
Preparation for Pt-Loaded Zeolite Catalysts Using w/o Microemulsion and Their Hydrocracking Behaviors on Fischer-Tropsch Product

Abstract
:1. Introduction
2. Results and Discussion
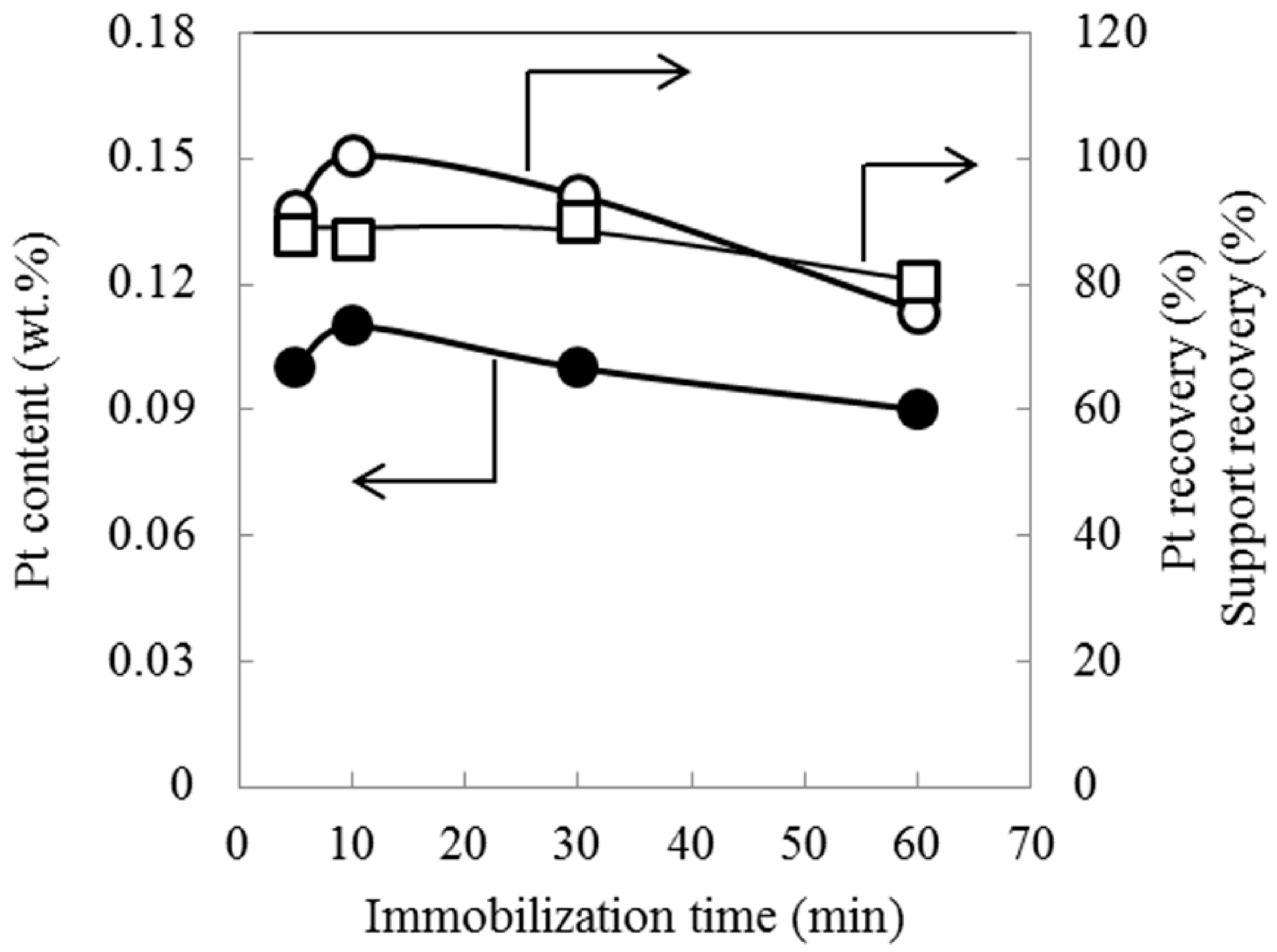
2.1. Effect of Immobilization Time

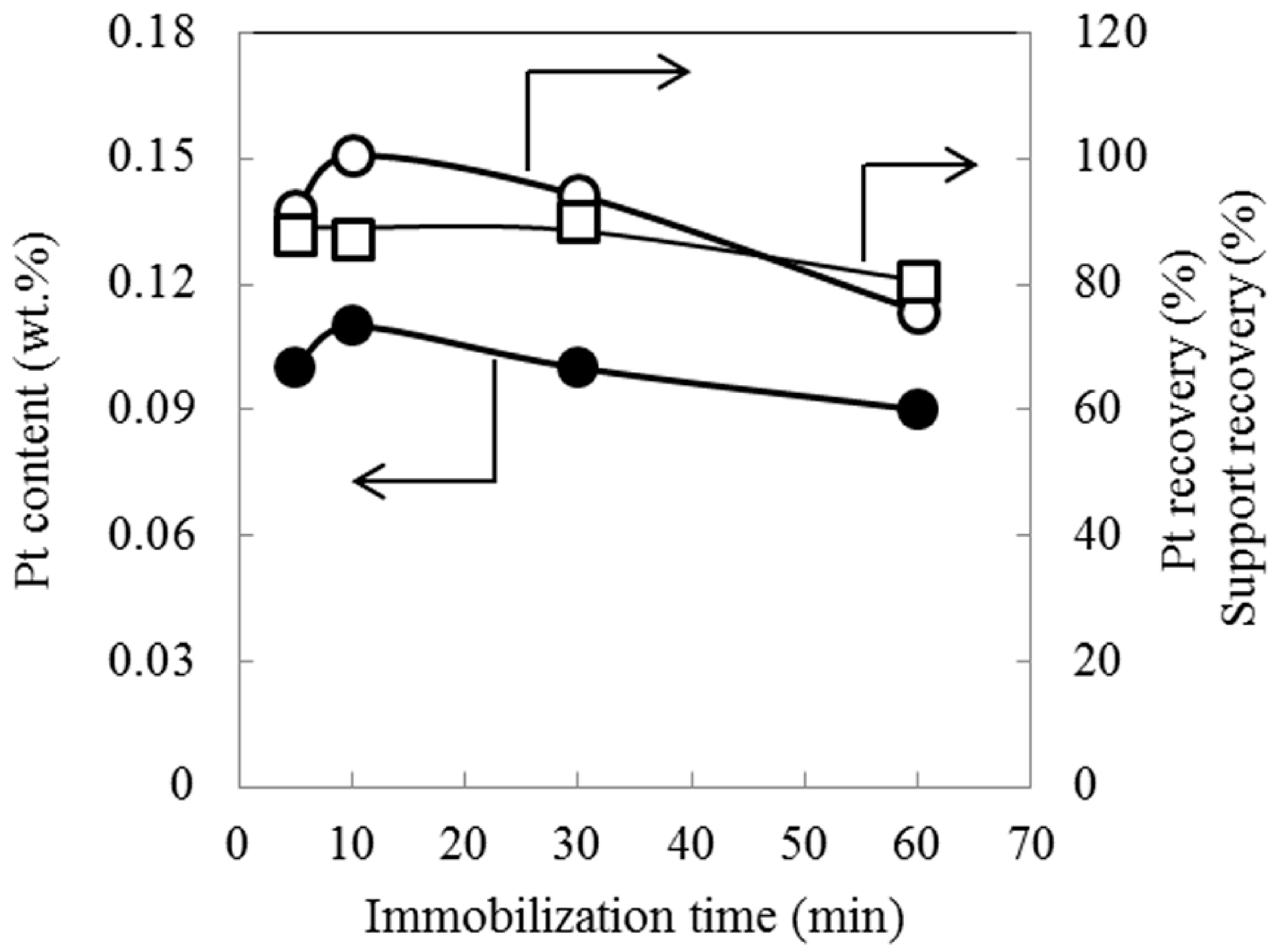{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle forming agent/reducing agent | Pt particle type | ||
|---|---|---|---|
| Name | Rational formula | Name | Rational formula |
| Hydrazine | N2H4 | Pt metal | - |
| TEAC | (C2H5)4N+Cl− | (TEA)2PtCl6 | [(C2H5)4N]2PtCl6 |
| TPAB | (C3H7)4N+Br− | (TPA)2PtCl6 | [(C3H7)4N]2PtCl6 |
| HTAB | C6H13(CH3)3N+Br− | (HTA)2PtCl6 | [C6H13(CH3)3N]2PtCl6 |
| CTAC | C16H33(CH3)3N+Cl− | (CTA)2PtCl6 | [C16H33(CH3)3N]2PtCl6 |
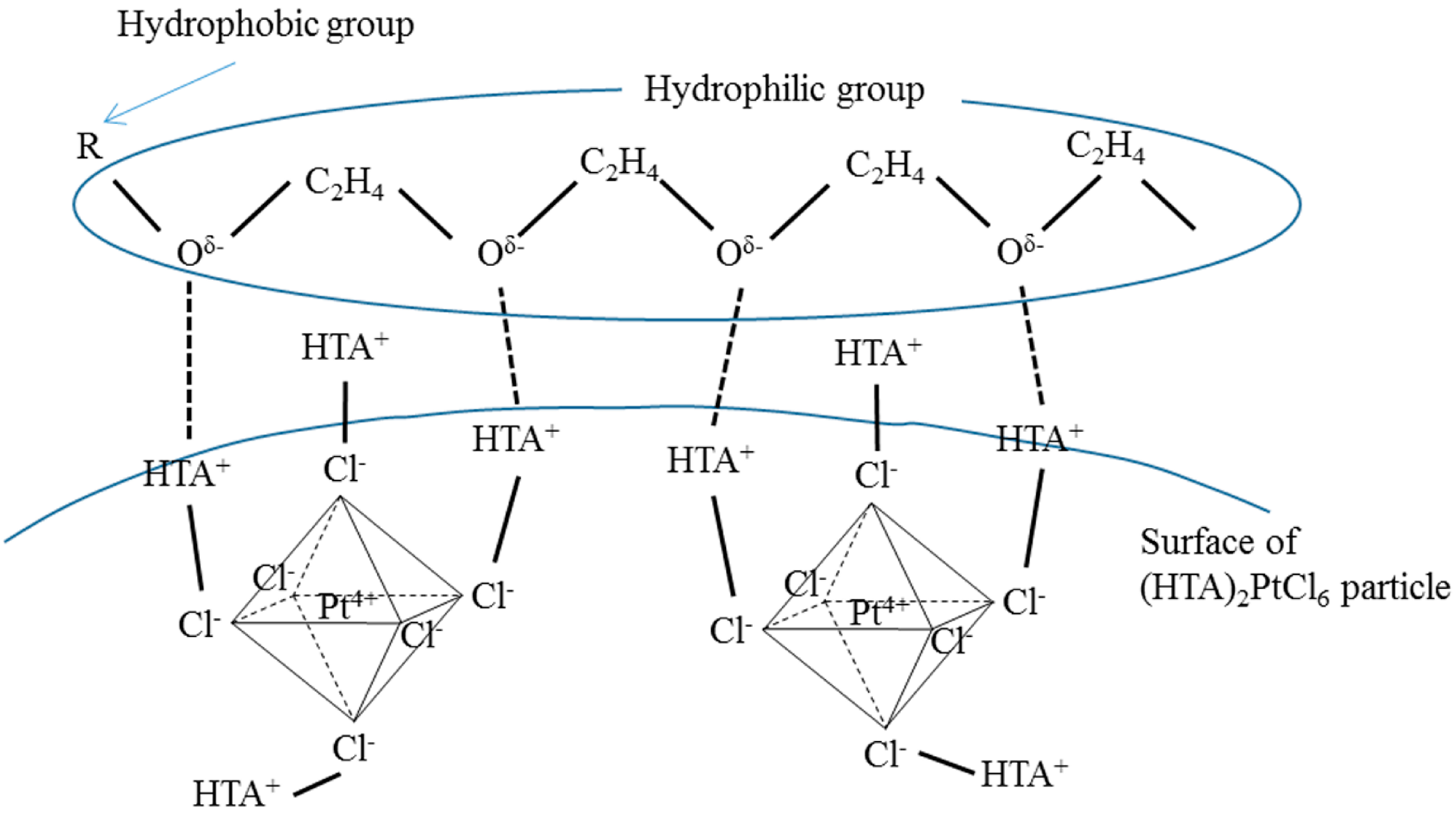
2.2. Pore Structure and Acid Amount of Pt-Loaded Zeolite Catalysts
| # | Catalyst | Remarks | BET surface area | Pore volume | Average pore diameter | Micropore area | Mesopore area |
|---|---|---|---|---|---|---|---|
| m2·g−1 | cm3·g−1 | nm | m2·g−1 | m2·g−1 | |||
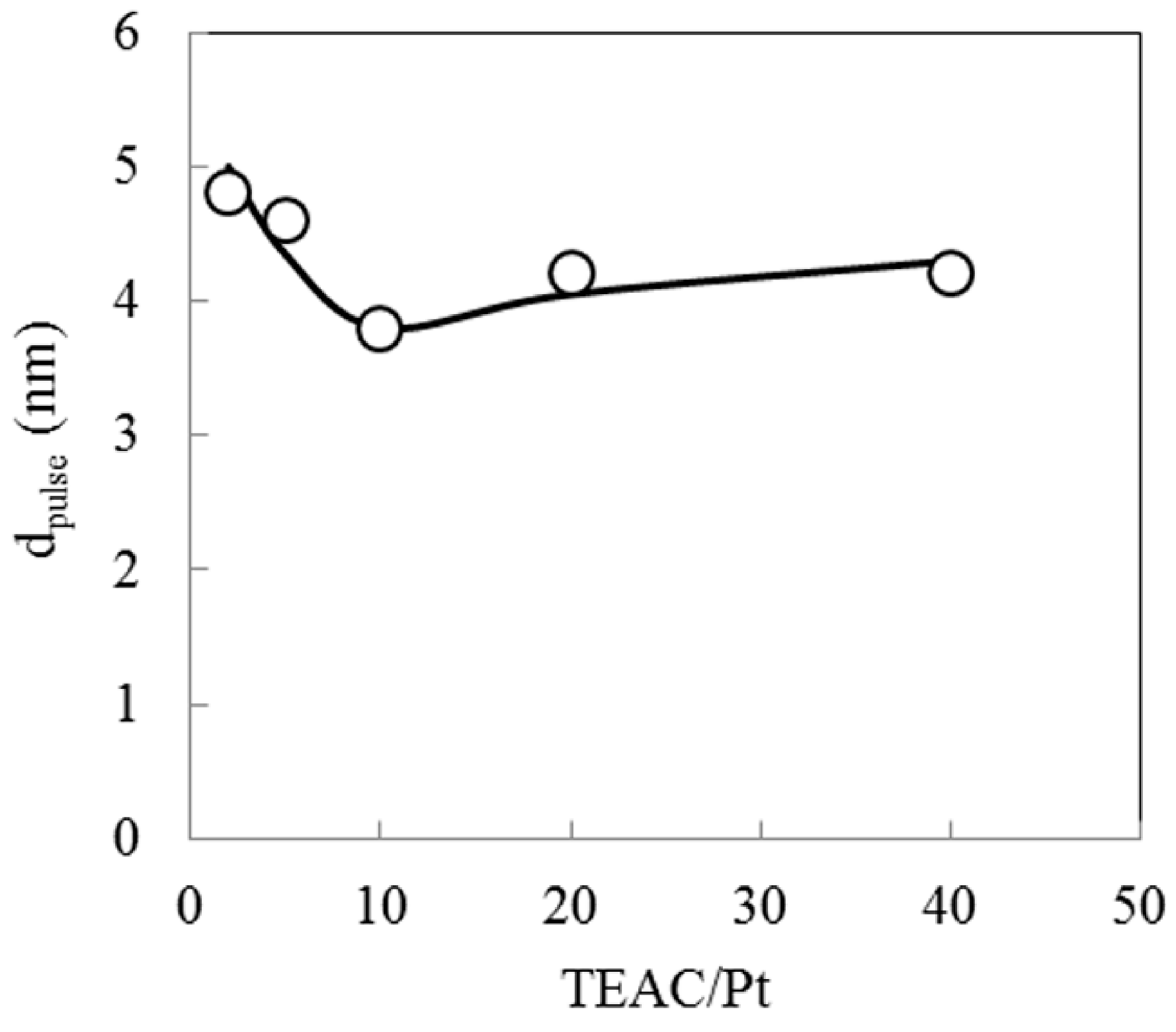
| A | M-Pt(TEAC)/940HOA | TEAC/Pt = 10 | 598 | 137 | 2.1 | 792 | 68 |
| B | M-Pt(CTAC)/940HOA | CTAC/Pt = 10 | 590 | 135 | 2.1 | 772 | 68 |
| C | M-Pt(TPAB)/940HOA | TPAB/Pt = 10 | 598 | 138 | 2.1 | 782 | 68 |
| D | M-Pt(HTAB)/940HOA | HTAB/Pt = 10 | 608 | 140 | 2.2 | 795 | 68 |
| E | M-Pt(N2H4)/940HOA | N2H4/Pt = 80 | 605 | 139 | 2.1 | 791 | 66 |
| F | M-Pt(TEAC)/940HOA | TEAC/Pt = 2 | 610 | 140 | 2.2 | 796 | 67 |
| G | M-Pt(TEAC)/940HOA | TEAC/Pt = 5 | 644 | 148 | 2.1 | 840 | 72 |
| H | M-Pt(TEAC)/940HOA | TEAC/Pt = 20 | 617 | 142 | 2.2 | 804 | 69 |
| I | M-Pt(TEAC)/940HOA | TEAC/Pt = 40 | 587 | 135 | 2.1 | 763 | 66 |
| J | I-Pt/940HOA | Impregnation | 622 | 143 | 2.2 | 807 | 67 |
| # | Catalyst | Remarks | Acid amount a | Weak b | Medium c | Strong d |
|---|---|---|---|---|---|---|
| mmol·g−1 | mmol·g−1 | |||||
| A | M-Pt(TEAC)/940HOA | TEAC/Pt = 10 | 0.74 | 0.32 | 0.34 | 0.08 |
| B | M-Pt(CTAC)/940HOA | CTAC/Pt = 10 | 0.77 | 0.36 | 0.35 | 0.05 |
| C | M-Pt(TPAB)/940HOA | TPAB/Pt = 10 | 0.75 | 0.37 | 0.35 | 0.03 |
| D | M-Pt(HTAB)/940HOA | HTAB/Pt = 10 | 0.68 | 0.33 | 0.31 | 0.05 |
| E | M-Pt(N2H4)/940HOA | N2H4/Pt = 80 | 0.74 | 0.37 | 0.34 | 0.03 |
| F | M-Pt(TEAC)/940HOA | TEAC/Pt = 2 | 0.74 | 0.33 | 0.33 | 0.08 |
| G | M-Pt(TEAC)/940HOA | TEAC/Pt = 5 | 0.70 | 0.34 | 0.31 | 0.04 |
| H | M-Pt(TEAC)/940HOA | TEAC/Pt = 20 | 0.72 | 0.34 | 0.32 | 0.06 |
| I | M-Pt(TEAC)/940HOA | TEAC/Pt = 40 | 0.77 | 0.35 | 0.34 | 0.08 |
| J | I-Pt/940HOA | Impregnation | 0.69 | 0.34 | 0.31 | 0.04 |
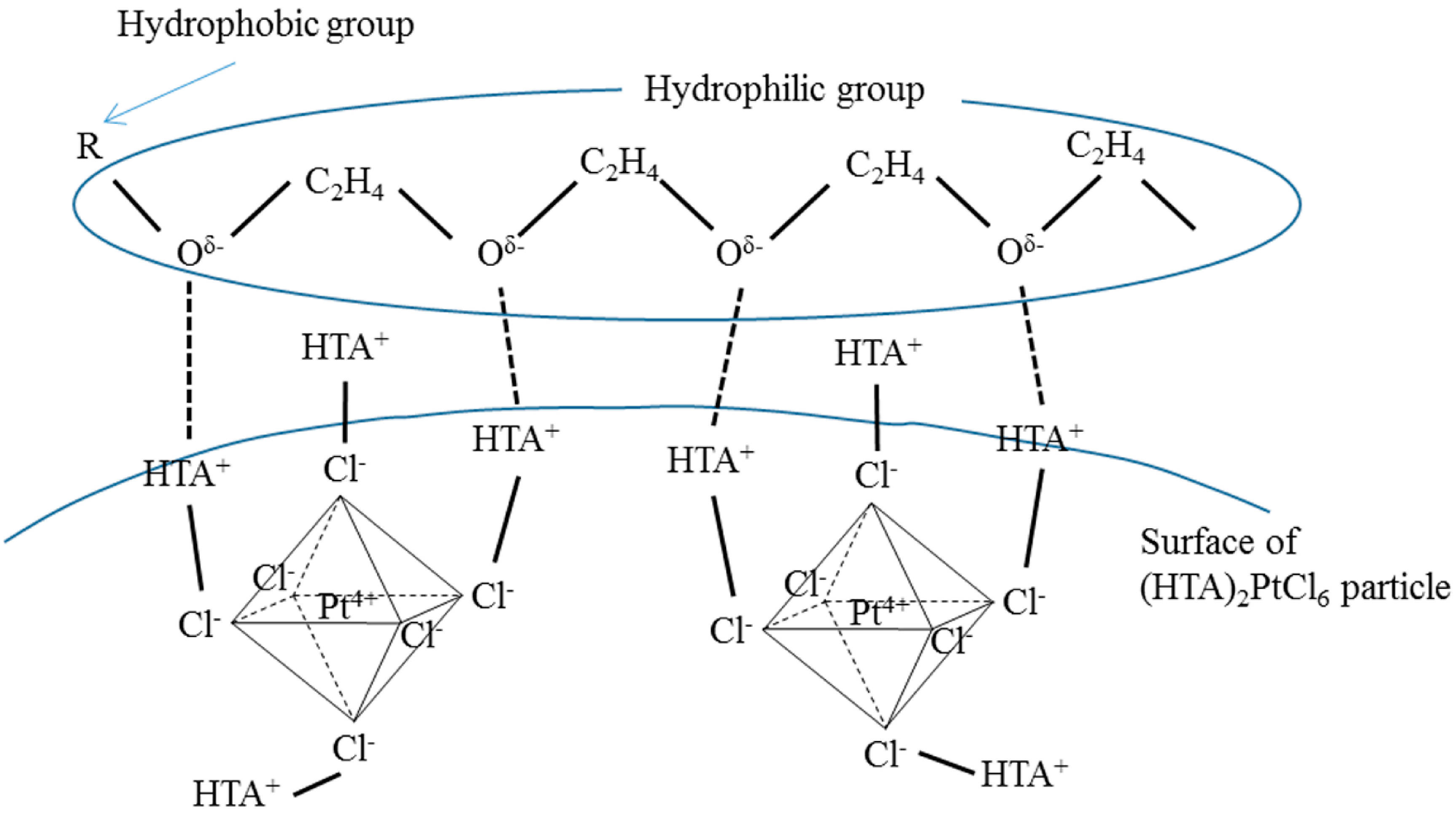
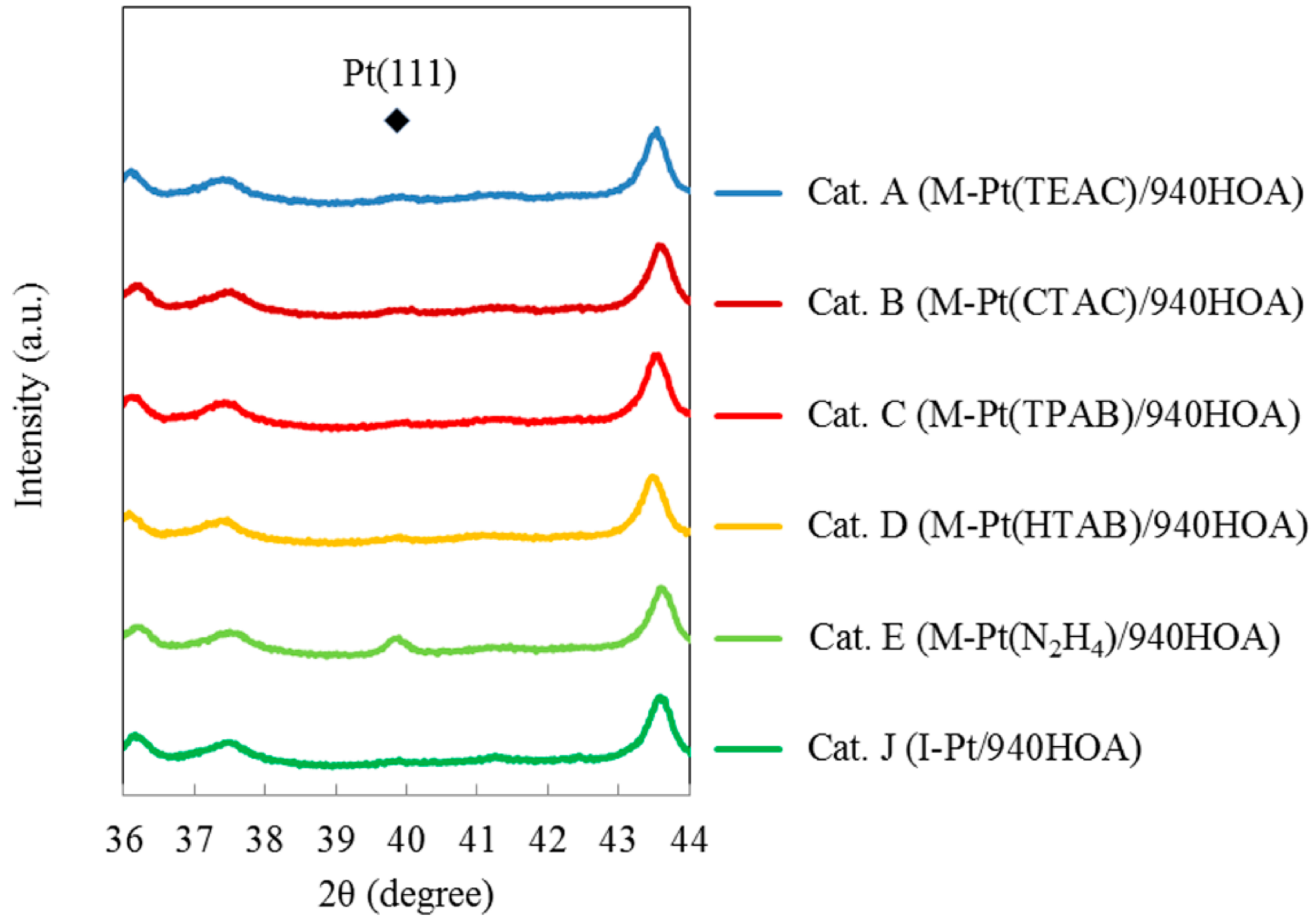
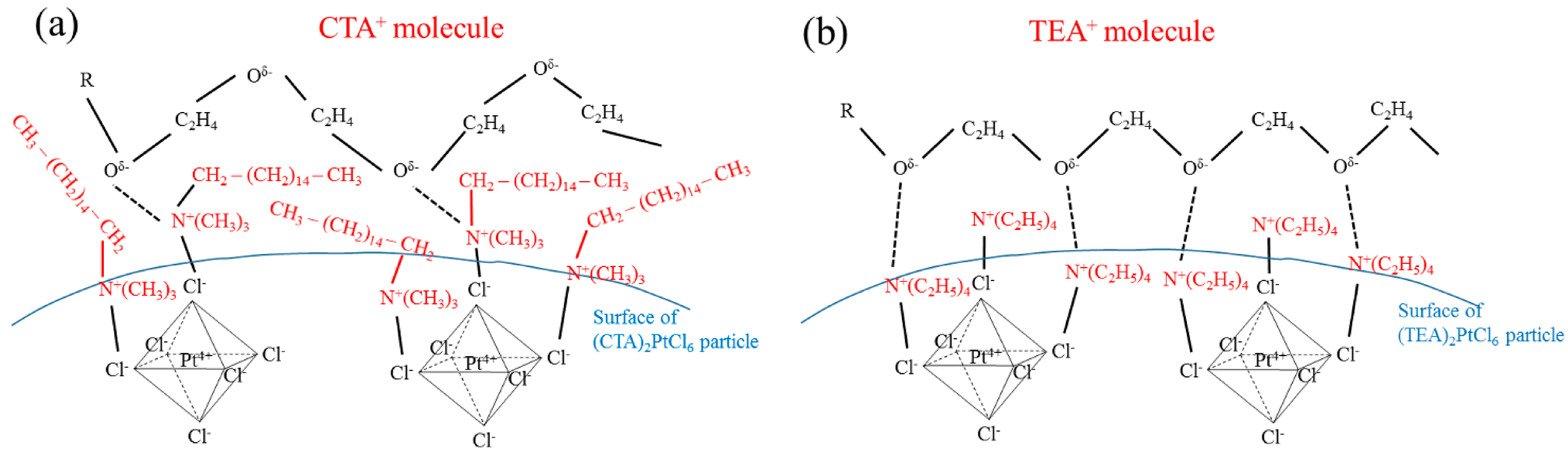
2.3. Effect of Type of Complex-Forming Agents

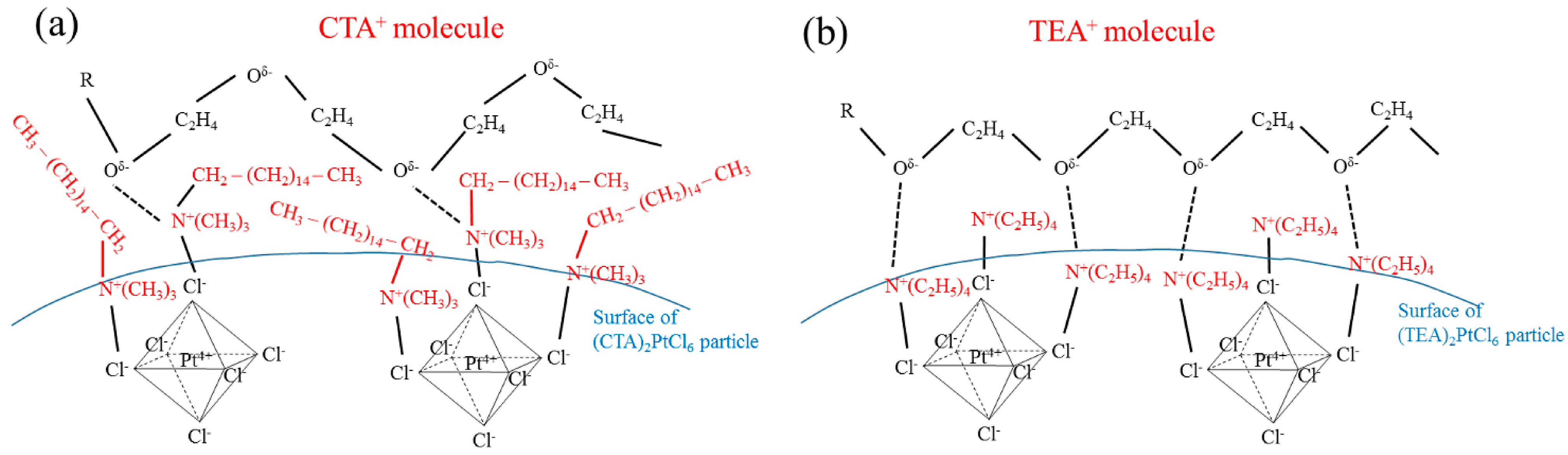
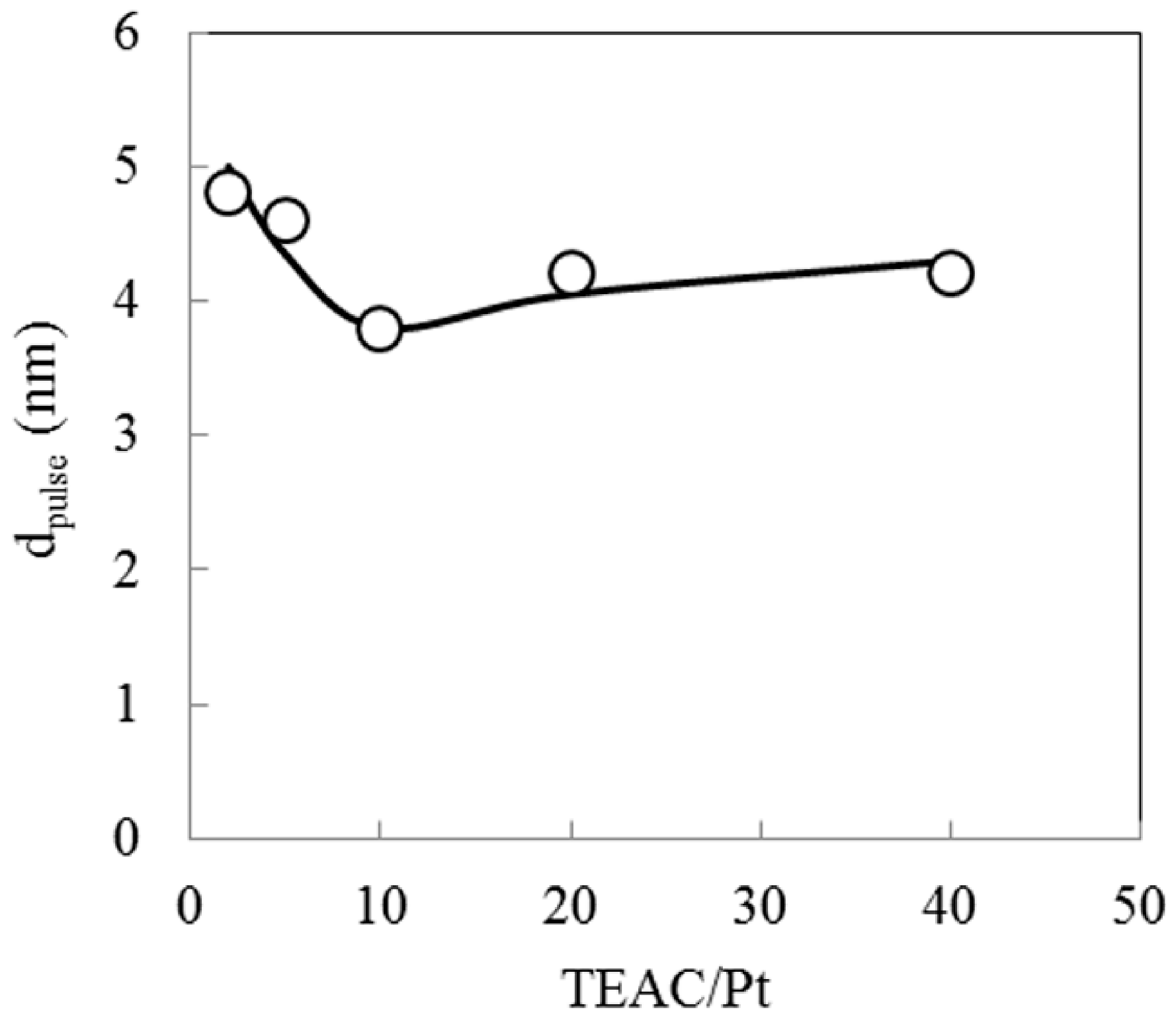
2.4. Effect of Complex-Forming Agent/Pt
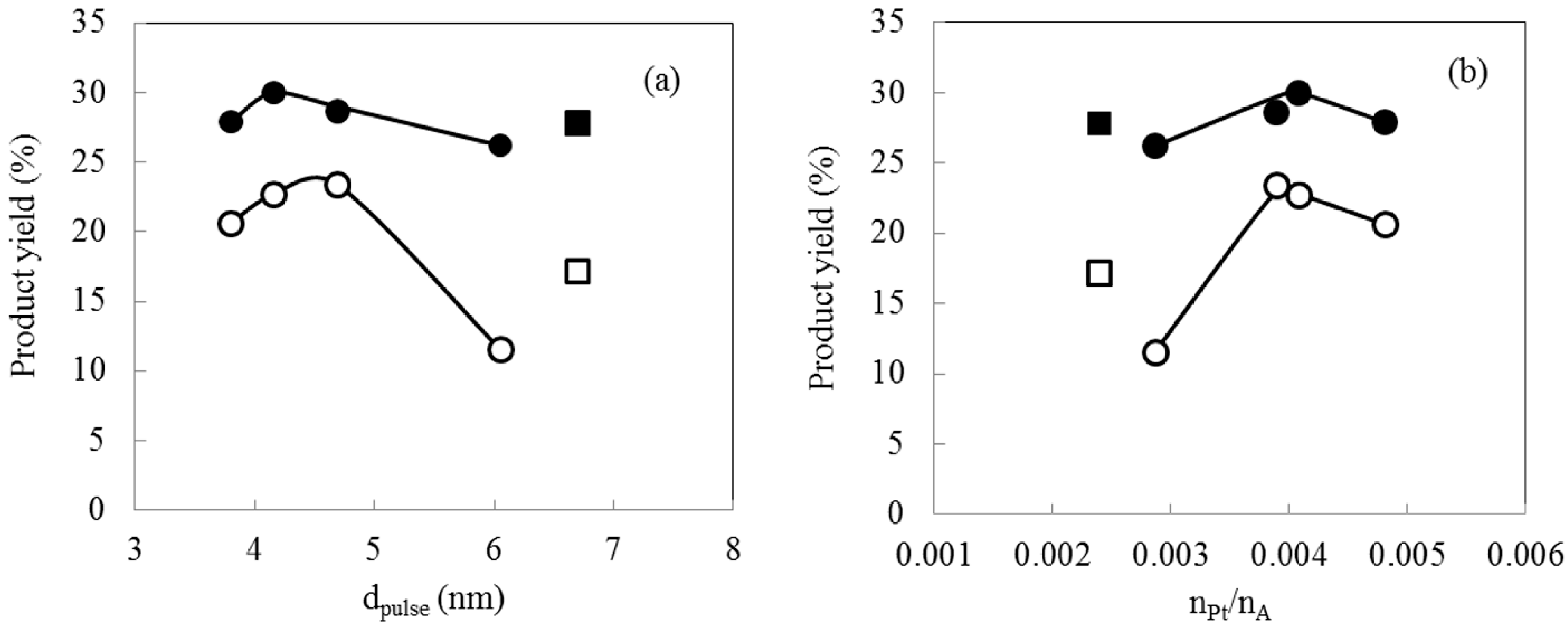
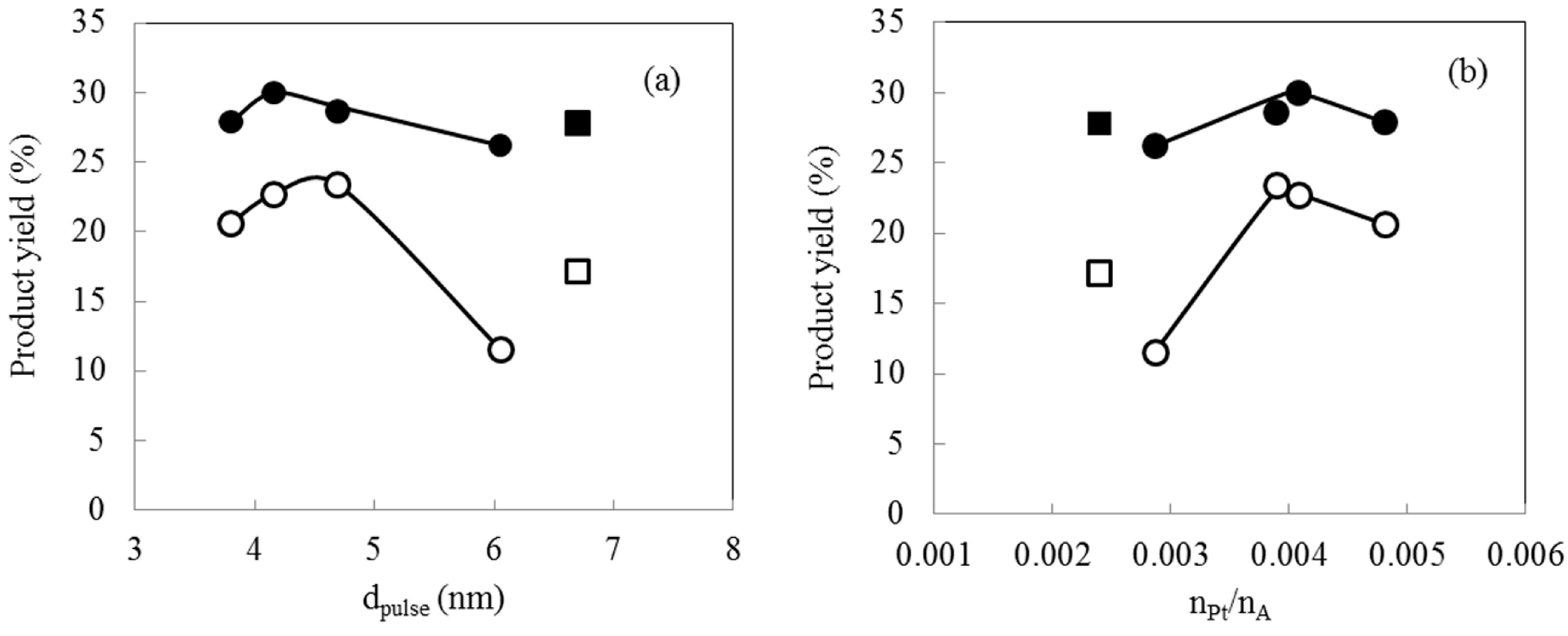
2.5. Hydrocracking Behaviors of Pt-Loaded Zeolite Catalysts
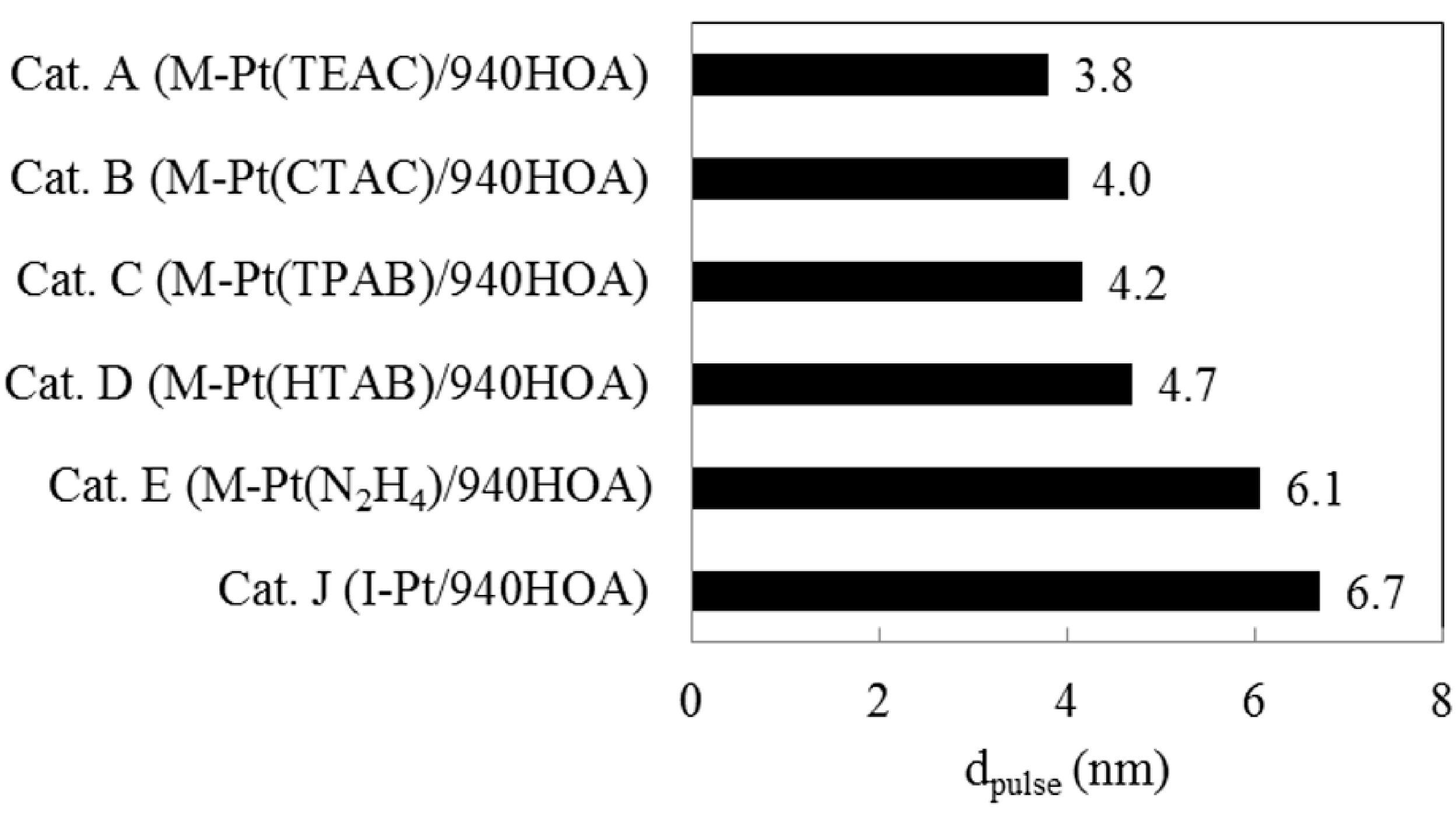
| # | Catalyst | Remarks | Product yield on a carbon basis (%) | dpulse | nPt/nA | |||
|---|---|---|---|---|---|---|---|---|
| YC1–C8 | YC9–C15a | YC16+ | Loss | nm | ||||
| A | M-Pt(TEAC)/940HOA | TEAC/Pt = 10 | 20.6 | 27.9 | 49.4 | 2.0 | 3.8 | 0.0048 |
| C | M-Pt(TPAB)/940HOA | TPAB/Pt = 10 | 22.7 | 30.0 | 48.9 | −1.7 | 4.2 | 0.0041 |
| D | M-Pt(HTAB)/940HOA | HTAB/Pt = 10 | 23.4 | 28.6 | 46.3 | 1.6 | 4.7 | 0.0039 |
| E | M-Pt(N2H4)/940HOA | N2H4/Pt = 80 | 11.5 | 26.2 | 62.2 | 0.1 | 6.1 | 0.0029 |
| J | I-Pt/940HOA | Impregnation | 17.1 | 27.8 | 53.1 | 1.9 | 6.7 | 0.0024 |
3. Experimental Section
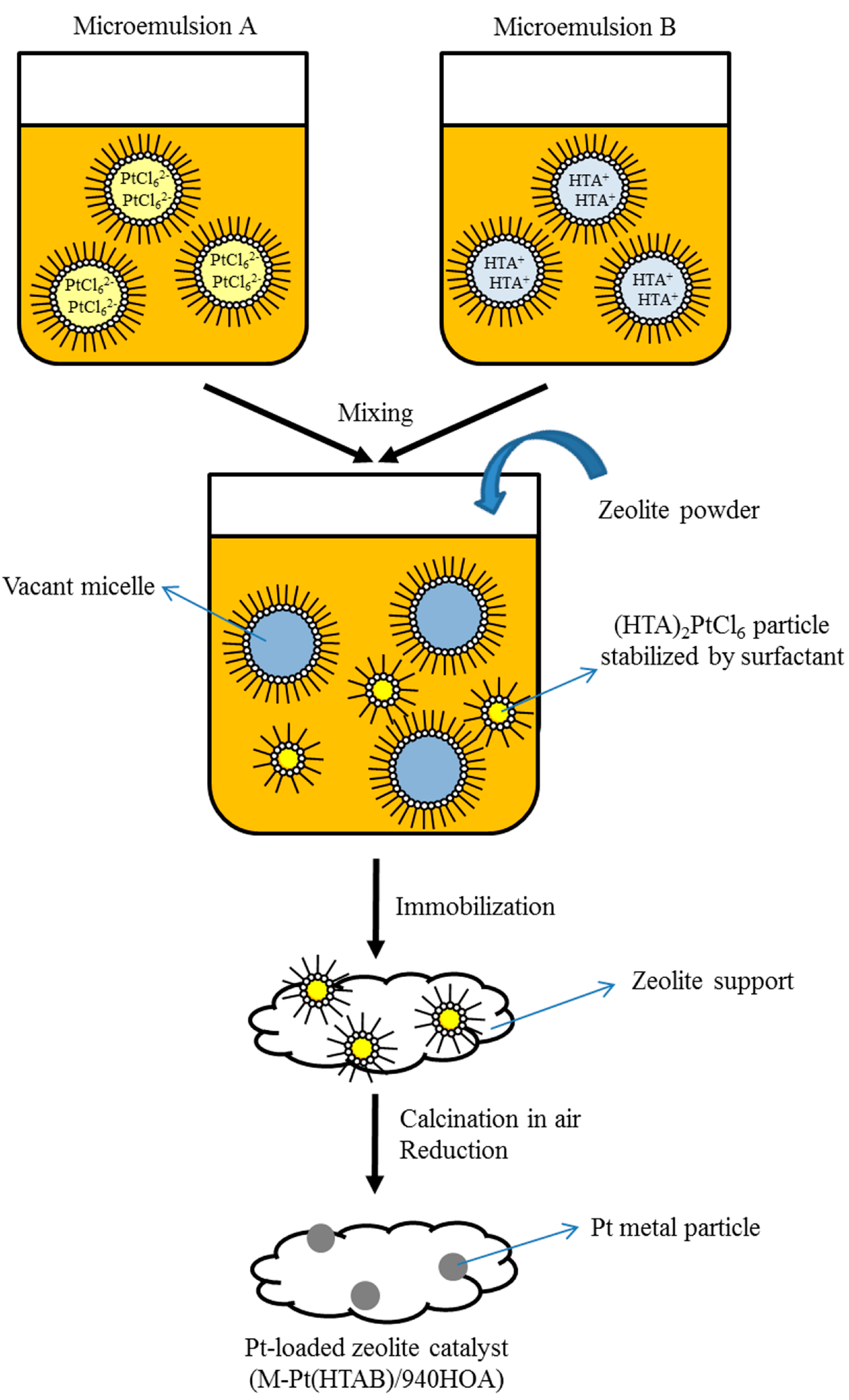
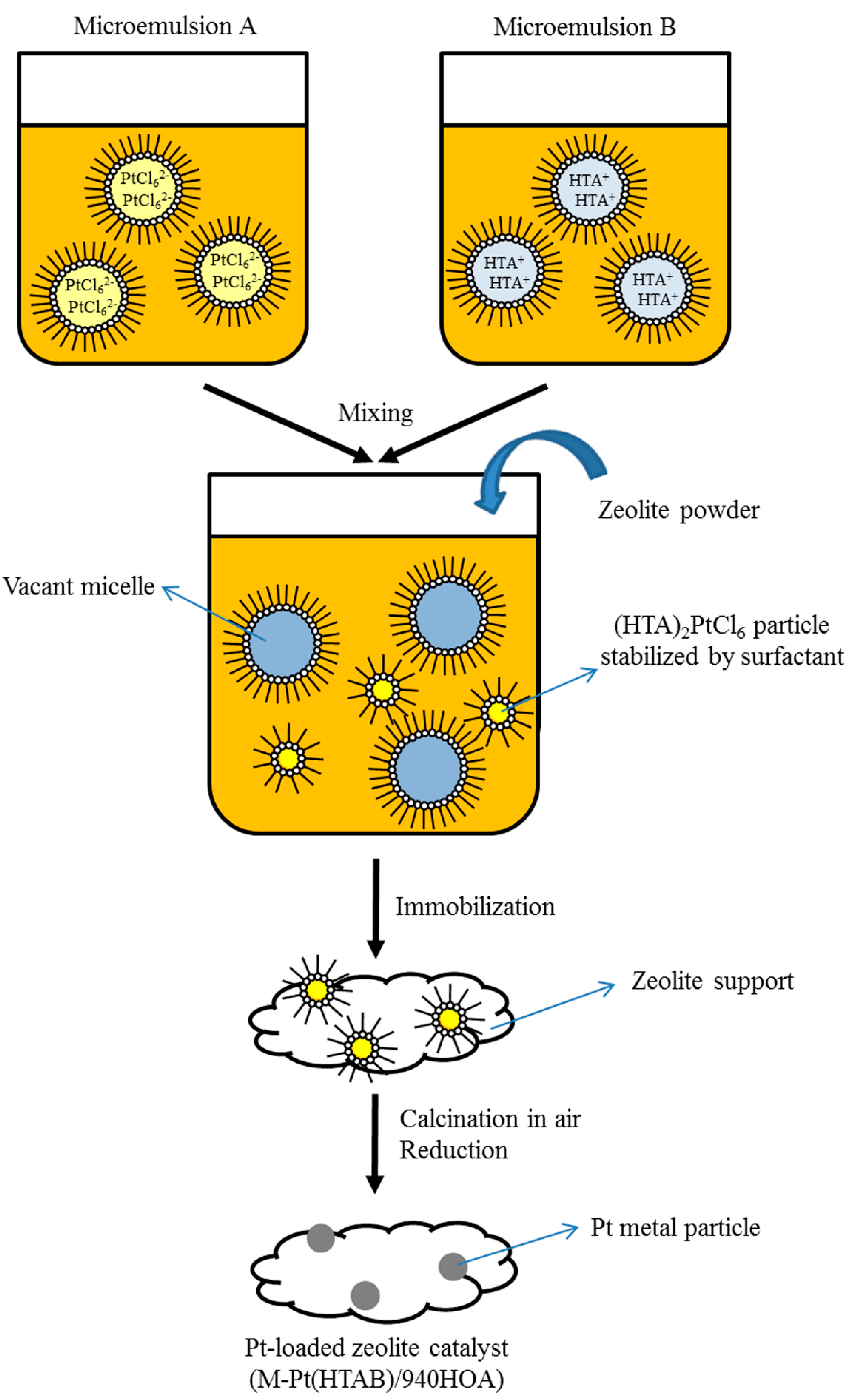
3.1. Catalyst Preparation
3.2. Characterization
3.3. Hydrocracking Test
3.3.1. Production of the FT Product as a Feedstock of Hydrocracking
| Composition on a carbon basis (%) | Elemental analysis (wt.%) | |||||||
|---|---|---|---|---|---|---|---|---|
| C5–C8 | C9–C15a | C16b | C17+ | C | H | N | S | O c |
| 0.0 | 23.1 | 20.2 | 56.7 | 85.0 | 14.9 | 0.0 | 0.0 | 0.2 |
3.3.2. Hydrocracking Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tijmensen, M.J.A.; Faaij, A.P.C.; Hamelinck, C.N.; Hardeveld, M.R.M. Exploration of the possibilities for production of Fischer-Tropsch liquids and power via biomass gasification. Biomass Bioenergy 2002, 23, 129–152. [Google Scholar] [CrossRef]
- Trippe, F.; Fröhling, M.; Schultmann, F.; Stahl, R.; Henrich, E. Techno-economic assessment of gasification as a process step within biomass-to-liquid (BtL) fuel and chemicals production. Fuel Process. Technol. 2011, 92, 2169–2184. [Google Scholar] [CrossRef]
- Vliet, O.P.R.; Faaij, A.P.C.; Turkenburg, W.C. Fischer-Tropsch diesel production in a well-to-wheel perspective: A carbon, energy flow and cost analysis. Energy Convers. Manag. 2009, 50, 855–876. [Google Scholar] [CrossRef]
- Sunde, K.; Brekke, A.; Solberg, B. Environmental impacts and costs of woody Biomass-to-Liquid (BTL) production and use—A review. Forest Policy Econ. 2011, 13, 591–602. [Google Scholar] [CrossRef]
- Krylova, A.Y.; Kozyukov, E.A. State-of-the-art processes for manufacturing synthetic liquid fuels via the Fischer-Tropsch synthesis. Solid Fuel Chem. 2007, 41, 335–341. [Google Scholar] [CrossRef]
- Fujimoto, S.; Yanagita, T.; Ogata, M.; Minowa, T. Evaluation of CO2 mitigation by BTL biofuels from woody biomass through simulated case studies. Int. Energy J. 2008, 9, 73–80. [Google Scholar]
- Hanaoka, T.; Miyazawa, T.; Nurunnabi, M.; Hirata, S.; Sakanishi, K. Liquid fuel production from woody biomass via oxygen-enriched air/CO2 gasification on a bench scale. J. Jpn. Inst. Energy 2011, 90, 1072–1080. [Google Scholar] [CrossRef]
- Coonradt, H.L.; Garwood, W.E. Mechanism of hydrocracking. I&EC Process. Des. Dev. 1964, 3, 38–45. [Google Scholar] [CrossRef]
- Chao, K.J.; Wu, H.C.; Leu, L.J. Hydroisomerization of light normal paraffins over series of platinum-loaded mordenite and beta catalysts. Appl. Catal. A 1996, 143, 223–243. [Google Scholar] [CrossRef]
- Alvarez, F.; Montes, A.; Perot, G.; Guisnet, M. Hydroisomerization and hydrocracking of alkanes. 4. Methylcyclohexane transformation on PtUSHY catalysts. Stud. Surface Sci. Catal. 1989, 49, 1367–1376. [Google Scholar]
- Maldonado, F.J.; Becue, T.; Silva, J.M.; Ribeiro, M.F.; Massiani, P.; Kermarec, M. Influence of the alkali in Pt/alkali-β zeolite on the Pt characteristic and catalytic activity in the transformation of n-hexane. J. Catal. 2000, 195, 342–351. [Google Scholar] [CrossRef]
- Guisnet, M.; Alvarez, F.; Giannetto, G.; Perot, G. Hydroisomerization and hydrocracking of n-heptane on PtH zeolites. Effect of the porosity and of the distribution of metallic and acid sites. Catal. Today 1987, 1, 415–433. [Google Scholar]
- Alvarez, F.; Ribeiro, F.R.; Perot, G.; Thomazeau, C.; Guisnet, M. Hydroisomerization and hydrocracking of alkanes. J. Catal. 1996, 162, 179–189. [Google Scholar] [CrossRef]
- Alvarez, F.; Giannetto, G.; Guisnet, M.; Perot, G. Hydroisomerization and hydrocracking of n-alkanes. 2. n-heptane transformation on a Pt-dealuminated Y zeolite—Comparison with a Pt-Y zeolite. Appl. Catal. 1987, 34, 353–365. [Google Scholar]
- Cho, K.M.; Park, S.; Seo, J.G.; Youn, M.H.; Nam, I.; Baeck, S.H.; Chung, J.S.; Jun, K.W.; Song, I.K. Effect of calcination temperature of alumina supports on the wax hydrocracking performance of Pd-loaded mesoporous alumina xerogel catalysts for the production of middle distillate. Chem. Eng. J. 2009, 146, 307–314. [Google Scholar] [CrossRef]
- Haan, R.; Joorst, G.; Mokoena, E.; Nicolaides, C.P. Non-sulfided nickel supported on silicate alumina as catalyst for the hydrocracking of n-hexadecane and of iron-based Fischer-Tropsch wax. Appl. Catal. A 2007, 327, 247–254. [Google Scholar] [CrossRef]
- Camblor, M.A.; Corma, A.; Martinez, A.; Martinez-Soria, V.; Valencia, S. Mild hydrocracking of vacuum gasoil over NiMo-β zeolite catalysts: The role of the location of the NiMo phases and the crystallite size of the zeolite. J. Catal. 1998, 179, 537–547. [Google Scholar] [CrossRef]
- Krar, M.; Kovacs, S.; Kallo, D.; Hancsok, J. Fuel purpose hydrotreating of sunflower oil on CoMo/Al2O3 catalyst. Bioresour. Technol. 2010, 101, 9287–9293. [Google Scholar] [CrossRef]
- Roussel, M.; Norsic, S.; Lemberton, J.L.; Guisnet, M.; Cseri, T.; Benazzi, E. Hydrocracking of n-decane on a bifunctional sulfide NiW/ailica-alumina catalyst: Effect of the operating conditions. Appl. Catal. A 2005, 279, 53–58. [Google Scholar] [CrossRef]
- Hoar, T.P.; Schulman, J.H. Transparent water-in-oil dispersions: The oleopathic hydro-micelle. Nature 1943, 152, 102–103. [Google Scholar] [CrossRef]
- Davis, H.T.; Bodet, J.F.; Scriven, L.E.; Miller, W.G. Microemulsions and their precursors. In Physics of Amphiphilic Layers; Springer-Verlag: Berlin & Heidelberg, Germany, 1987; Volume 21, pp. 310–327. [Google Scholar]
- Boutonnet, M.; Kizling, J.; Stenius, P.; Maire, G. The preparation of monodisperse colloidal metal particles from microemulsions. Colloid Surface 1982, 5, 209–223. [Google Scholar] [CrossRef]
- Nagy, J.B.; Derouane, E.G.; Gourgue, A.; Lufimpadio, N.; Ravet, I.; Verfaillie, J.P. Physico-chemical characterization of microemulsions: Preparation of monodisperse colloidal metal boride particles. In Surfactants in Solutions, 1st ed.; Mittal, K.L., Ed.; Springer: New York, NY, USA, 1989; Volume 10, pp. 1–43. [Google Scholar]
- Boutonnet, M.; Kizling, J.; Mintsa-Eya, V.; Choplin, A.; Touroude, R. Monodisperse colloidal metal particles from nonaqueous solutions: Catalytic behavior in hydrogenation of but-1-ene of platinum, palladium, and rhodium particles supported on pumice. J. Catal. 1987, 103, 95–104. [Google Scholar] [CrossRef]
- Hanaoka, T.; Hatsuta, T.; Tago, T.; Kishida, M.; Wakabayashi, K. Control of the rhodium particle size of the silica-supported catalysts by using microemulsion. Appl. Catal. A 2000, 190, 291–296. [Google Scholar] [CrossRef]
- Ikeda, M.; Takeshima, S.; Tago, T.; Kishida, M.; Wakabayashi, K. Preparation of size-controlled Pt catalysts supported alumina. Catal. Lett. 1999, 58, 195–197. [Google Scholar] [CrossRef]
- Ikeda, M.; Tago, T.; Kishida, M.; Wakabayashi, K. Thermal stability of Pt particles of Pt/Al2O3 catalysts prepared using microemulsion and catalytic activity in NO–CO reaction. Catal. Commun. 2001, 2, 261–267. [Google Scholar] [CrossRef]
- Kishida, M.; Umakoshi, K.; Kim, W.Y.; Hanaoka, T.; Nagata, H.; Wakabayashi, K. A novel preparation method of supported metal catalysts using microemulsion. J. Chem. Eng. Jpn. 1995, 21, 990–996. [Google Scholar]
- Luisi, P.L.; Giomini, M.; Pileni, M.P.; Robinson, B.H. Reverse micelle as hosts for proteins and small molecules. Biochim. Biophys. Acta 1988, 947, 209–246. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Hardeveld, R.; Hartog, F. The statistics of surface atoms and surface sites on metal crystals. Surface Sci. 1969, 15, 189–230. [Google Scholar] [CrossRef]
- Dauscher, A.; Garin, F.; Maire, G. Correlations between the surface structure of platinum single crystals and hydrocarbon skeletal rearrangement mechanisms: Approach to the nature of the active sites. J. Catal. 1987, 105, 233–244. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lippens, B.C.; Boer, J.H. Studies on pore systems in catalysts. V. The t method. J. Catal. 1965, 4, 319–323. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar]
- Liu, Y.; Hanaoka, T.; Miyazawa, T.; Murata, K.; Okabe, K.; Sakanishi, K. Fischer-Tropsch synthesis in slurry-phase reactors over Mn- and Zr-modified Co/SiO2 catalysts. Fuel Process. Technol. 2009, 90, 901–908. [Google Scholar] [CrossRef]
- Hanaoka, T.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Hot and dry cleaning of biomass-gasified gas using activated carbons with simultaneous removal of tar, particles, and sulfur compounds. Catalysts 2012, 2, 281–298. [Google Scholar] [CrossRef]
- Miyazawa, T.; Hanaoka, T.; Shimura, K.; Hirata, S. Mn and Zr modified Co/SiO2 catalysts development in slurry-phase Fischer-Tropsch synthesis. Appl. Catal. A 2013, 467, 47–54. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanaoka, T.; Miyazawa, T.; Shimura, K.; Hirata, S. Preparation for Pt-Loaded Zeolite Catalysts Using w/o Microemulsion and Their Hydrocracking Behaviors on Fischer-Tropsch Product. Catalysts 2015, 5, 88-105. https://doi.org/10.3390/catal5010088
Hanaoka T, Miyazawa T, Shimura K, Hirata S. Preparation for Pt-Loaded Zeolite Catalysts Using w/o Microemulsion and Their Hydrocracking Behaviors on Fischer-Tropsch Product. Catalysts. 2015; 5(1):88-105. https://doi.org/10.3390/catal5010088
Chicago/Turabian StyleHanaoka, Toshiaki, Tomohisa Miyazawa, Katsuya Shimura, and Satoshi Hirata. 2015. "Preparation for Pt-Loaded Zeolite Catalysts Using w/o Microemulsion and Their Hydrocracking Behaviors on Fischer-Tropsch Product" Catalysts 5, no. 1: 88-105. https://doi.org/10.3390/catal5010088
APA StyleHanaoka, T., Miyazawa, T., Shimura, K., & Hirata, S. (2015). Preparation for Pt-Loaded Zeolite Catalysts Using w/o Microemulsion and Their Hydrocracking Behaviors on Fischer-Tropsch Product. Catalysts, 5(1), 88-105. https://doi.org/10.3390/catal5010088