
Molybdenum (VI) Imido Complexes Derived from Chelating Phenols: Synthesis, Characterization and ɛ-Caprolactone ROP Capability



Abstract
:1. Introduction
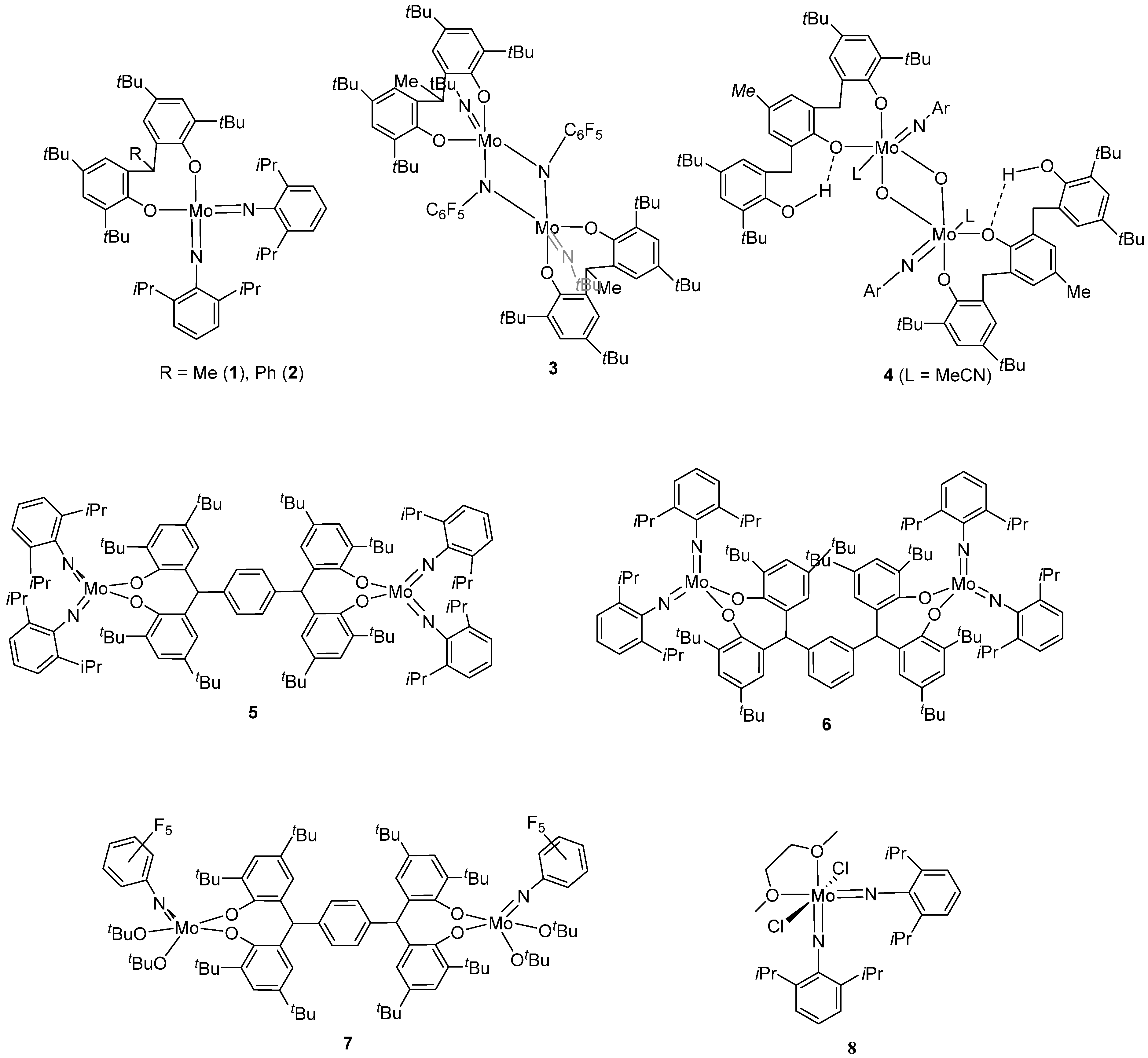
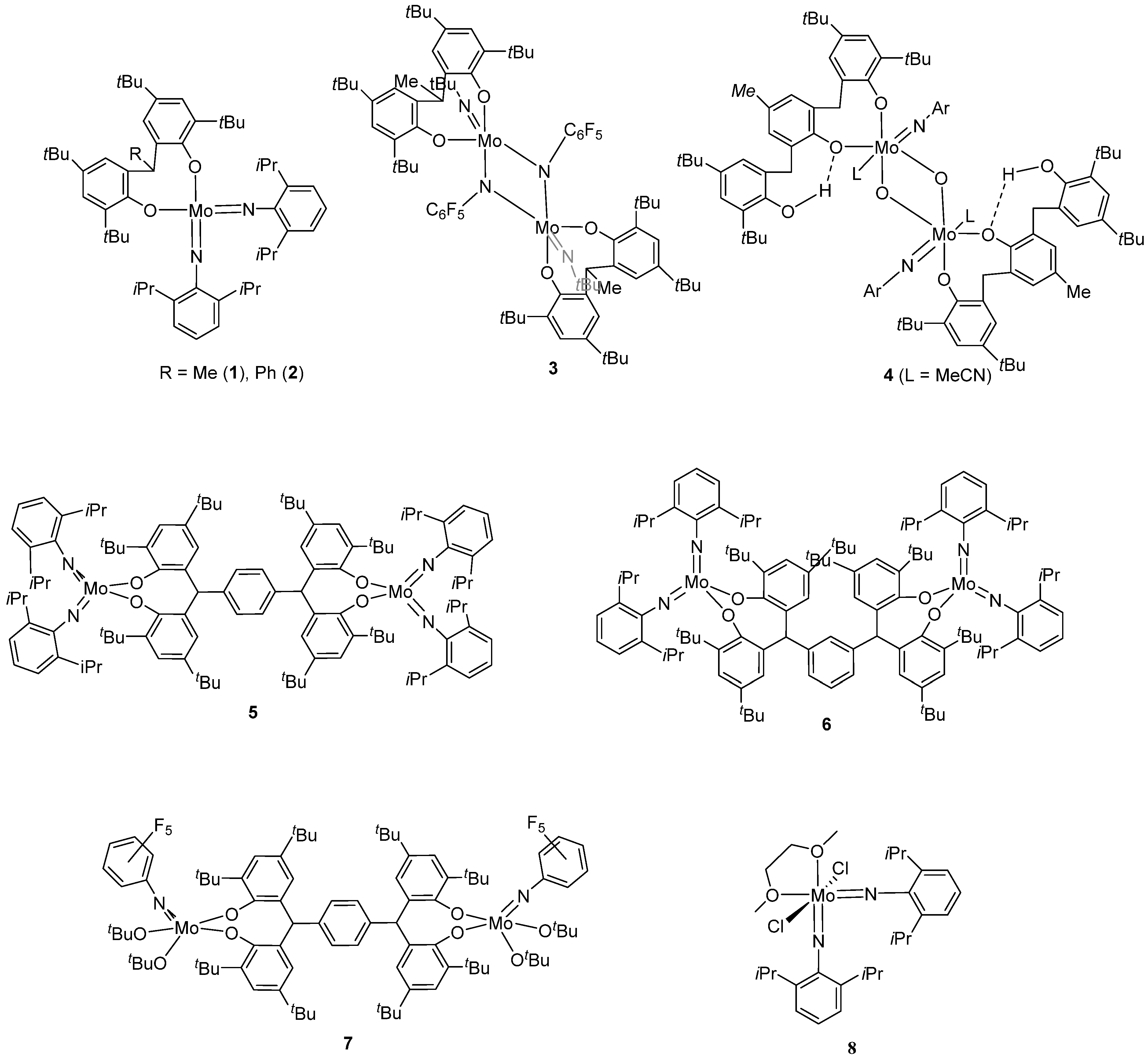
2. Results and Discussion
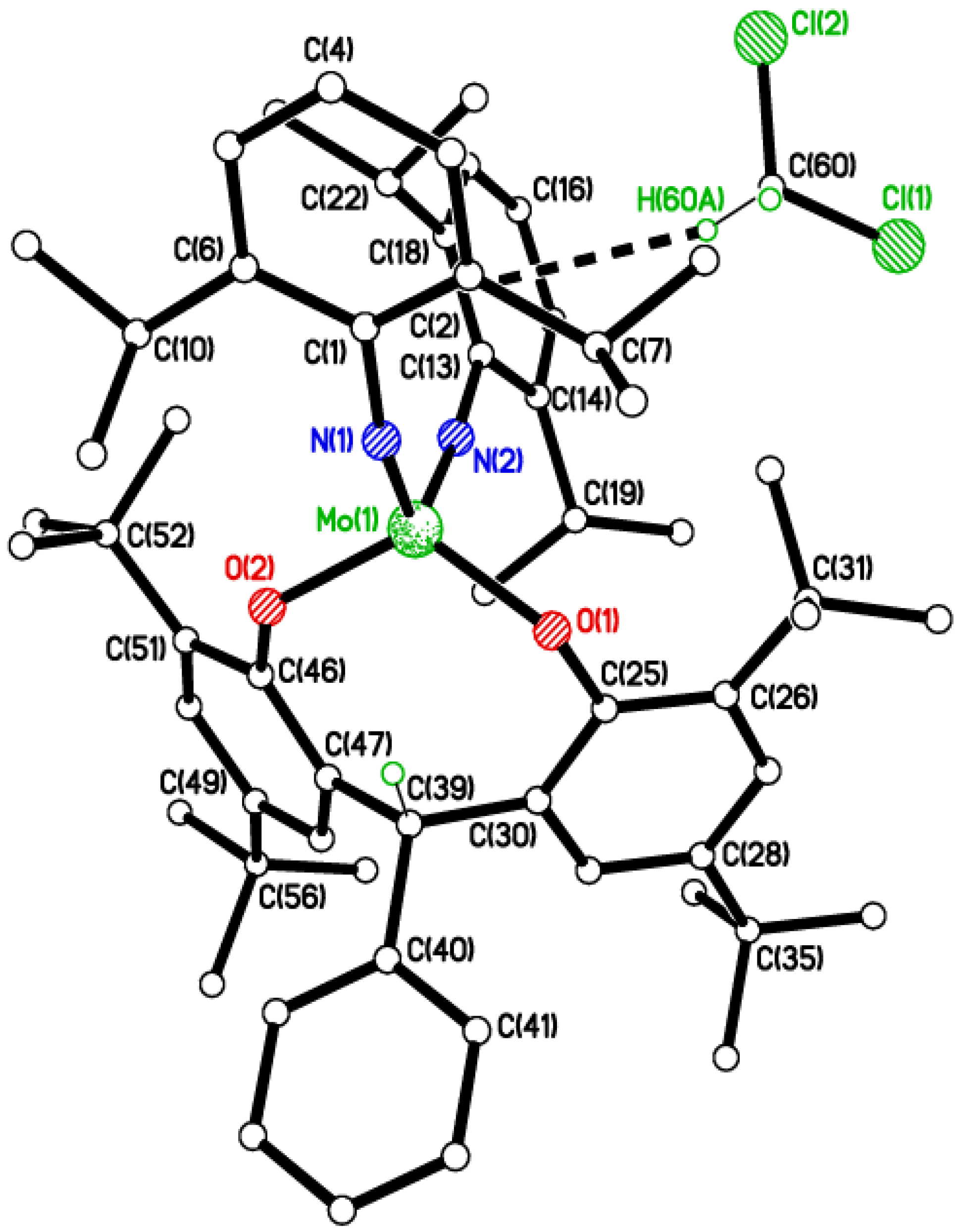
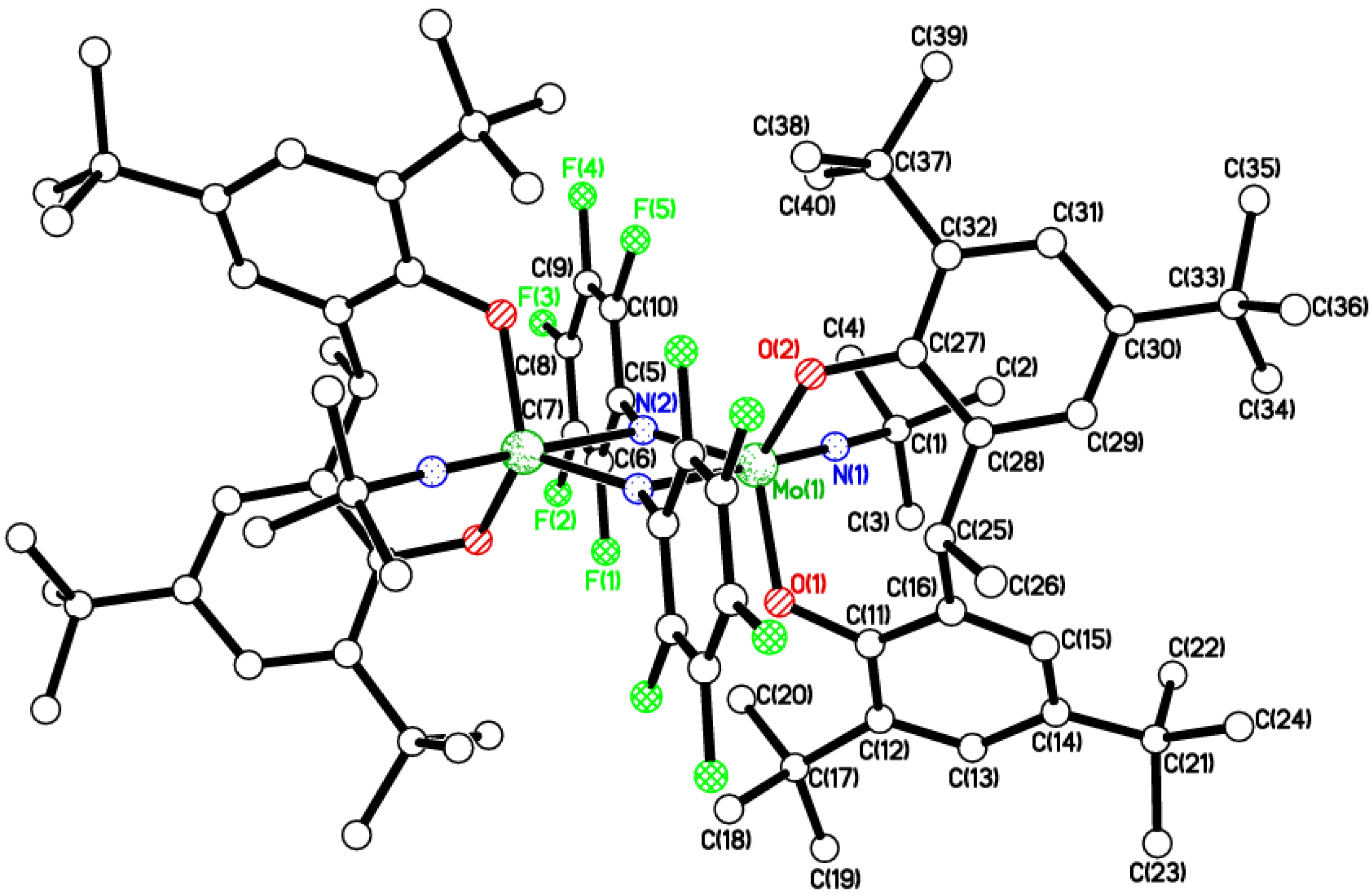
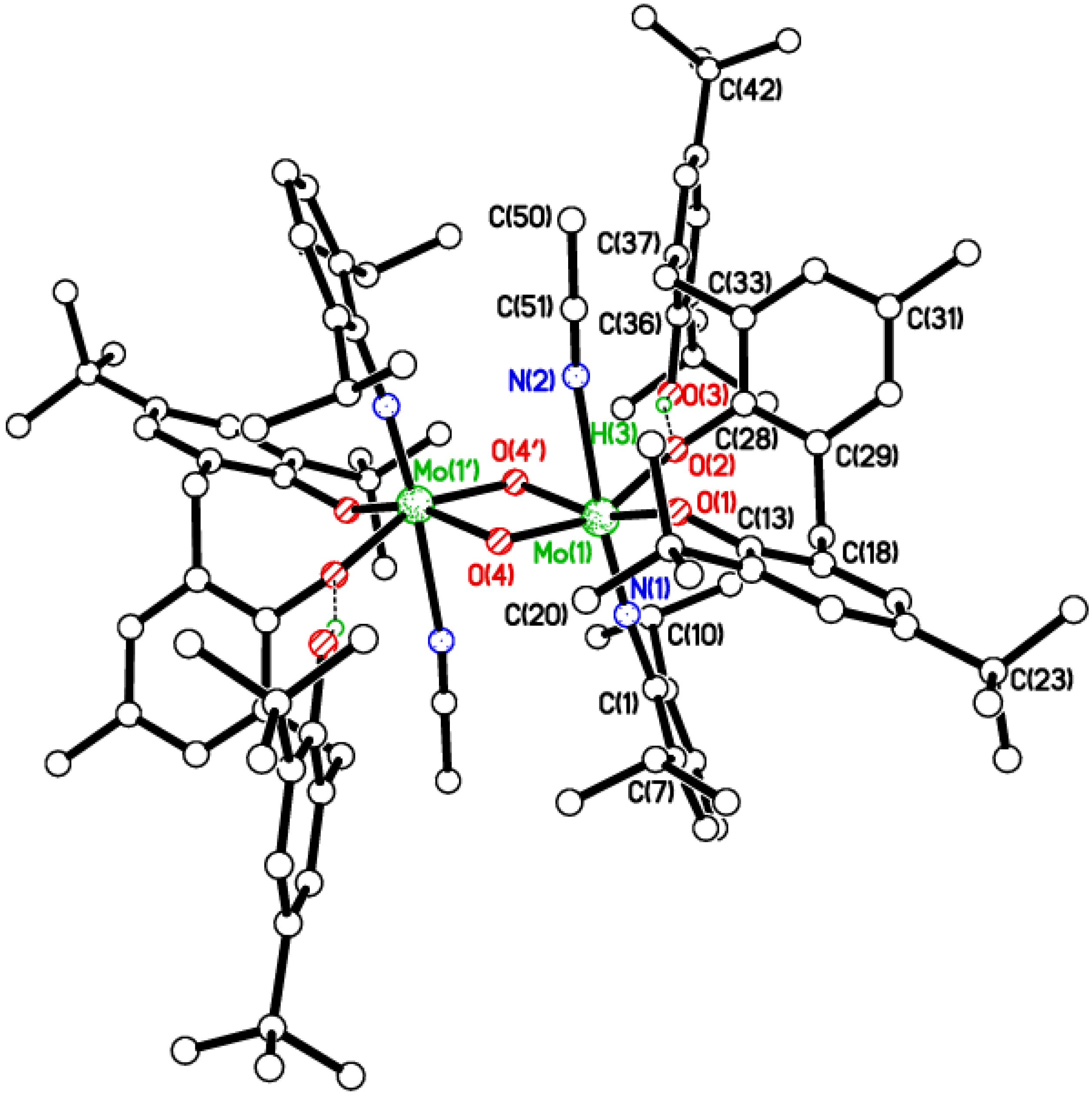
2.1. Di-Phenolate Compounds
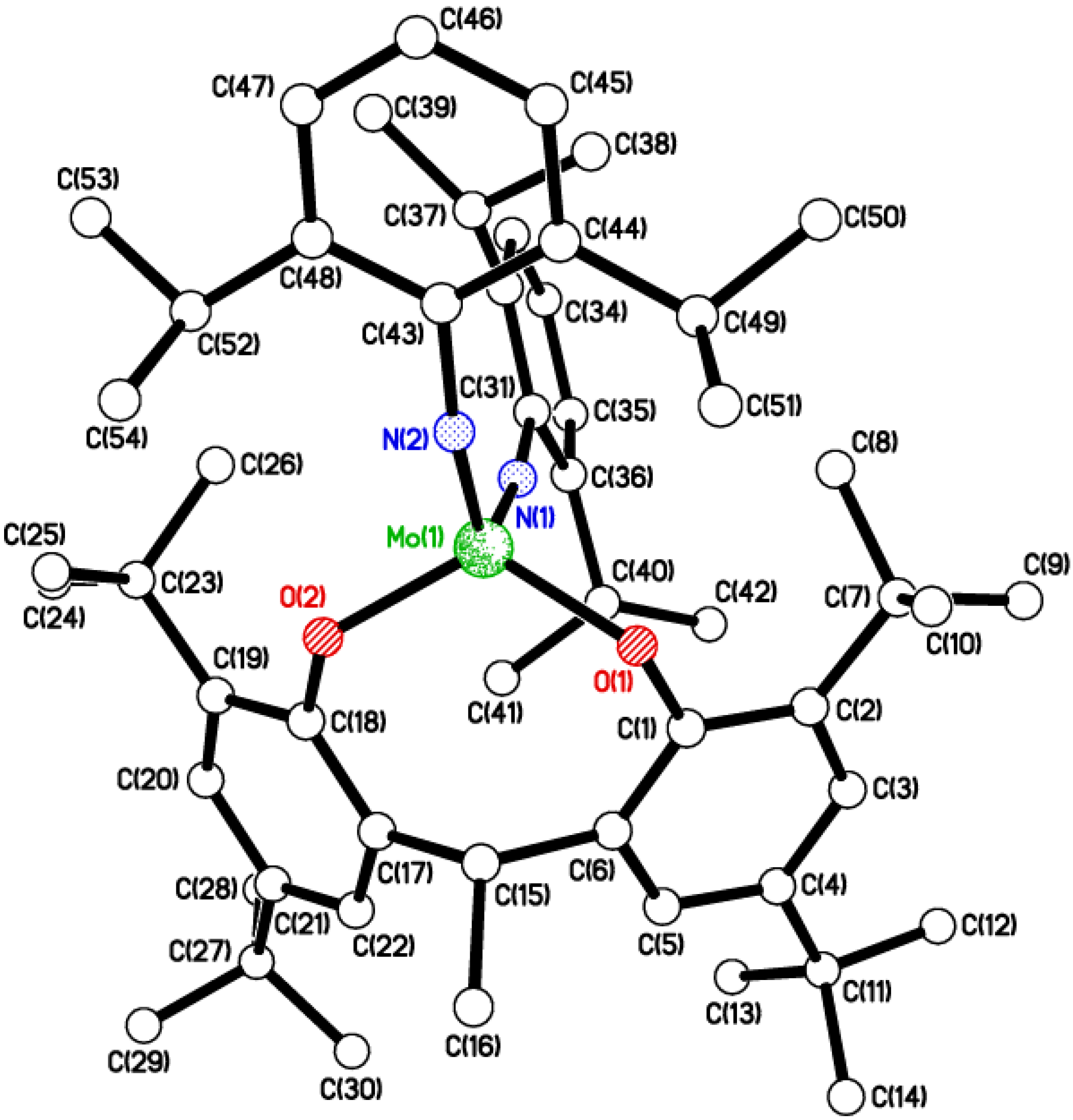
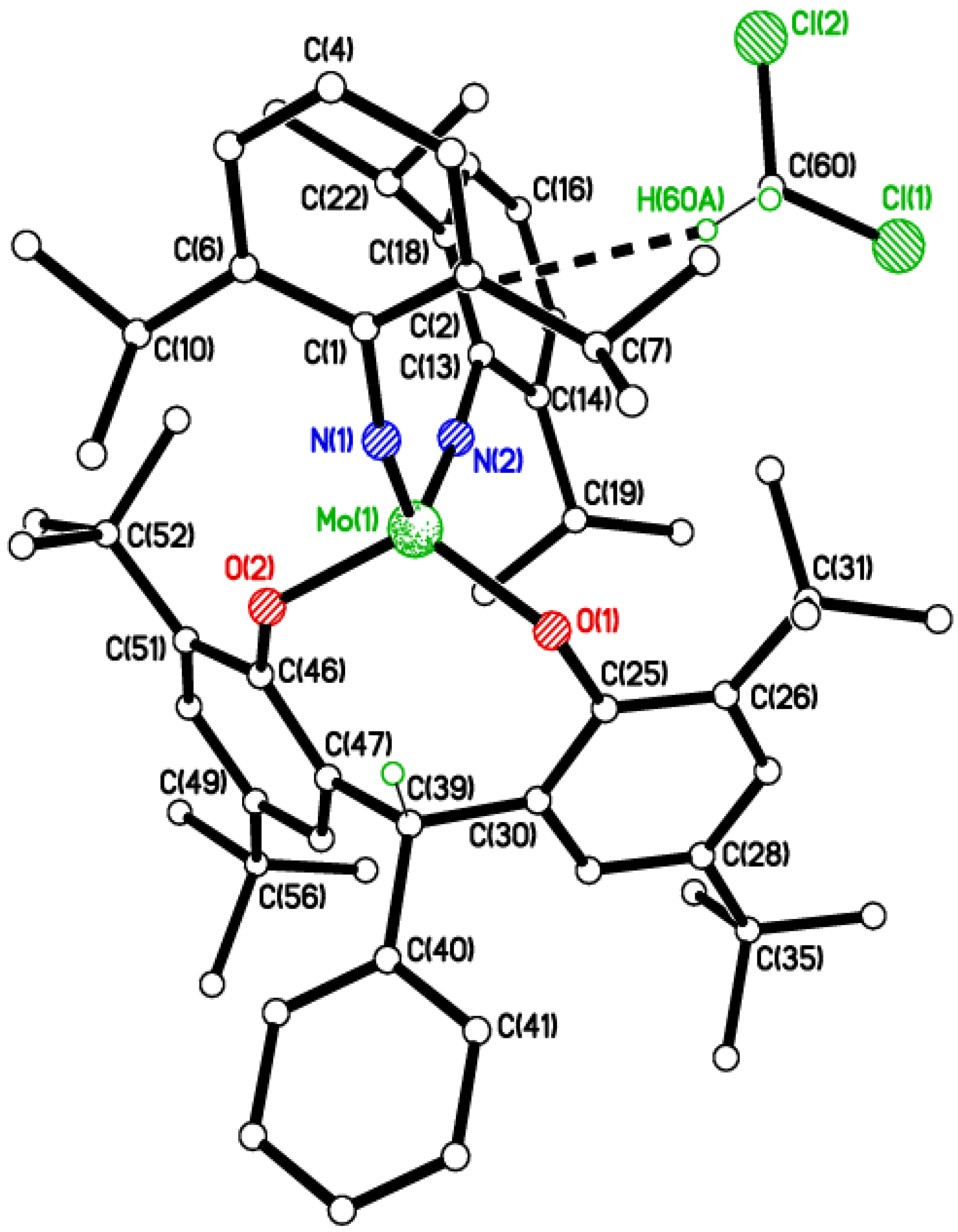
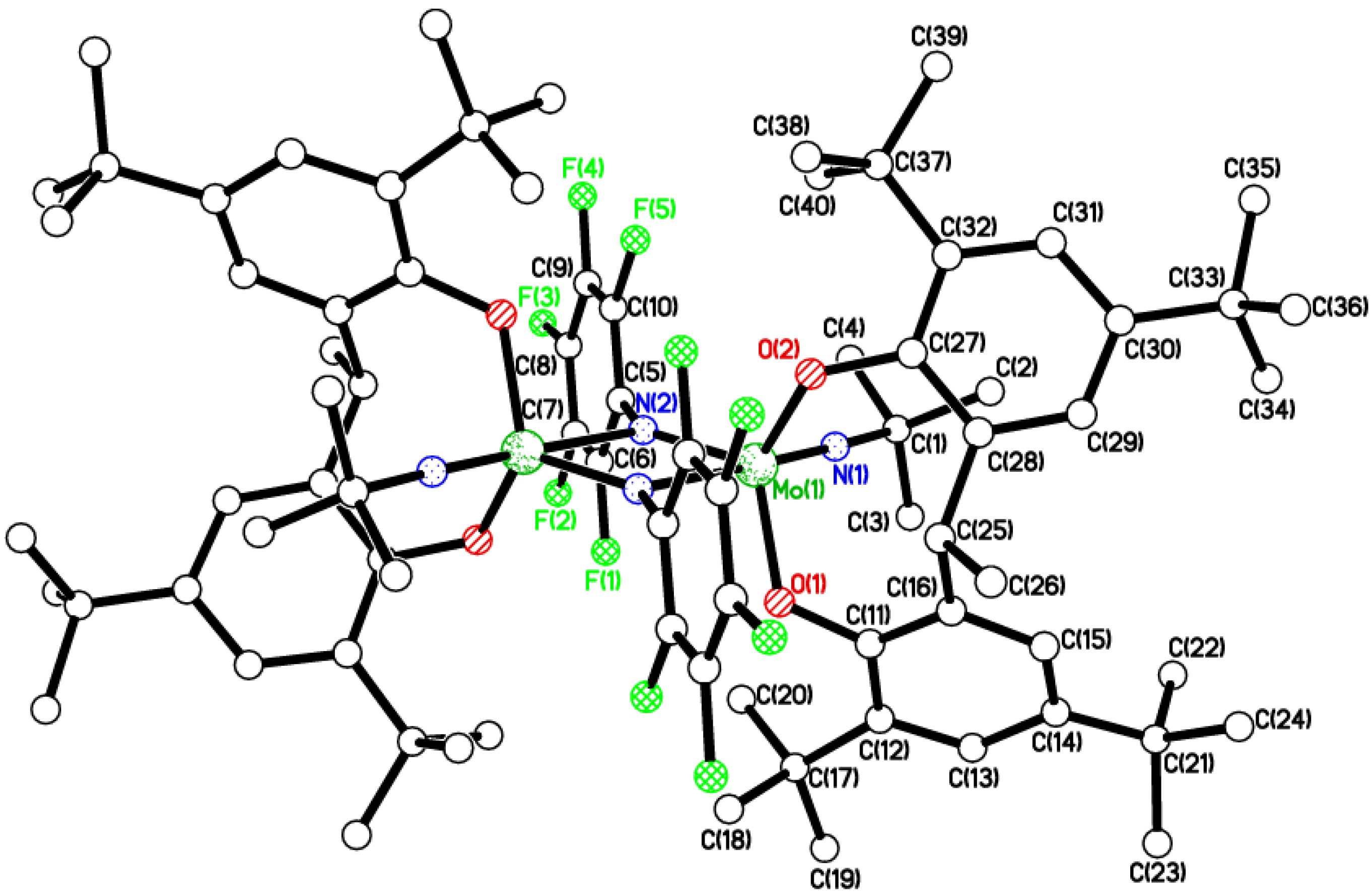
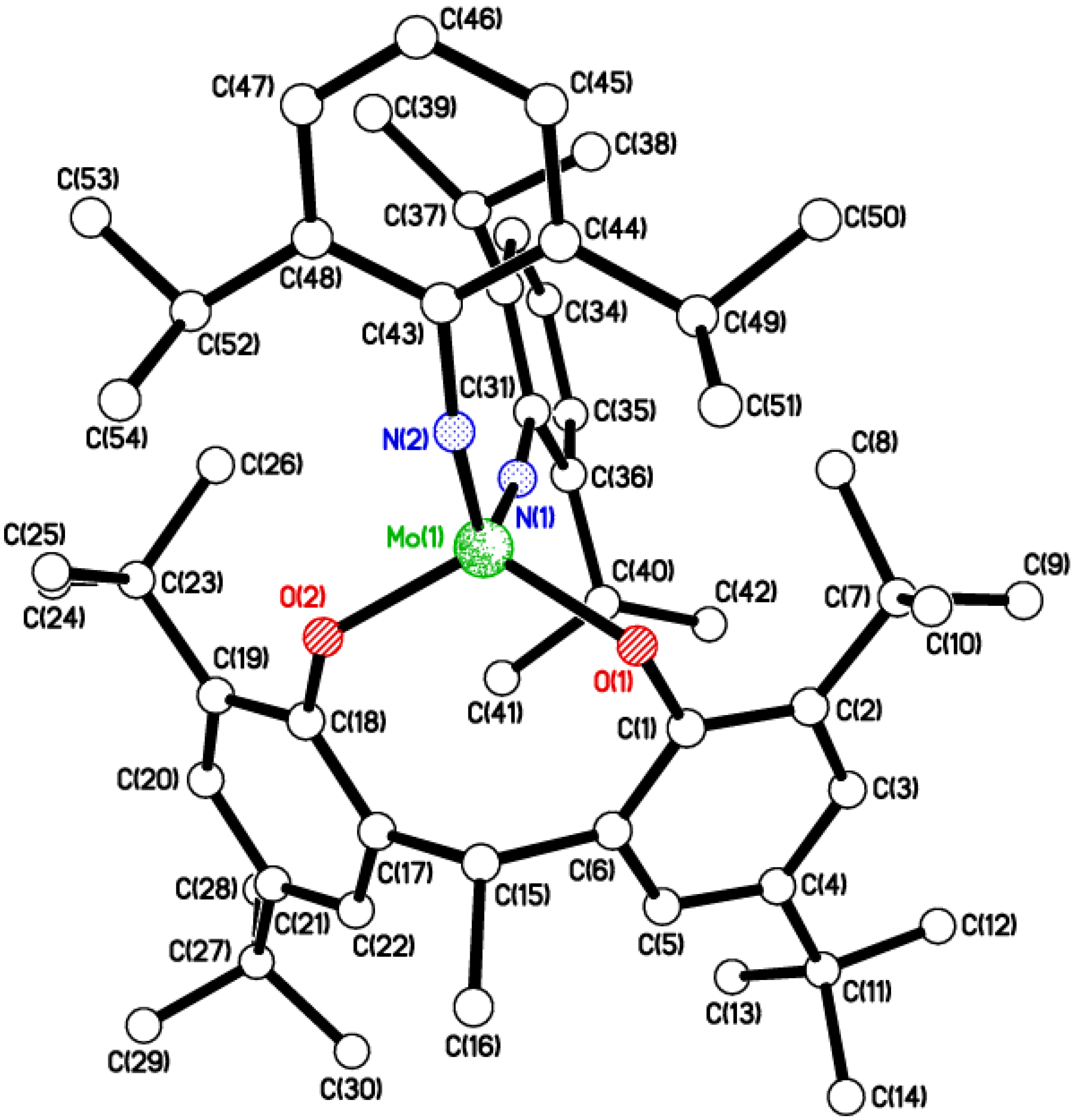
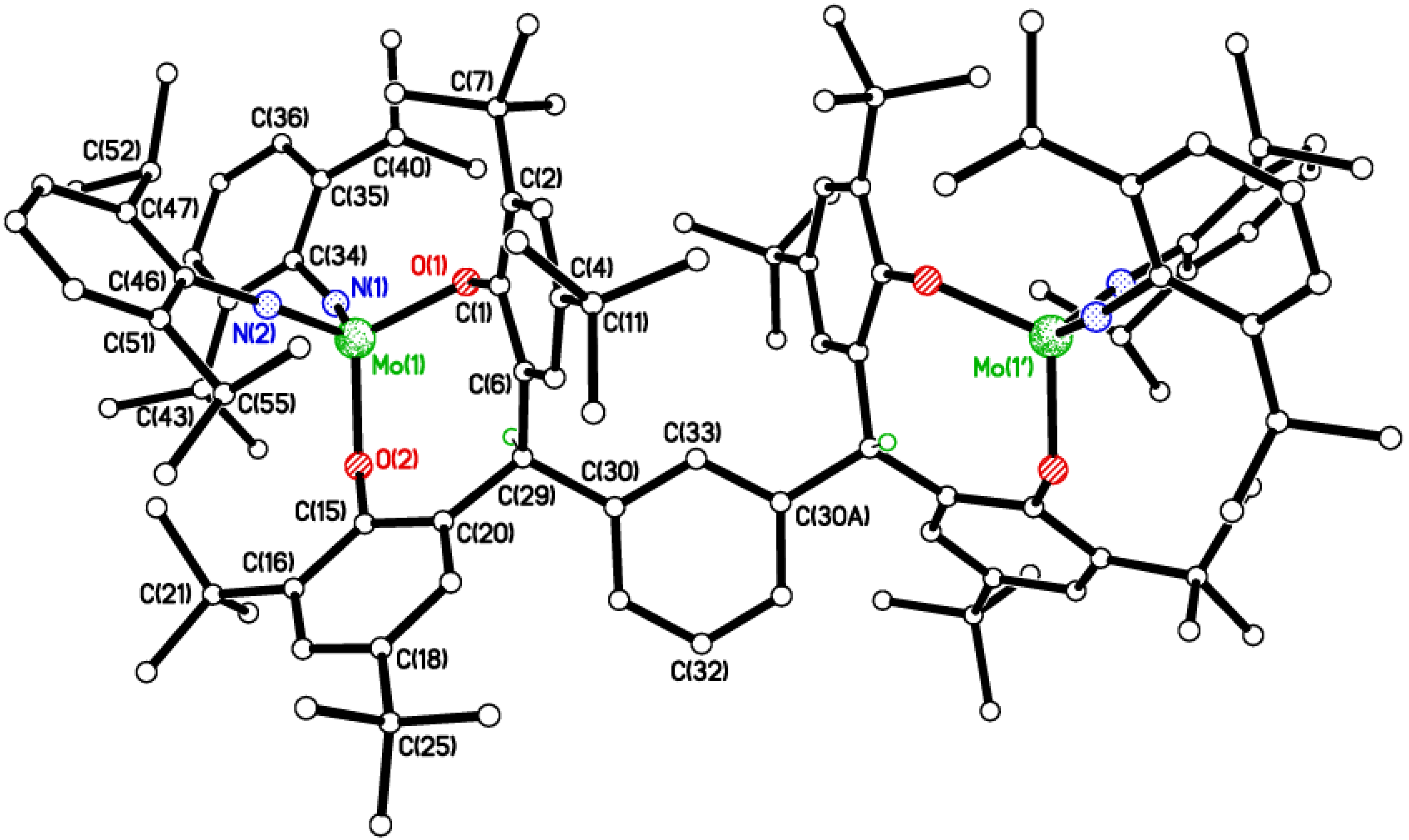
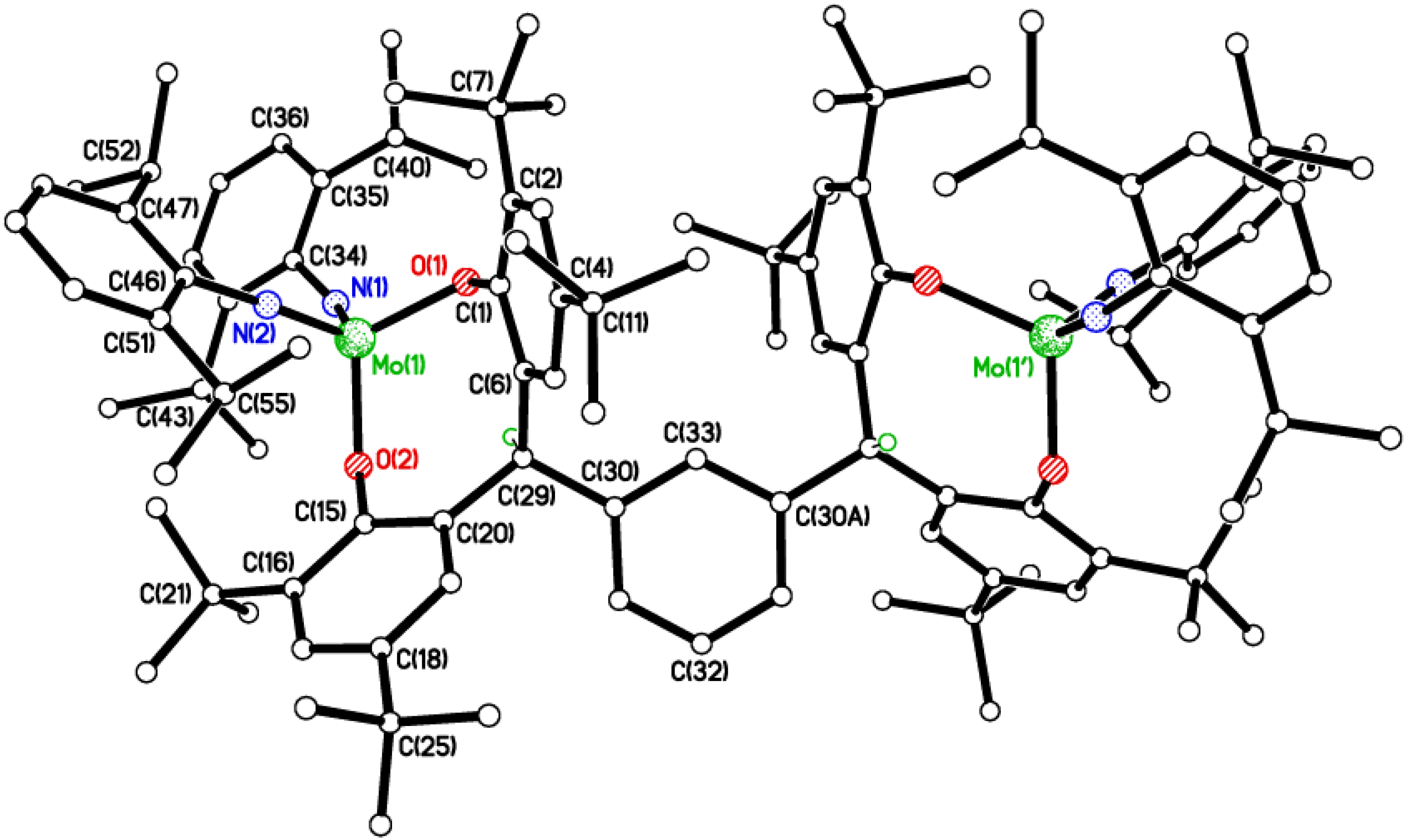
2.2. Tri-Phenolate Compound
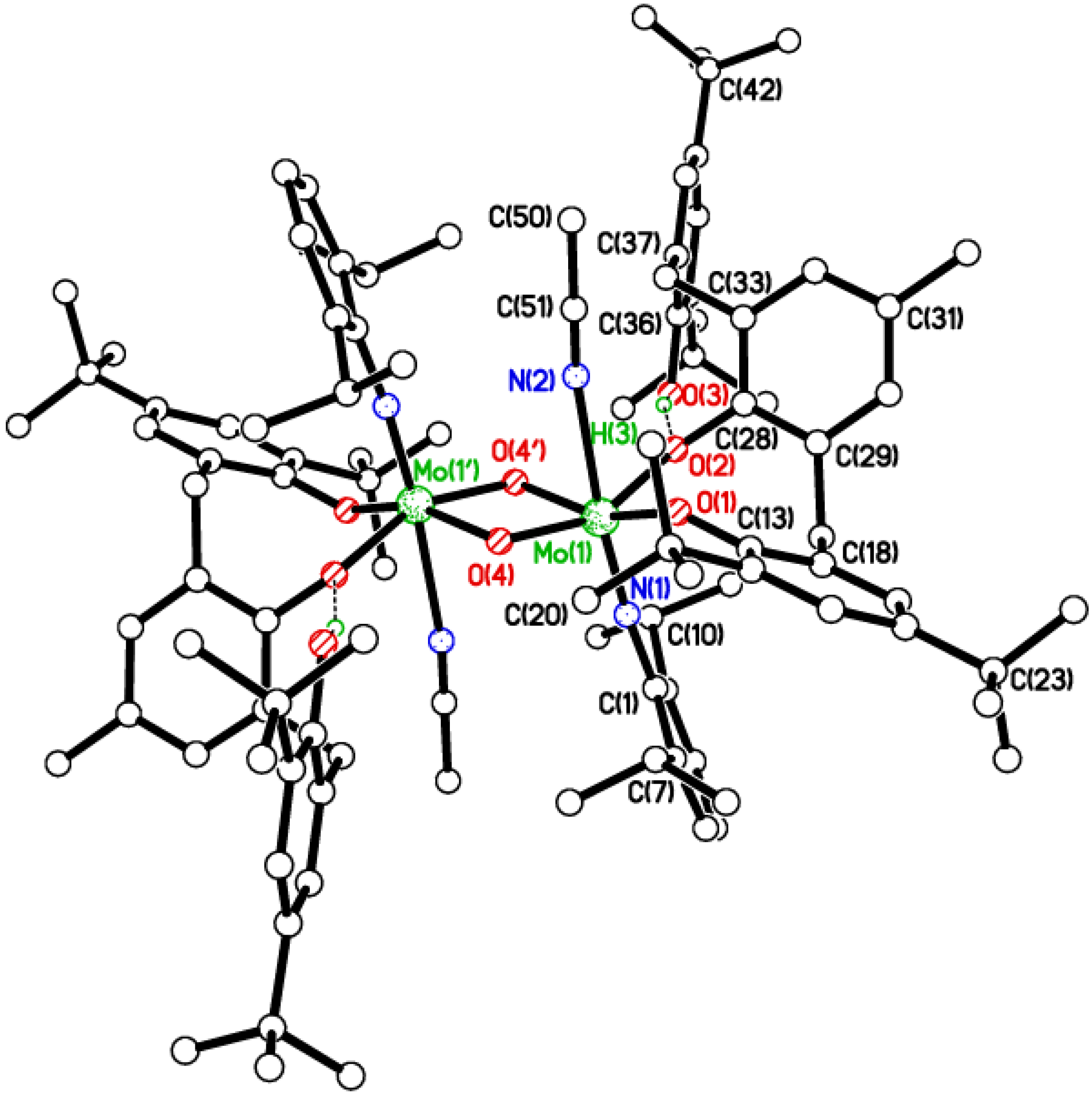
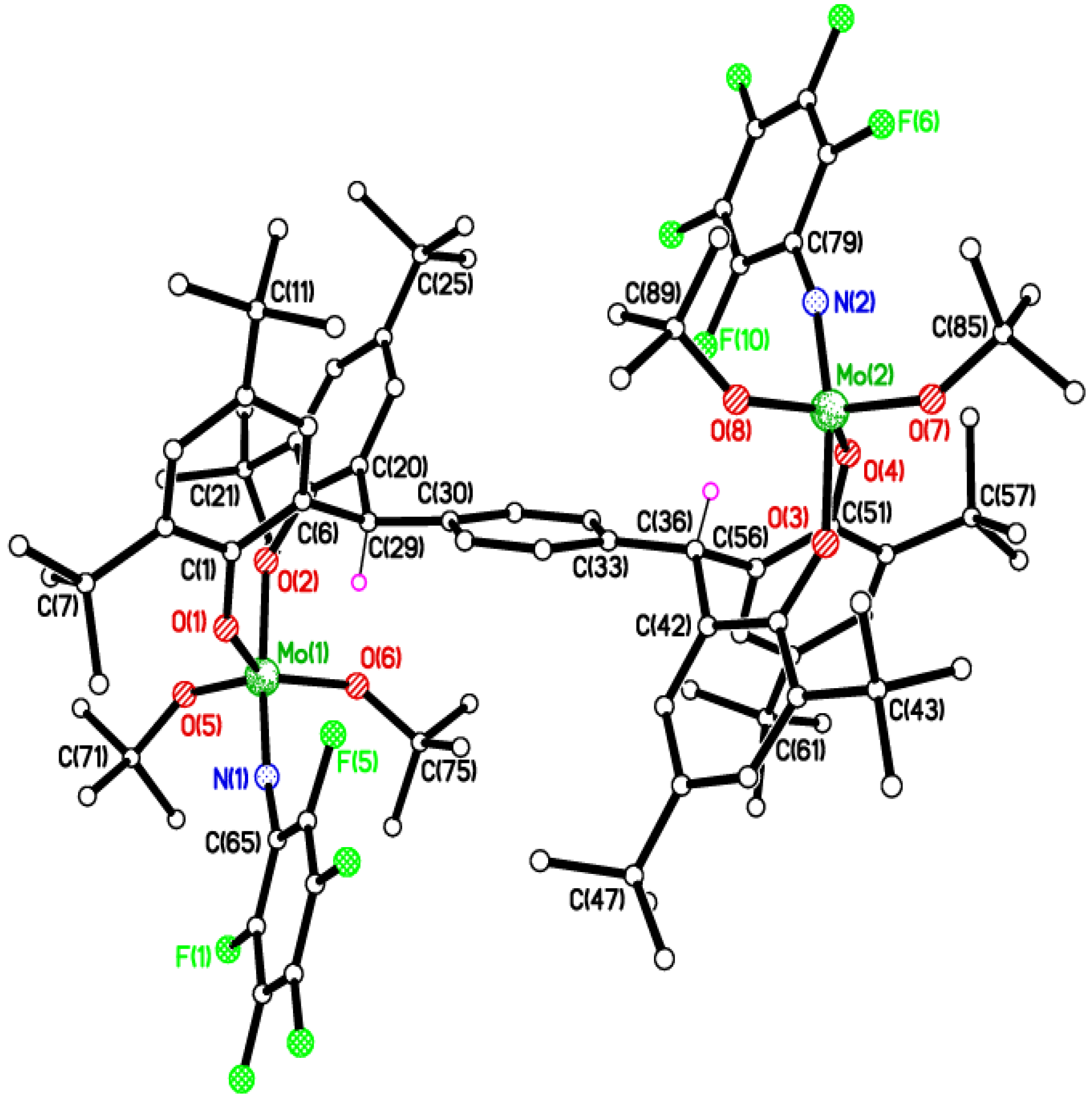
2.3. Tetra-Phenolate Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2·CH2Cl2 | 3 | 4·6MeCN | 6·2C6H14 | 7·6MeCN | 8 |
|---|---|---|---|---|---|---|---|
| Formula | C54H78MoN2O2 | C59H80MoN2O2·CH2Cl2 | C80H106F10Mo2N4O4 | C102H140Mo2N4O8·6(C2H3N) | C112H154Mo2N4O4·2(C6H14) | C92H122F10Mo2N2O8·6(C2H3N) | C28H44Cl2MoN2O2 |
| Formula weight | 883.12 | 1030.11 | 1569.56 | 1988.37 | 1984.61 | 2012.11 | 607.49 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P1 | P-1 | P21/n | P-1 | C2/c | Pī | Pī |
| Unit cell dimensions | |||||||
| a (Å) | 10.0435(8) | 11.7691(7) | 14.2727(8) | 13.0178(8) | 40.815(3) | 18.7522(10) | 10.0491(10) |
| b (Å) | 10.3143(8) | 14.1045(9) | 15.4944(8) | 13.4853(8) | 17.0938(11) | 22.6704(12) | 10.6022(11) |
| c (Å) | 12.8862(11) | 18.0657(12) | 18.0242(10) | 16.9177(10) | 16.3488(11) | 25.8469(18) | 15.6133(19) |
| α (º) | 74.5019(14) | 83.543(5) | - | 106.1133(10) | 90 | 79.933(6) | 92.902(9) |
| β (º) | 87.3130(14) | 81.066(5) | 98.4505(10) | 96.4972(10) | 94.8200(10) | 81.162(6) | 90.577(9) |
| γ (º) | 78.6415(14) | 74.829(5) | - | 92.7062(10) | 90 | 81.434(6) | 112.932(8) |
| V (Å3) | 1261.15(18) | 2851.1(3) | 3942.7(4) | 2825.3(3) | 11365.9(13) | 10605.9(11) | 1529.2(3) |
| Z | 1 | 2 | 2 | 1 | 4 | 4 | 2 |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) | 100(2) | 100(2) | 150(2) |
| Wavelength (Å) | 0.6861 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Calculated density (g·cm−3) | 1.163 | 1.200 | 1.322 | 1.169 | 1.160 | 1.260 | 1.319 |
| Absorption coefficient (mm−1) | 0.27 | 0.37 | 0.39 | 0.28 | 0.27 | 0.31 | 0.63 |
| Transmission factors (min./max.) | 0.960 and 0.995 | 0.912 and 0.857 | 0.958 and 0.985 | 0.936 and 0.970 | 1.000 and 0.634 | 0.976 and 0.994 | 0.981 and 0.855 |
| Crystal size (mm3) | 0.15 × 0.12 × 0.02 | 0.50 × 0.45 × 0.45 | 0.11 × 0.07 × 0.04 | 0.24 × 0.15 × 0.11 | 0.18 × 0.09 × 0.04 | 0.08 × 0.05 × 0.02 | 0.40 × 0.38 × 0.06 |
| θ (max) (°) | 29.2 | 29.3 | 28.9 | 27.5 | 25.0 | 26.4 | |
| Reflections measured | 13007 | 30297 | 24317 | 25167 | 67463 | 101749 | 11706 |
| Unique reflections | 11751 | 15207 | 9273 | 13076 | 12964 | 36898 | 6145 |
| Rint | 0.031 | 0.0572 | 0.048 | 0.027 | 0.0710 | 0.182 | 0.0965 |
| Reflections with F2 > 2σ(F2) | 11618 | 11245 | 6286 | 10128 | 9692 | 14715 | 3429 |
| Number of parameters | 554 | 615 | 498 | 659 | 566 | 2420 | 326 |
| R1 (F2 > 2σ(F2)) | 0.038 | 0.042 | 0.044 | 0.043 | 0.044 | 0.084 | 0.063 |
| wR2 (all data) | 0.094 | 0.102 | 0.110 | 0.107 | 0.126 | 0.212 | 0.156 |
| GOOF, S | 1.01 | 0.91 | 1.03 | 1.03 | 1.02 | 0.86 | 0.89 |
| Largest difference peak and hole (e Å−3) | 0.75 and −0.49 | 1.11 and −1.31 | 0.46 and −0.46 | 0.70 and −0.54 | 0.67 and −0.47 | 0.88 and −2.06 | 0.55 and −0.95 |

2.4. Polymerization Screening
| Entry | Cat. | CL:Mo:BnOH | Temp./°C | Time/h | Conversion (%) a | Mn,GPC b | Mn,Cal c | PDI d |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 250:1:1 | 80 | 0.5 | - | - | - | - |
| 2 | 1 | 250:1:1 | 100 | 0.5 | - | - | - | - |
| 3 | 1 | 250:1:1 | 100 | 1 | 70 | 4.62 | 20.08 | 1.41 |
| 4 | 1 | 250:1:0 | 80 | 3 | 45 | 3.77 | 12.84 | 1.17 |
| 5 | 1 | 250:1:1 | 100 | 3 | 86 | 5.53 | 24.64 | 1.22 |
| 6 | 1 | 250:1:1 | 100 | 6 | 99 | 6.40 | 28.35 | 1.40 |
| 7 | 1 | 250:1:1 | 100 | 12 | 99.5 | 10.67 | 28.50 | 1.40 |
| 8 | 2 | 250:1:1 | 100 | 0.5 | - | - | - | - |
| 9 | 2 | 250:1:1 | 100 | 1 | 96 | 5.23 | 27.50 | 1.29 |
| 10 | 2 | 250:1:0 | 80 | 3 | 51 | 2.19 | 14.55 | 1.19 |
| 11 | 2 | 250:1:1 | 100 | 3 | 89 | 7.14 | 25.50 | 1.25 |
| 12 | 2 | 250:1:1 | 100 | 6 | 95 | 7.24 | 27.21 | 1.22 |
| 13 | 2 | 250:1:1 | 100 | 12 | 99 | 10.33 | 28.35 | 1.57 |
| 14 | 3 | 250:1:0 | 80 | 3 | - | - | - | - |
| 15 | 3 | 250:1:1 | 100 | 3 | 55 | 2.14 | 15.80 | 1.11 |
| 16 | 3 | 250:1:1 | 100 | 6 | 99 | 6.74 | 28.35 | 1.17 |
| 17 | 3 | 125:1:1 | 100 | 12 | 99 | 8.51 | 14.23 | 1.60 |
| 18 | 3 | 250:1:1 | 100 | 12 | 99 | 9.73 | 28.35 | 1.46 |
| 19 | 3 | 500:1:1 | 100 | 12 | 99.5 | 13.00 | 56.89 | 1.72 |
| 20 | 3 | 1000:1:1 | 100 | 12 | 98 | 16.14 | 111.96 | 1.72 |
| 21 | 4 | 250:1:1 | 100 | 12 | 99 | 7.80 | 28.35 | 1.25 |
| 22 | 5 | 250:1:0 | 80 | 3 | - | 0.68 | - | 1.22 |
| 23 | 5 | 250:1:1 | 30 | 24 | - | - | - | - |
| 24 | 5 | 250:1:1 | 100 | 1 | 45 | 0.822 | 12.94 | 1.23 |
| 25 | 5 | 250:1:1 | 100 | 3 | 98 | 2.84 | 28.07 | 1.23 |
| 26 | 5 | 250:1:1 | 100 | 6 | 93 | 3.13 | 26.64 | 1.17 |
| 27 | 5 | 250:1:1 | 100 | 12 | 98 | 4.39 | 28.07 | 1.17 |
| 28 | 6 | 200:1:1 | 100 | 0.5 | - | - | - | - |
| 29 | 6 | 200:1:1 | 100 | 1 | 68 | 5.43 | 19.51 | 1.13 |
| 30 | 6 | 250:1:0 | 80 | 3 | 50 | 1.93 | 14.26 | 1.24 |
| 31 | 6 | 250:1:1 | 80 | 3 | 48 | 3.75 | 13.80 | 1.22 |
| 32 | 6 | 250:1:1 | 100 | 6 | 100 | 7.15 | 28.64 | 1.34 |
| 33 | 6 | 250:1:1 | 100 | 12 | 100 | 7.27 | 28.64 | 1.26 |
| 34 | 7 | 250:1:0 | 80 | 3 | - | - | - | - |
| 35 | 7 | 250:1:1 | 100 | 0.5 | - | - | - | - |
| 36 | 7 | 250:1:0 | 100 | 1 | 95 | 2.14 | 27.10 | 1.11 |
| 37 | 7 | 250:1:0 | 100 | 3 | 98 | 2.25 | 27.96 | 1.08 |
| 38 | 7 | 250:1:0 | 100 | 6 | 99.5 | 8.79 | 28.39 | 1.33 |
| 39 | 7 | 250:1:0 | 100 | 12 | 99 | 9.11 | 28.24 | 1.37 |
| 40 | 7 | 250:1:1 | 100 | 12 | 99 | 6.10 | 28.35 | 1.46 |
| 41 | 8 | 250:1:1 | 80 | 3 | 85 | 2.64 | 24.36 | 1.21 |
| 42 | 8 | 250:1:1 | 100 | 3 | 99 | 5.97 | 28.35 | 1.19 |
| 43 | 8 | 250:1:1 | 100 | 6 | 98 | 8.21 | 28.07 | 1.36 |
| 44 | 8 | 250:1:1 | 100 | 12 | 100 | 14.32 | 28.64 | 1.46 |
3. Experimental Section
3.1. General
3.2. Synthesis of [Mo(NC6H3i-Pr2-2,6)2L1Me] (1)
3.3. Synthesis of [Mo(NC6H3i-Pr2-2,6)2L1Ph] (2)
3.4. Synthesis of [Mo(Nt-Bu)(μ-NC6F5)(L1Me)]2 (3)
3.5. Synthesis of [Mo(NC6H3i-Pr2-2,6)(NCMe)(μ-O)L2H]2 (4)
3.6. Synthesis of {[Mo(NC6H3i-Pr2-2,6)]2(μ-L3p)} (5)
3.7. Synthesis of {[Mo(NC6H3i-Pr2-2,6))]2(μ-L3m)} (6)
3.8. Synthesis of {[Mo(NC6F5)(Ot-Bu)2]2(μ-L3p)} (7)
3.9. Procedure for ROP
3.10. Crystallography
4. Conclusions
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484–3504. [Google Scholar] [CrossRef] [PubMed]
- Arbaoui, A.; Redshaw, C. Metal catalysts for ε-caprolactone polymerization. Polym. Chem. 2010, 1, 801–826. [Google Scholar] [CrossRef]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Schrock, R.R. Recent advances in olefin metathesis by molybdenum and tungsten imido alkylidene complexes. J. Mol. Catal. A 2004, 213, 21–30. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, K.-Q.; Redshaw, C.; Elsegood, M.R.J. Molybdenum complexes derived from the oxydianiline [(2-NH2C6H4)2O]: Synthesis, characterization and ε-caprolactone ROP capability. Dalton Trans. 2015, 44, 13133–13140. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.M.; Nakanishi, M.; Kimura, M. Process for the Preparation of Lactone Polyesters. US Patent DE2947978, 15 September 1981. [Google Scholar]
- Báez, J.E.; Martῐnez-Rosales, M.; Martῐnez-Richa, A. Ring-opening polymerization of lactones catalyzed by decamolybdate anion. Polymer 2003, 44, 6767–6772. [Google Scholar] [CrossRef]
- Báez, J.E.; Martῐnez-Richa, A. Synthesis and characterization of poly(ε-caprolactone) and copolyesters by catalysis with molybdenum compounds: Polymers with acid-functional asymmetric telechelic architecture. Polymer 2005, 46, 12118–12129. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, A.; Martῐnez-Richa, A. One-step route to α-hydroxyl-ω-(carboxylic acid) polylactones using catalysis by decamolybdate anion. Macromolecules 2005, 38, 1599–1608. [Google Scholar] [CrossRef]
- Mahha, Y.; Atlamsani, A.; Blais, J.-C.; Tessier, M.; Brégeault, J.M.; Salles, L. Oligomerization of ε-caprolactone and δ-valerolactone using heteropolyacid initiators and vanadium or molybdenum complexes. J. Mol. Catal. A 2005, 234, 63–73. [Google Scholar] [CrossRef]
- Maruta, Y.; Abiko, A. Bis(salicyldehydato)dioxomolybdenum complexes: Catalysis for ring opening polymerization. Polym. Bull. 2014, 71, 1413–1440. [Google Scholar] [CrossRef]
- Haymore, B.L.; Maatta, E.A.; Wentworth, R.A.D. A Bisphenylnitrene Complex of Molybdenum with a Bent Nitrene Ligand. Preparation and Structure of cis-Mo(NC6F5)2(S2CN(C2H5)2)2. J. Am. Chem. Soc. 1979, 101, 2063–2068. [Google Scholar] [CrossRef]
- Gibson, V.C.; Redshaw, C.; Clegg, W.; Elsegood, M.R.J. “Electronic bending” of imido ligands and the effect on the coordination mode of a tridentate ancillary ligand. Polyhedron 2007, 26, 3161–3167. [Google Scholar] [CrossRef]
- Floriani, C.; Corazza, F.; Lesueur, W.; Chiesi-Villa, A.; Guastini, C. A sensitive spectroscopic probe for monitoring changes in the coordination sphere of titanium: Eight-membered dioxatitanacycles and their organometallic derivatives. Angew. Chem. Int. 1989, 28, 66–67. [Google Scholar] [CrossRef]
- Toscano, P.J.; Schermerhorn, E.J.; Dettelbacher, C.; Macherone, D. Monomeric four-coordinate vanadium(V) oxo complexes containing a labile ligand: Synthesis and X-ray structural characterization of [(C23H30O2)V(O)Cl]. Chem. Commun. 1991, 933–934. [Google Scholar] [CrossRef]
- Corazza, F.; Floriani, C.; Chiesi-Villa, A.; Guastini, C. Titanium(IV) and zirconium(IV) tetrahydroborate moieties supported by a phenoxo ligand. Inorg. Chem. 1991, 30, 145–148. [Google Scholar] [CrossRef]
- Okuda, J.; Fokken, S.; Kaug, H.-C.; Massa, W. Synthesis and characterization of mononuclear titanium complexes containing a bis(phenoxyl) ligand derived from 2,2′-methylene-bis (6-tert-butyl-4-methylphenol). Chem. Ber. 1995, 128, 221–227. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Huang, J.-H.; Huffmann, J.C.; Streib, W.E.; Tiedtke, D. Synthesis and structural characterization of 2,2′-methylene-bis(6-t-butyl-4-methyl-phenoxide) complexes of titanium, zirconium and tantalum. Polyhedron 1997, 16, 2941–2949. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Huang, J.-H.; Huffmann, J.C.; Parkin, I.P. Dinuclear (d3-d3) diolate complexes of molybdenum and tungsten. 2. Derivatives of 2,2′-methylenebis(6-tert-butyl-4-methylphenoxide). Direct observation of the conversion of bridged to chelate isomers (M = Mo) and reversible carbon-hydrogen bond oxidative addition (M = W). Inorg. Chem. 1997, 36, 1642–1651. [Google Scholar] [PubMed]
- Mulford, D.R.; Fanwick, P.E.; Rothwell, I.P. Group 4 and 5 metal derivatives of 2,2′-methylene -bis(6-phenylphenoxide). Polyhedron 2000, 19, 35–42. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta. Crystallogr. Sect. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Maestri, A.G.; Brown, S.N. Titanatranes derailed: Static and dynamic triethanolamine slippage induced by polyphenoxide chelation. Inorg. Chem. 2004, 43, 6995–7004. [Google Scholar] [CrossRef] [PubMed]
- Redshaw, C.; Gibson, V.C.; Elsegood, M.R.J.; Clegg, W. New coordination modes at molybdenum for 2-diphenylphosphinoaniline derived ligands. Chem. Commun. 2007, 1951–1953. [Google Scholar] [CrossRef]
- Gordon, B.W.F.; Scott, M.J. Linked aryloxides ligands: Synthesis and characterization of alkali metal clusters of a partial calix[n]arene. Inorg. Chim. Acta 2000, 297, 206–216. [Google Scholar] [CrossRef]
- Matsuo, T.; Kawaguchi, H. Synthesis and structures of Niobium(V) complexes stabilized by linear-linked aryloxide trimers. Inorg. Chem. 2002, 41, 6090–6098. [Google Scholar] [CrossRef] [PubMed]
- Redshaw, C.; Humphrey, S.M. Complexes of tungsten (IV, VI) derived from linked aryloxide ligands. Polyhedron 2006, 25, 1946–1954. [Google Scholar] [CrossRef]
- Tang, L.; Wasserman, E.P.; Neithamer, D.R.; Krystosek, R.D.; Cheng, Y.; Price, P.C.; He, Y.; Emge, T.J. Highly active catalysts for the ring opening polymerization of ethylene oxide and propylene oxide based on products of alkylaluminum compounds with bulky tetraphenol ligands. Macromolecules 2008, 41, 7306–7315. [Google Scholar] [CrossRef]
- Cundari, T.R.; Gordon, M.S. Principal Resonance Contributors to High-Valent Transition-Metal Alkylidene Complexes. J. Am. Chem. Soc. 1991, 113, 5231–5243. [Google Scholar] [CrossRef]
- Fox, H.H.; Schofield, M.H.; Schrock, R.R. Electronic Structure of Mo(VI) Alkylidene Complexes and an Examination of Reactive Intermediates Using the SCF-Xα-SW Method. Organometallics 1994, 13, 2804–2815. [Google Scholar] [CrossRef]
- Poater, A.; Solans-Monfort, X.; Clot, E.; Copéret, C.; Eisenstein, O. DFT calculations of d° M(NR)(CHtBu)(X)(Y) (M = Mo, W; R = CPh3, 2,6-iPr2-C6H3; X and Y = CH2tBu, OtBu, OSi(OtBu)3) olefin metathesis catalysts: Structural, spectroscopic and electronic properties. Dalton Trans. 2006, 25, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Oskam, J.H.; Fox, H.H.; Yap, K.B.; McConville, D.H.; O’Dell, R.; Lichtenstein, B.J.; Schrock, R.R. Ligand variation in alkylidene complexes of the type Mo(CHR)(NR′)(OR′′)2. J. Organomet. Chem. 1993, 459, 185–198. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Cotton, F.A.; Folting, K.; Huffman, J.C.; Raterman, A.L.; Shamshoum, E.S. Insertion reactions of hexaalkoxydimolybdenum and –ditungsten compounds with organic isocyanates. Syntheses and structures of W2(OCMe3)4[N(C6H5)C(O)OCMe3]2 and Mo2(O-i-Pr)4 [N(C6H5)C(O)O-i-Pr]. Inorg. Chem. 1984, 23, 4423–4427. [Google Scholar] [CrossRef]
- Dyer, P.W.; Gibson, V.C.; Howard, J.A.K.; Whittle, B.; Wilson, C. Four coordinate bis(imido) alkene complexes of molybdenum(IV): Relatives of the zirconocene family. J. Chem. Soc. Chem. Commun. 1992, 1666–1668. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. A 1990, A46, 194–201. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Khafaji, Y.; Prior, T.J.; Elsegood, M.R.J.; Redshaw, C. Molybdenum (VI) Imido Complexes Derived from Chelating Phenols: Synthesis, Characterization and ɛ-Caprolactone ROP Capability. Catalysts 2015, 5, 1928-1947. https://doi.org/10.3390/catal5041928
Al-Khafaji Y, Prior TJ, Elsegood MRJ, Redshaw C. Molybdenum (VI) Imido Complexes Derived from Chelating Phenols: Synthesis, Characterization and ɛ-Caprolactone ROP Capability. Catalysts. 2015; 5(4):1928-1947. https://doi.org/10.3390/catal5041928
Chicago/Turabian StyleAl-Khafaji, Yahya, Timothy J. Prior, Mark R. J. Elsegood, and Carl Redshaw. 2015. "Molybdenum (VI) Imido Complexes Derived from Chelating Phenols: Synthesis, Characterization and ɛ-Caprolactone ROP Capability" Catalysts 5, no. 4: 1928-1947. https://doi.org/10.3390/catal5041928
APA StyleAl-Khafaji, Y., Prior, T. J., Elsegood, M. R. J., & Redshaw, C. (2015). Molybdenum (VI) Imido Complexes Derived from Chelating Phenols: Synthesis, Characterization and ɛ-Caprolactone ROP Capability. Catalysts, 5(4), 1928-1947. https://doi.org/10.3390/catal5041928