
Alkaline Earth Carbonate Engineered Pt Electronic States for High-Efficiency Propylene Oxidation at Low Temperatures


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Result and Discussion
2.1. Synthesis and Characterization of Pt-MCO3
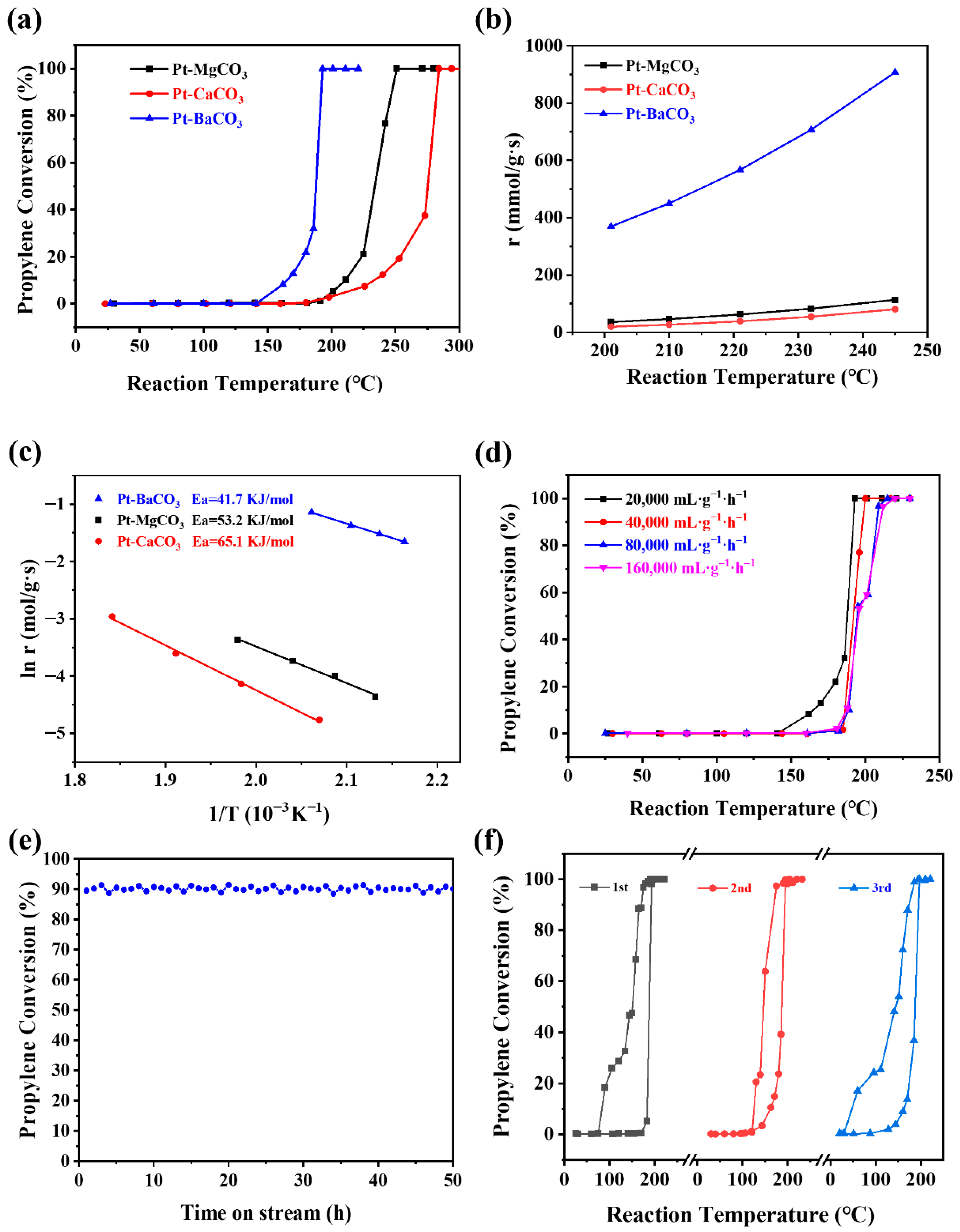
2.2. The Catalytic Performance Tests
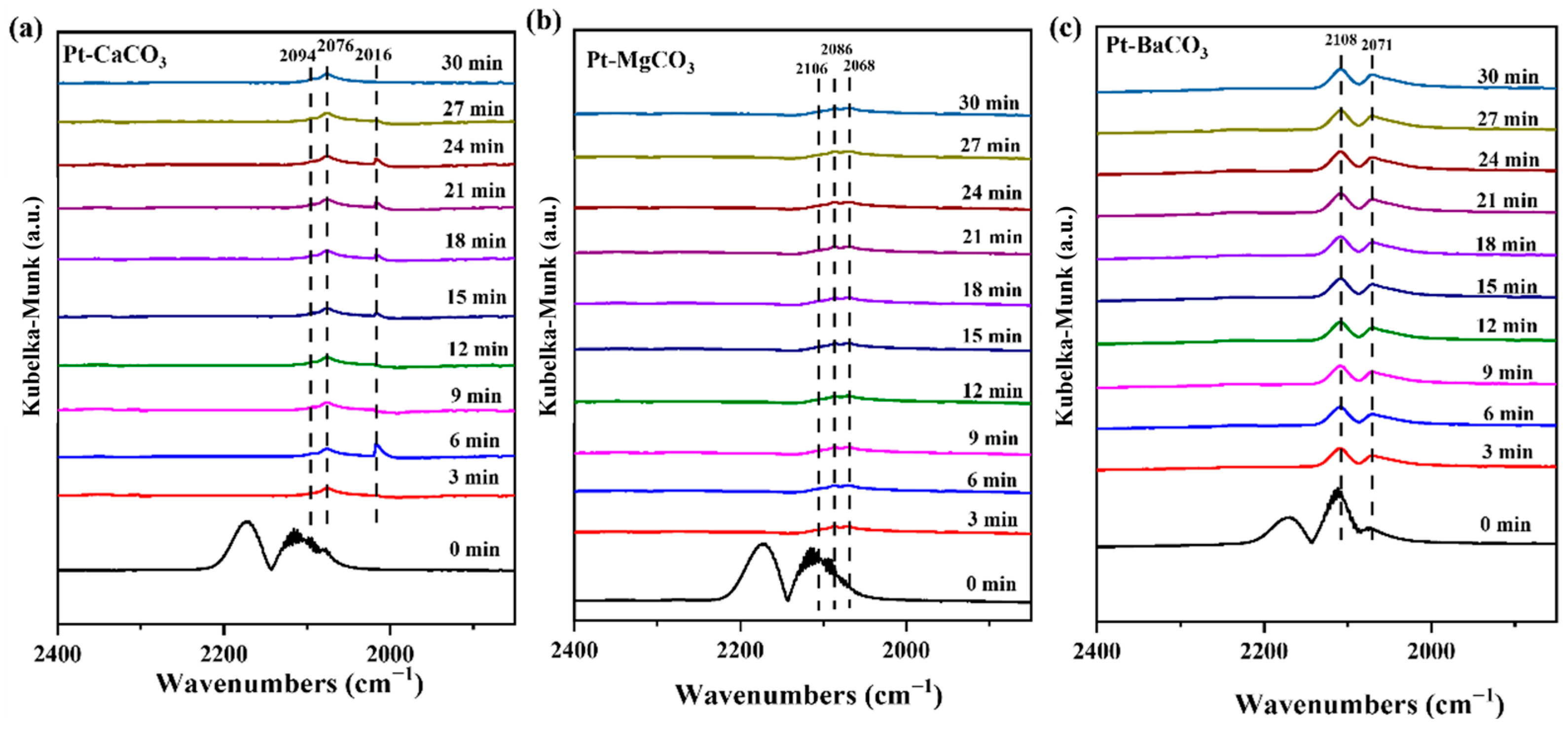
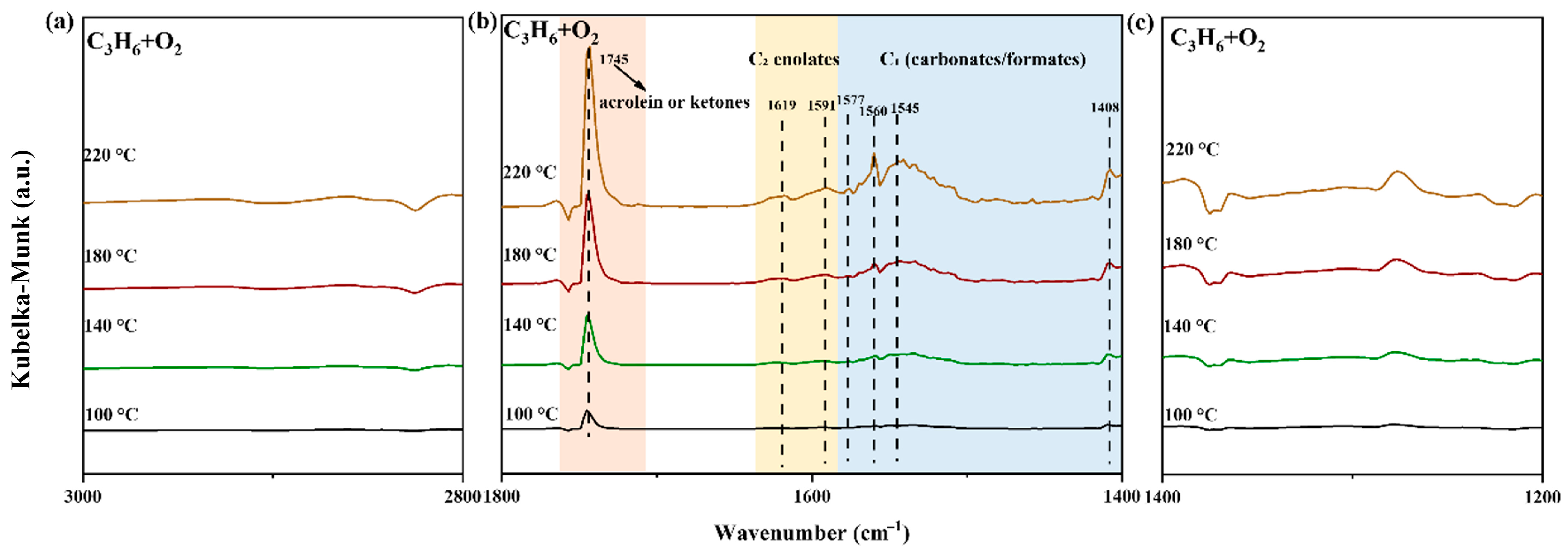
2.3. The Reaction Path in Pt/MCO3
2.4. Understanding the Structure–Performance Relationship in Pt/MCO3
3. Experimental
3.1. Preparation of Catalysts
3.2. Catalyst Performance Evaluation
3.3. Characterization of Catalysts
3.4. Isotope-Labeling Experiments
3.5. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Wang, Z.; Wei, L.; Liu, Y.; Dai, H.; Deng, J. Recent progress on VOC pollution control via the catalytic method. Chin. J. Catal. 2024, 61, 71–96. [Google Scholar] [CrossRef]
- Yang, C.; Miao, G.; Pi, Y.; Xia, Q.; Wu, J.; Li, Z.; Xiao, J. Abatement of various types of VOCs by adsorption/catalytic oxidation: A review. Chem. Eng. J. 2019, 370, 1128–1153. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, J.; Mu, Y.; Ji, X.; Cai, Y.; Jiang, N.; Xie, S.; Qian, Q.; Liu, F.; Tan, W.; et al. Fabrication of Highly Dispersed Ru Catalysts on CeO2 for Efficient C3H6 Oxidation. Environ. Sci. Technol. 2024, 58, 19533–19544. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, S.; Shibano, T.; Koga, H.; Matsui, M.; Asakura, H.; Teramura, K.; Okumura, M.; Tanaka, T. Excellent Catalytic Activity of a Pd-Promoted MnOx Catalyst for Purifying Automotive Exhaust Gases. ChemCatChem 2020, 12, 4276–4280. [Google Scholar] [CrossRef]
- Chenglin, S.; Yonghui, B.; Guangsuo, Y.; Xiaqing, L.; Li, M.; Pengcheng, F.; Fang, L. Co-combustion Characteristic and Gas Emission Characteristic of Entrained Flow Gasification Fine Slag Residual Carbon and Coal. J. Ningxia Univ. (Nat. Sci. Ed.) 2024, 45, 282–289. [Google Scholar] [CrossRef]
- Lee, S.; Lin, C.; Kim, S.; Mao, X.; Kim, T.; Kim, S.-J.; Gorte, R.J.; Jung, W. Manganese Oxide Overlayers Promote CO Oxidation on Pt. ACS Catal. 2021, 11, 13935–13946. [Google Scholar] [CrossRef]
- Park, J.; Kim, D.; Byun, S.W.; Shin, H.; Ju, Y.; Min, H.; Kim, Y.J.; Heo, I.; Hazlett, M.J.; Kim, M.; et al. Impact of Pd:Pt ratio of Pd/Pt bimetallic catalyst on CH4 oxidation. Appl. Catal. B Environ. Energy 2022, 316, 121623. [Google Scholar] [CrossRef]
- Liccardo, G.; Cendejas, M.C.; Mandal, S.C.; Stone, M.L.; Porter, S.; Nhan, B.T.; Kumar, A.; Smith, J.; Plessow, P.N.; Cegelski, L.; et al. Unveiling the Stability of Encapsulated Pt Catalysts Using Nanocrystals and Atomic Layer Deposition. J. Am. Chem. Soc. 2024, 146, 23909–23922. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, Y.; Zheng, J.; Zhang, D.; Chen, W.; Yuan, W.; Chen, X.; Liu, R.; Chen, B.; Li, L.; et al. Diverse Effects of SO2-Induced Pt–O–SO3 on the Catalytic Oxidation of C3H6 and C3H8. Environ. Sci. Technol. 2024, 58, 18020–18032. [Google Scholar] [CrossRef]
- You, Y.; Xu, A.; Tang, X.; Guo, Y.; Zhan, W.; Wang, L.; Dai, S.; Guo, Y. Refining Metal–Support Interactions via Surface Modification of Irreducible Oxide Support for Enhanced Complete Propane Oxidation. ACS Catal. 2024, 14, 11457–11467. [Google Scholar] [CrossRef]
- An, Y.; Chen, S.-Y.; Wang, B.; Zhou, L.; Hao, G.; Wang, Y.; Chen, J.; Tsung, C.-K.; Liu, Z.; Chou, L.-Y. Controlling the metal–support interaction with steam-modified ceria to boost Pd activity towards low-temperature CO oxidation. J. Mater. Chem. A 2023, 11, 16838–16845. [Google Scholar] [CrossRef]
- Fang, Q.; Du, H.; Zhang, X.; Ding, Y.; Zhang, Z.C. Alteration of Hydrodeoxygenation Pathways of Ni/TiO2-A Catalyst through Controlled Regulation of Strong Metal–Support Interactions and Surface Acidity. ACS Catal. 2024, 14, 5047–5063. [Google Scholar] [CrossRef]
- Song, Y.; Beyazay, T.; Tüysüz, H. Effect of Alkali- and Alkaline-Earth-Metal Promoters on Silica-Supported Co−Fe Alloy for Autocatalytic CO2 Fixation. Angew. Chem. Int. Ed. 2023, 63, e202316110. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sun, Z.; Shao, B.; Zhu, Y.; Ma, R.; Li, S.; Li, J.; Chen, Y.; Liu, H.; Hu, J. Highly efficient hydrogenation of carbonate to methanol for boosting CO2 mitigation. Chem. Eng. J. 2024, 495, 153465. [Google Scholar] [CrossRef]
- Naicheng, L.; Mingxiao, M.; Mingiie, M. Research on lnfluencing Factors af Early Strength of CaO Fly Ash Geopolymer. J. Ningxia Univ. (Nat. Sci. Ed.) 2022, 43, 378–382. [Google Scholar]
- Zhang, W.; Li, D.; Liu, C.; Jiang, Z.; Gao, C.; Shen, L.; Liu, Q.; Qiu, R.; Gu, H.; Lian, C.; et al. Facilitated CO2 hydrogenation by strong metal-support interaction between Ni and BaCO3. Chem Catal. 2024, 4, 101113. [Google Scholar] [CrossRef]
- Xiong, P.; Xu, Z.; Wu, T.-S.; Yang, T.; Lei, Q.; Li, J.; Li, G.; Yang, M.; Soo, Y.-L.; Bennett, R.D.; et al. Synthesis of core@shell catalysts guided by Tammann temperature. Nat. Commun. 2024, 15, 420. [Google Scholar] [CrossRef]
- Wang, P.; Shi, M.; Shu, Y.; Niu, Q.; Zhang, P. Porous CaCO3 fabricated by Ca-containing solid wastes for effective CO2 adsorption. J. Environ. Chem. Eng. 2025, 13, 115815. [Google Scholar] [CrossRef]
- Ren, H.; Yu, W.; Lv, M.; Gao, J.; Hu, H.; Wang, M.; Cui, X.; Liu, J.; Jiang, L. Carbon-Encapsulated Single-Atom Platinum Nickel Alloy for Efficient and Durable Alkaline Hydrogen Oxidation Through Enhanced Charge Polarization. Adv. Funct. Mater. 2024, 35, 2413754. [Google Scholar] [CrossRef]
- Hou, Z.; Sun, Z.; Cui, C.; Zhu, D.; Yang, Y.; Zhang, T. Ru Coordinated ZnIn2S4 Triggers Local Lattice-Strain Engineering to Endow High-Efficiency Electrocatalyst for Advanced Zn-Air Batteries. Adv. Funct. Mater. 2022, 32, 2110572. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; He, D.; Gao, X.; Jiang, W.; Li, D.; Sun, Y.; Lin, X.; Xu, J.; Wang, H.; et al. Carbonate shell regulates CuO surface reconstruction for enhanced CO2 electroreduction. Nat. Synth. 2024, 4, 53–66. [Google Scholar] [CrossRef]
- Zhu, H.; Lv, X.; Wu, Y.; Wang, W.; Wu, Y.; Yan, S.; Chen, Y. Carbonate-carbonate coupling on platinum surface promotes electrochemical water oxidation to hydrogen peroxide. Nat. Commun. 2024, 15, 8846. [Google Scholar] [CrossRef]
- Cui, H.; Cheng, Z.; Zhou, Z. Unravelling the role of alkaline earth metal carbonates in intermediate temperature CO2 capture using alkali metal salt-promoted MgO-based sorbents. J. Mater. Chem. A 2020, 8, 18280–18291. [Google Scholar] [CrossRef]
- Khan, H.A.; Natarajan, P.; Jung, K.-D. Stabilization of Pt at the inner wall of hollow spherical SiO2 generated from Pt/hollow spherical SiC for sulfuric acid decomposition. Appl. Catal. B Environ. Energy 2018, 231, 151–160. [Google Scholar] [CrossRef]
- Xinxin, H.; Lijun, H.; Yanming, D.; Jiankang, Z.; Ting, L.; Yaojun, G.; Mingze, S. Numerical Analysis of the Influence of Residual Stress and Grain Size on the Indentation Behavior of Beryllium. J. Ningxia Univ. (Nat. Sci. Ed.) 2024, 45, 31–35. [Google Scholar]
- Lachkov, P.T.; Chin, Y.-H. Catalytic consequences of reactive oxygen species during C3H6 oxidation on Ag clusters. J. Catal. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Hazlett, M.J.; Moses-Debusk, M.; Parks, J.E.; Allard, L.F.; Epling, W.S. Kinetic and mechanistic study of bimetallic Pt-Pd/Al2O3 catalysts for CO and C3H6 oxidation. Appl. Catal. B Environ. Energy 2016, 202, 404–417. [Google Scholar] [CrossRef]
- Shu, Y.; Wang, M.; Duan, X.; Liu, D.; Yang, S.; Zhang, P. Low-temperature total oxidation of methane by pore- and vacancy-engineered NiO catalysts. AIChE J. 2022, 68, e17664. [Google Scholar] [CrossRef]
- Bae, W.B.; Kim, D.Y.; Byun, S.W.; Lee, S.J.; Kuk, S.K.; Kwon, H.J.; Lee, H.C.; Hazlett, M.J.; Liu, C.; Kim, Y.J.; et al. Direct NO decomposition over Rh-supported catalysts for exhaust emission control. Chem. Eng. J. 2023, 475, 146005. [Google Scholar] [CrossRef]
- Xia, H.; Dang, C.; Zhou, D.; Cai, W. Lamellar cross-linking Ni/CeO2 as an efficient and durable catalyst for dry reforming of methane. Chem. Eng. J. 2024, 489, 151365. [Google Scholar] [CrossRef]
- Chai, Y.; Chen, S.; Chen, Y.; Wei, F.; Cao, L.; Lin, J.; Li, L.; Liu, X.; Lin, S.; Wang, X.; et al. Dual-Atom Catalyst with N-Colligated Zn1Co1 Species as Dominant Active Sites for Propane Dehydrogenation. J. Am. Chem. Soc. 2023, 146, 263–273. [Google Scholar] [CrossRef]
- Hu, X.; Xu, D.; Jiang, J. Strong Metal-Support Interaction between Pt and TiO2 over High-Temperature CO2 Hydrogenation. Angew. Chem. Int. Ed. 2024, 64, e202419103. [Google Scholar] [CrossRef]
- Shaodi, S.; Xiaomin, W.; Zhiwei, H.; Huazheng, S.; Huawang, Z.; Guohua, J. Engineering stable Pt nanoclusters on defective two-dimensional TiO2 nanosheets by introducing SMSI for efficient ambient formaldehyde oxidation. Chem. Eng. J. 2022, 435, 135035. [Google Scholar] [CrossRef]
- Haus, M.O.; Meledin, A.; Leiting, S.; Louven, Y.; Roubicek, N.C.; Moos, S.; Weidenthaler, C.; Weirich, T.E.; Palkovits, R. Correlating the Synthesis, Structure, and Catalytic Performance of Pt–Re/TiO2 for the Aqueous-Phase Hydrogenation of Carboxylic Acid Derivatives. ACS Catal. 2021, 11, 5119–5134. [Google Scholar] [CrossRef]
- Guo, M.; Meng, Q.; Gao, M.-L.; Zheng, L.; Li, Q.; Jiao, L.; Jiang, H.-L. Single-Atom Pt Loaded on MOF-Derived Porous TiO2 with Maxim-Ized Pt Atom Utilization for Selective Hydrogenation of Halonitro-benzene. Angew. Chem. Int. Ed. 2024, 64, e202418964. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Li, H.; Zhang, Q.; Wang, C.; Xu, J.; Shen, H.; Yang, J.; Pan, C.; Zhu, Y.; Luo, Z.; et al. Oxygen Vacancy-Governed Opposite Catalytic Performance for C3H6 and C3H8 Combustion: The Effect of the Pt Electronic Structure and Chemisorbed Oxygen Species. Environ. Sci. Technol. 2022, 56, 3245–3257. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Tian, X.; Shu, Y.; Zhang, Z.; Liu, Q.; Zhang, P.; Luo, Z. Ideal Co-NiO solid solution with excellent low-temperature propene catalytic combustion performance. Chem. Eng. Sci. 2024, 302, 120923. [Google Scholar] [CrossRef]
- Liu, P.; Zheng, C.; Liu, W.; Wu, X.; Liu, S. Oxidative Redispersion-Derived Single-Site Ru/CeO2 Catalysts with Mobile Ru Complexes Trapped by Surface Hydroxyls Instead of Oxygen Vacancies. ACS Catal. 2024, 14, 6028–6044. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.; Xie, Y.-D.; Lu, A.-H. Evolution of the Active Phase of Pt/Sn–Al2O3 Catalysts During Acidic Impregnation and Their Use in Propane Dehydrogenation. ACS Appl. Mater. Interfaces 2024, 16, 47773–47783. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Yu, H.; Wang, X. The functions of Pt located at different positions of HZSM-5 in H2-SCR. Chem. Eng. J. 2018, 355, 470–477. [Google Scholar] [CrossRef]
- Xiao, Z.; Lin, X.; Feng, W.; Chen, B.; Meng, Q.; Wang, T. Efficient Hydrogen Production from the Aqueous-Phase Reforming of Biomass-Derived Oxygenated Hydrocarbons over an Ultrafine Pt Nanocatalyst. Catalysts 2023, 13, 1428. [Google Scholar] [CrossRef]
- Singh Pal, R.; Rana, S.; Kumar Sharma, S.; Khatun, R.; Khurana, D.; Suvra Khan, T.; Kumar Poddar, M.; Sharma, R.; Bal, R. Enhancement of oxygen vacancy sites of La2-xMxCe2O7-δ (M = Ca, Ba, Sr) catalyst for the low temperature oxidative coupling of Methane: A combined DFT and experimental study. Chem. Eng. J. 2023, 458, 141379. [Google Scholar] [CrossRef]
- Xiaohui, R.; Fei, L.; Qi, W.; Hui, S.; Shunqin, L.; Sijie, L.; Gaoliang, Y.; Bowen, D.; Zongyu, H.; Xu-Sheng, W.; et al. Engineering interfacial charge transfer channel for efficient photocatalytic H2 evolution: The interplay of CoPx and Ca2+ dopant. Appl. Catal. B Environ. Energy 2021, 303, 120887. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Lv, Y.; Shu, Y.; Guo, Y.; Zhang, P. Alkaline Earth Carbonate Engineered Pt Electronic States for High-Efficiency Propylene Oxidation at Low Temperatures. Catalysts 2025, 15, 696. https://doi.org/10.3390/catal15080696
Sun X, Lv Y, Shu Y, Guo Y, Zhang P. Alkaline Earth Carbonate Engineered Pt Electronic States for High-Efficiency Propylene Oxidation at Low Temperatures. Catalysts. 2025; 15(8):696. https://doi.org/10.3390/catal15080696
Chicago/Turabian StyleSun, Xuequan, Yishu Lv, Yuan Shu, Yanglong Guo, and Pengfei Zhang. 2025. "Alkaline Earth Carbonate Engineered Pt Electronic States for High-Efficiency Propylene Oxidation at Low Temperatures" Catalysts 15, no. 8: 696. https://doi.org/10.3390/catal15080696
APA StyleSun, X., Lv, Y., Shu, Y., Guo, Y., & Zhang, P. (2025). Alkaline Earth Carbonate Engineered Pt Electronic States for High-Efficiency Propylene Oxidation at Low Temperatures. Catalysts, 15(8), 696. https://doi.org/10.3390/catal15080696