 | 99 | 9.1 | 1.45 |
| 4 | 80 | 70 | 86 | 1224 | 96 | 60 | 98 | 12.5 | 1.38 |
| 5 | 80 | 110 | 84 | 1176 | 98 | 65 | 98 | 7.8 | 1.37 |
| 6 | 80 | 150 | 65 | 936 | 97 | 47 | 98 | 5.0 | 1.63 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrilho, R.M.B.; Dias, L.D.; Rivas, R.; Pereira, M.M.; Claver, C.; Masdeu-Bultó, A.M. Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins. Catalysts 2017, 7, 210. https://doi.org/10.3390/catal7070210
Carrilho RMB, Dias LD, Rivas R, Pereira MM, Claver C, Masdeu-Bultó AM. Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins. Catalysts. 2017; 7(7):210. https://doi.org/10.3390/catal7070210
Chicago/Turabian StyleCarrilho, Rui M. B., Lucas D. Dias, Raquel Rivas, Mariette M. Pereira, Carmen Claver, and Anna M. Masdeu-Bultó. 2017. "Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins" Catalysts 7, no. 7: 210. https://doi.org/10.3390/catal7070210
APA StyleCarrilho, R. M. B., Dias, L. D., Rivas, R., Pereira, M. M., Claver, C., & Masdeu-Bultó, A. M. (2017). Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins. Catalysts, 7(7), 210. https://doi.org/10.3390/catal7070210