2.2.2. Catalyst Evaluation in Dodecane
Table 3 summarizes the results of experiments performed to evaluate the synthesized catalysts and the bare supports in the upgrading of guaiacol in dodecane.
Table 3.
Guaiacol upgrading in dodecane over different catalysts a.
Table 3.
Guaiacol upgrading in dodecane over different catalysts a.
| Catalyst | Guaiacol conversion (%) | Selectivity to [Yield of] catechol (%) | Selectivity to [Yield of] phenol (%) | Gravimetric mass balance b (%) |
|---|
| None | 7 | 15 [1] | 0 [0] | 98 |
| AC | 12 | 10 [1.2] | 3 [0.4] | 99 |
| 7.5% Ac/AC | 26 | 21 [5.5] | 12 [3.1] | 98 |
| 20% Ac/AC | 61 | 7 [4] | 9 [6] | 70 |
| 7.5% Am/AC | 37 | 20 [7.4] | 1 [0.4] | 98 |
| 20% Am/AC | 53 | 31 [16] | 3 [2] | 95 |
| CNF | 6 | 19 [1] | 3 [0.2] | 97 |
| 7.5% Ac/CNF | 40 | 8 [0.3] | 12 [4.8] | 85 |
| 20% Ac/CNF | 56 | 9 [5] | 35 [20] | 96 |
| 7.5% Am/CNF | 33 | 9 [3] | 38 [12] | 98 |
| 20% Am/CNF | 53 | 11 [5.8] | 37 [20] | 96 |
| CNT | 16 | 11 [1.8] | 2 [0.3] | 97 |
| 7.5% Ac/CNT | 72 | 36 [26] | 3 [2] | 95 |
| 20% Ac/CNT | 91 | 24 [22] | 5 [4] | 98 |
| 7.5% Am/CNT | 33 | 33 [11] | 3 [1] | 97 |
| 20% Am/CNT | 24 | 9 [2] | 36 [8.6] | 96 |
| GNT | 13 | 17 [2.2] | 4 [0.5] | 97 |
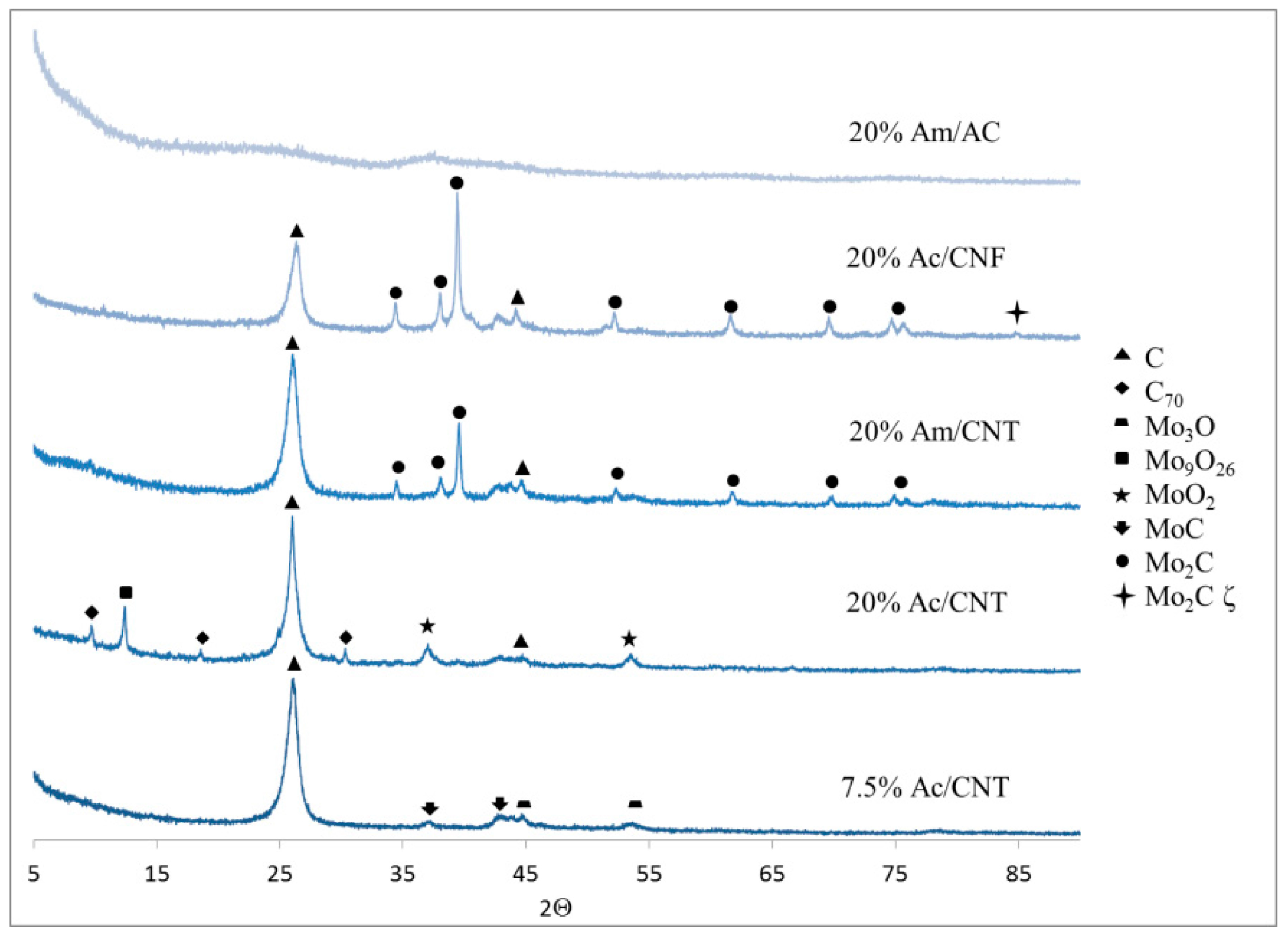
Albeit the conversion and selectivity values observed in the experiment performed without catalyst can be attributed to thermal effects, it is interesting to note that some of the bare carbon supports display additional activity. Given that carbon nanofibers and carbon nanotubes are respectively grown using Ni and Fe catalysts and that the hydrodeoxygenation of guaiacol has been reported to proceed over both Ni [
19,
20,
21] and Fe [
22,
23,
24] catalysts, the activity of the bare supports could conceivably be caused by the residual presence of these metals. Indeed, the Ni and Fe content of the carbon supports was measured by means of Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) and determined to be non-negligible, as shown in
Table 4.
Table 4.
Results of ICP-OES analysis of the carbon supports used in this study.
Table 4.
Results of ICP-OES analysis of the carbon supports used in this study.
| Carbon support | Fe content | Ni content |
|---|
| AC | 5022 ppm | 666 ppm |
| CNF | 334 ppm | 1.9% |
| CNT | 2.35% | 40 ppm |
| GNT | 837 ppm | 18 ppm |
However, the activity of Ni present in the CNF can be deemed to be minimal, since the conversion and selectivity values obtained using CNF are almost identical to those obtained in the blank (sans catalyst) run. Carbon nanotubes afforded higher guaiacol conversion than the blank run, albeit this cannot be entirely attributed to the presence of iron since a sample of graphitized nanotubes (GNT)—in which Fe content is reduced to 837 ppm by means of a thermal treatment (in helium) exceeding 2700 °C [
25]—showed almost identical results (see
Table 3). Similarly, the additional activity displayed by the bare supports relative to the blank run cannot be entirely attributed to the presence of acid sites on the surface of these materials since the graphitized nanotubes—in which these sites have been removed by the high temperatures employed during the graphitization process [
26]—shows similar results (see
Table 3). In a recent contribution by Jongerius
et al., the surface of the carbon supports in carbide catalysts were deemed inert since the high temperature employed in the carburization process effectively removes acidic functional groups from the supports [
2]. However, the conversion and selectivity values obtained over graphitized nanotubes suggest that bare supports display some activity which must be taken into account during the interpretation of the results obtained using the Mo-containing catalysts.
Notably, in contrast with the results obtained in aqueous medium, higher guaiacol conversions were invariably obtained over the Mo-containing catalysts relative to the bare supports in an organic environment (using dodecane as solvent). The most abundant reaction products were typically catechol and phenol, albeit in some instances significant amounts of cyclohexane were produced (a more comprehensive account of the results of product analysis is given in Table S2 of the Supplementary Materials accompanying this article). The fact that catechol was observed in all reaction mixtures suggests that guaiacol is converted into phenol via sequential demethylation and HDO [
7], which contrasts with the direct demethoxylation pathway proposed by Jongerius
et al. [
2], albeit the possibility for these two pathways to be operating in parallel cannot be discounted.
Among the aforementioned products—namely catechol, phenol, and cyclohexane—phenol represents the most desirable product, as it offers a good balance between depth of deoxygenation and value (an upgraded bio-oil or lignin depolymerization stream rich in catechol would still be unacceptably unstable and corrosive while a cyclohexane-rich product would be less valuable that a product rich in phenol). Therefore, based on the yield of phenol the best catalysts are 20% Ac/CNF, 7.5% Am/CNF, 20% Am/CNF and 20% Am/CNT (see
Table 3). Tellingly, the only crystalline phase detected in all these formulations is Mo
2C (or Mo
2C-ζ, which does not seem to behave inherently differently to Mo
2C). This suggests that crystalline Mo
2C is particularly selective to phenol, which is consistent with the fact that catalysts in which Mo
2C is accompanied by MoC—as well as catalysts comprising MoC and/or different molybdenum oxides and catalysts with no crystalline Mo-containing phases—all show lower selectivity to phenol than the catalysts containing Mo
2C or Mo
2C-ζ as their only crystalline phase. Indeed, while the samples containing Mo oxides (
i.e., 7.5% Ac/CNT and 20% Ac/CNT) showed high guaiacol conversions, the selectivity to phenol was extremely low, the preferred products being catechol and coke (see Table S2).
Of the samples containing Mo2C or Mo2C-ζ as their only crystalline phase, a comparison between the results obtained using 7.5% Am/CNF and 20% Am/CNF is particularly informative, since the only difference between these two catalysts is their metal loading (Mo precursor, carbon support, Mo2C phase and Mo2C particle size being all the same). Interestingly, the selectivity to phenol of these two catalysts is virtually identical, the only difference in their behavior being the lower guaiacol conversion shown by 7.5% Am/CNF, which is consistent with the fact that a lower metal loading (at similar dispersion) should translate into a reduced number of active sites.
It is also instructive to compare the results obtained with the 20% Am/CNF and 20% Am/CNT catalysts, these affording very different conversion values (at similar selectivity). In an effort to assess the intrinsic activity of these catalysts, CO chemisorption was applied in order to quantify the concentration of adsorption sites present (see
Table 5). Based on their very similar CO uptake, both catalysts have a similar quantity of active sites. Taking this into account and the fact that the guaiacol conversion obtained over 20% Am/CNF was over twice that obtained over 20% Am/CNT, the former formulation appears to be more intrinsically active, although this ignores possible differences in catalyst deactivation due to coke formation.
Table 5.
Results of CO chemisorption and intrinsic activity of 20% Am/CNF and 20% Am/CNT.
Table 5.
Results of CO chemisorption and intrinsic activity of 20% Am/CNF and 20% Am/CNT.
| Catalyst | CO uptake (μmol g−1) | Average rate (s−1) a |
|---|
| 20% Am/CNF | 49 | 0.06 |
| 20% Am/CNT | 53 | 0.03 |
As noted above, the use of CNT as support results in a bimodal particle size distribution containing both micro and nanoparticles in catalysts where carbides are formed, whereas the use of CNF affords solely well-dispersed carbide nanoparticles. In turn, given that these catalysts show a very similar amount of active sites (see
Table 5), it follows that the difference in the activity of the 20% Am/CNF and 20% Am/CNT catalysts can be attributed to the effect of particle morphology. Tellingly, spent 20% Am/CNT displayed higher amounts of coke on its surface relative to spent CNF-supported catalysts including 20% Am/CNF (see Table S2), which suggests that this morphology effect can be explained in terms of the catalyst resistance to coking—coke can cover more active sites (and arguably form more effectively) on a microparticle showing a high concentration of adjacent Mo
2C sites. Therefore, the optimization of particle morphology seems to be a promising way to further improve the performance of molybdenum carbide catalysts.
Finally, a 5% Ru/C reference catalyst was tested under identical reaction conditions for comparison purposes. Albeit this catalyst afforded quantitative guaiacol conversion (the mass balance being 94%), the catalyst displayed 100% selectivity to cyclohexane. Given that (as mentioned above) a cyclohexane-rich product would be less valuable that a product rich in phenol, the CNF-supported Mo2C catalysts employed in this study represent a promising alternative to the costly Ru-based catalyst commonly used to catalyze this reaction.
2.2.3. Catalyst Testing in an Organic Environment at Different Temperatures
In order to study the effect of temperature on the upgrading of guaiacol in dodecane over Mo
2C/CNF—the formulation showing the best performance (in terms of yield of phenol) at 300 °C—representative CNF-supported catalysts were tested at 350 °C. The catalysts employed in these tests, namely 7.5% Am/CNF and 20% Am/CNF, were chosen in order to study the effect of temperature on two similar catalysts (their only difference being the metal loading) showing noticeably different conversion. In turn, the reaction temperature of 350 °C was chosen based on a recent report by Jongerius
et al. [
2] in which the best results in terms of both guaiacol conversion and phenol selectivity were obtained at this temperature. The results of these experiments are shown in
Table 6 and a more comprehensive account of the results of product analysis is given in Table S3 of the Supplementary Materials accompanying this article.
Table 6.
Guaiacol upgrading in an organic environment at different temperatures a.
Table 6.
Guaiacol upgrading in an organic environment at different temperatures a.
| Catalyst | Temperature (°C) | Guaiacol conversion (%) | Selectivity to [Yield of] catechol (%) | Selectivity to [Yield of] phenol (%) | Gravimetric mass balance b (%) |
|---|
| None | 300 | 7 | 15 [1] | 0 [0] | 98 |
| None | 350 | 79 | 5 [4] | 2 [2] | 59 |
| 7.5% Am/CNF | 300 | 33 | 9 [3] | 38 [12] | 98 |
| 7.5% Am/CNF | 350 | 99 | 0 [0] | 49 [48] | 99 |
| 20% Am/CNF | 300 | 53 | 11 [5.8] | 37 [20] | 96 |
| 20% Am/CNF | 350 | 98 | 0 [0] | 48 [47] | 98 |
As mentioned above, the conversion and selectivity values observed in the experiments performed without catalyst can be attributed to thermal effects. Although the guaiacol conversion is considerably higher at 350 °C than at 300 °C in the absence of a catalyst, the selectivity to catechol and phenol remains minimal. Given that very small amounts of products were detected in the liquid product mixture (see Table S3) and a considerably lower mass balance was obtained from the blank experiment performed at 350 °C, it appears that most of the converted guaiacol afforded products in the gas phase which were not counted in the mass balance calculation. Other authors have explained low mass balances invoking the formation of high molecular weight condensation products not detectable through GC-based methods [
2]; however, the fact that in our case mass balances were calculated gravimetrically can be used to rule out this possibility. These results contrast with those obtained in the presence of a catalyst, in which conversions of ≥98% and selectivities to phenol approaching 50% were achieved at 350 °C. Tellingly, the mass balances of these reactions were also ≥98%, which suggests that approximately half of the guaiacol was converted to the coke observed on the spent catalyst surface (see Table S3), to products that were detectable but unidentifiable via GC—which cannot be quantified using GC since their response factors are unknown—and/or to soluble higher molecular weight products undetectable via GC [
2], the amount of these products being non-negligible according to a molar mass balance (see Table S4 of the Supplementary Materials). The fact that the performance of both CNF-supported catalysts was almost identical at 350 °C indicates that at this temperature the reaction is fast enough as to be driven to completion by the lower number of active sites present in the 7.5% Am/CNF catalyst. Similarly, the fact that no catechol was detected in the products of the reactions catalyzed at 350 °C suggests that if the conversion of guaiacol into phenol is intermediated by catechol, this reaction was also driven to completion (since phenol presents the most difficult C-O bond to cleave, it is particularly resistant to HDO [
7]). Notably, the conversion and selectivity to phenol values obtained at 350 °C over the CNF-supported Mo
2C catalysts are among the best in the literature for carbide catalysts, being comparable to those reported by Jongerius
et al. [
2].




{kind=link}
{kind=link}
