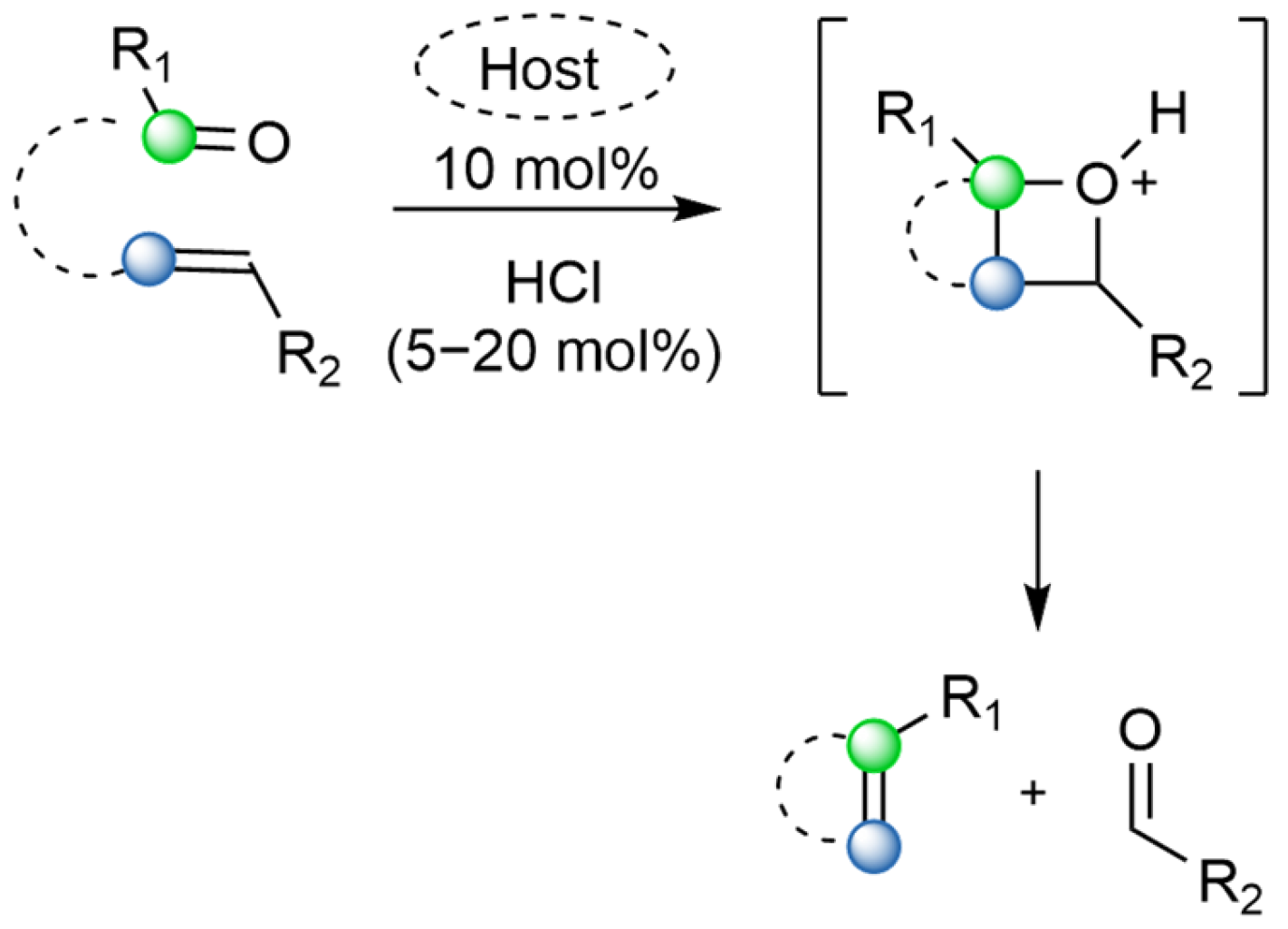
1. Introduction
Metathesis reactions are not new and have been widely used for decades. They are commonly used in the total synthesis of natural products and polymerization reactions. However, they are often perceived as reactions between two carbon–carbon double bonds either inter- or intramolecularly (
Scheme 1) [
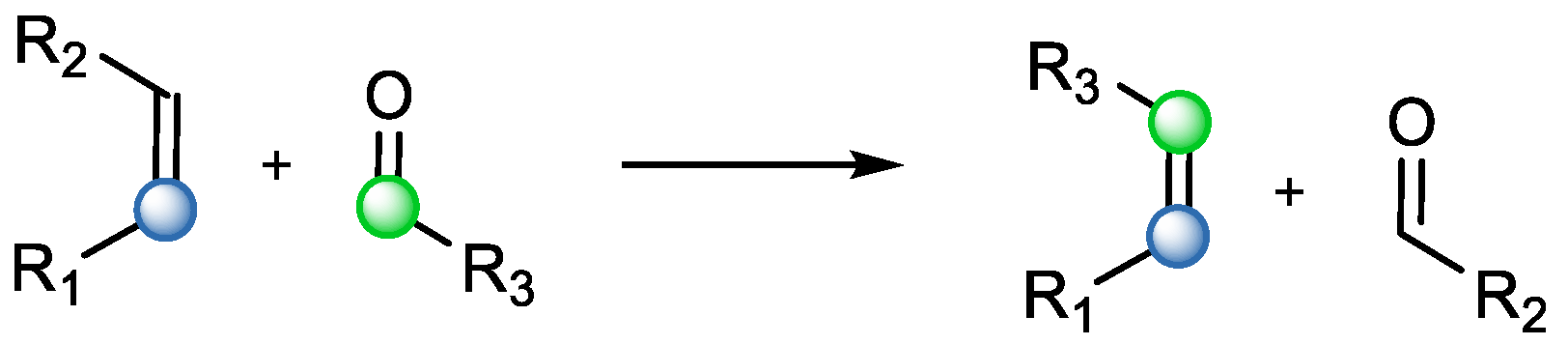
1]. In reality, metathesis can go beyond reactions involving only carbon–carbon bonds. One example of such versatility are carbonyl–olefin metathesis (COM) reactions (
Scheme 2) [
2]. COM reactions offer an elegant solution for natural product synthesis. Their advantage is that they do not require prefunctionalized moieties introduced in the molecule. Additionally, they has high atom economy [
3,
4]. For a long time, COM has not been as popular as olefin metathesis; however, this changed rather recently with the development of different catalytic methods that allowed for its broad application.
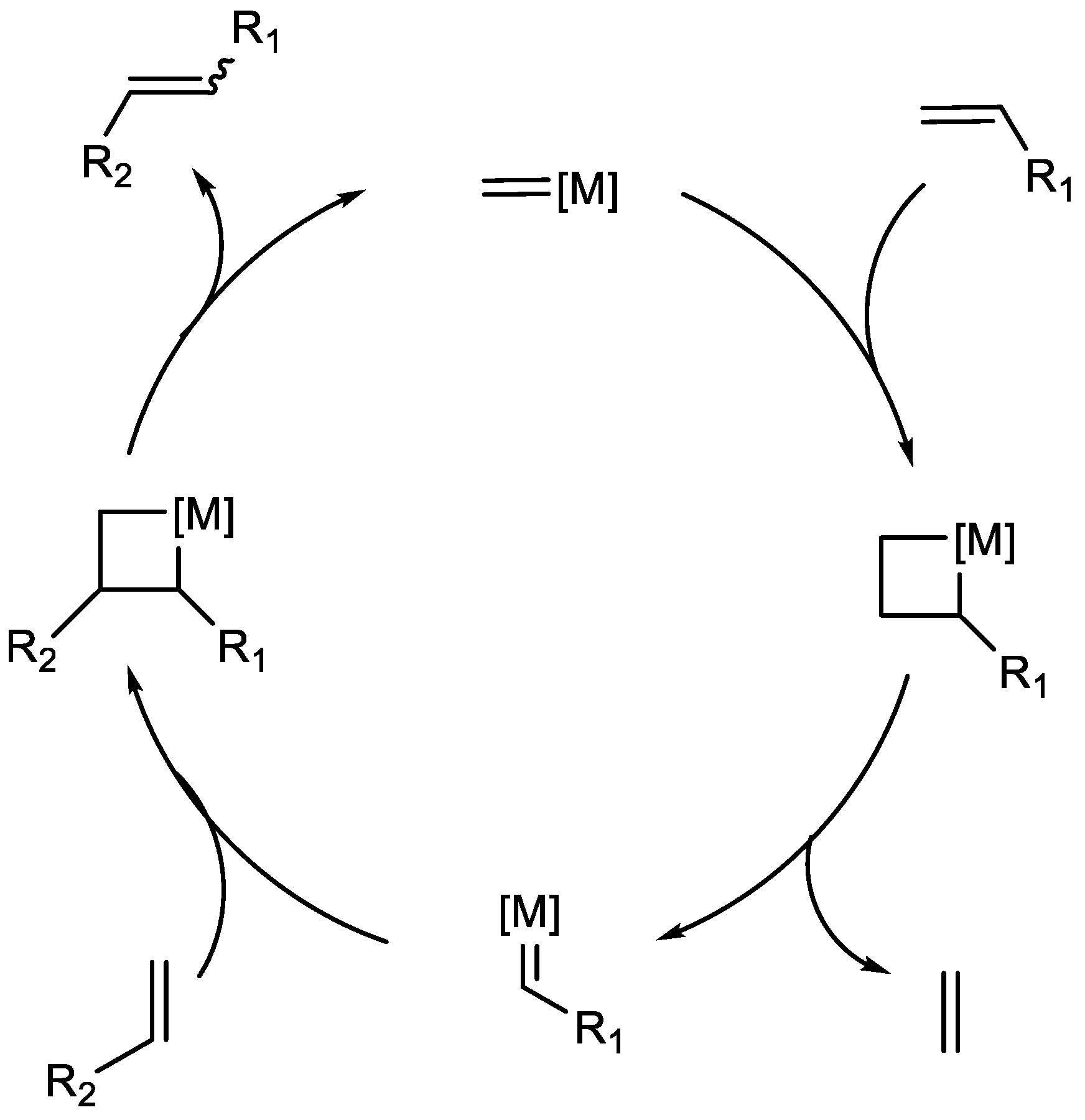
The mechanism of metathesis that is widely accepted was proposed by Yves Chauvin, thus called the Chauvin mechanism, for which he received a Nobel Prize together with Robert H. Grubbs and Richard R. Schrock in 2005. Grubbs and Schrock’s work was predominantly defined by their catalysts to promote metathesis. The Chauvin mechanism is based on a [2+2] cycloaddition with subsequent cycloreversion (
Scheme 3). However, this mechanism does not explain all the metathesis transformations outside the scope of olefin metathesis [
1]. Metathesis is also applied to alkanes, with dehydrogenation being the first step. A similar but reverse approach with hydrogenation can also be applied to alkynes, which offer certain extra possibilities regarding stereoselectivity, since alkynes can be stereoselectively reduced to either
cis or
trans alkenes using different catalysts. Furthermore, alkynes can undergo metathesis without prior transformation to alkenes [
5].
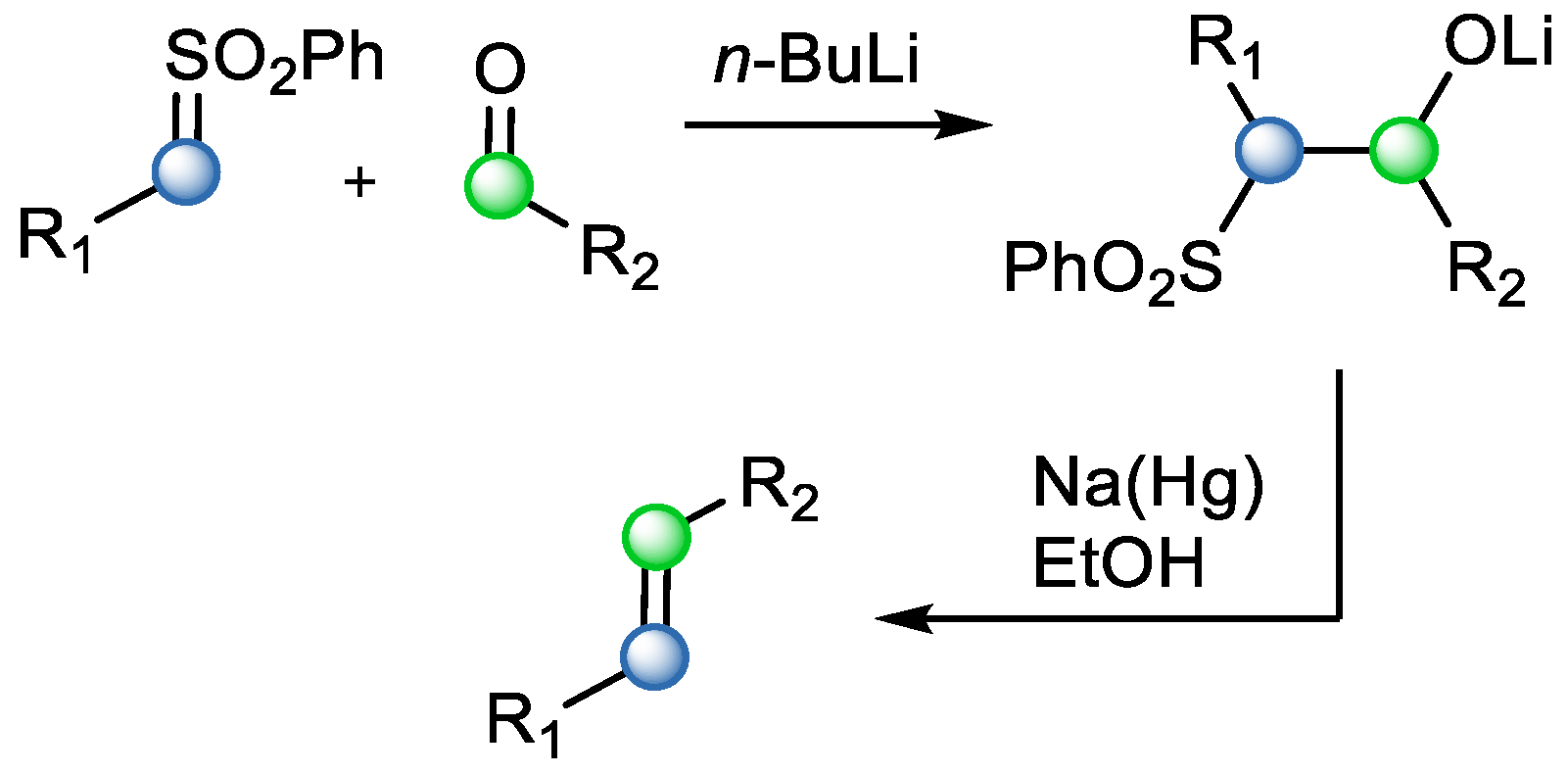
Carbonyl–olefin metathesis offers an alternative to some popular name reactions. It employs a similar principle to the Wittig reaction between ketones or aldehydes and phosphorus ylides, leading to a new carbon–carbon double bond (
Scheme 4). An advantage of carbonyl–olefin metathesis is that it does not require an additional moiety (phosphonium ylide), which enables higher atom economy [
6]. A similar scenario is valid for the case of Julia olefination or other modifications of it (Julia–Kocienski olefination, modified Julia olefination). In principle, these methods are based on sulfones reacting with either aldehydes or ketones to obtain different, mostly
trans olefins (
Scheme 5). Despite their versatility, functional group tolerance, and mild reaction conditions, these methods still involve metals (such as sodium amalgam) and atom waste originating in sulfonic moieties [
7]. Furthermore, classic olefination methods like the Wittig or Julia reactions often require the separate introduction of ylide or sulfone moieties, which can limit their efficiency and functional group compatibility, particularly in intramolecular settings. Since intramolecular cyclizations are crucial in the synthesis of complex natural products, the directness and functional group tolerance of COM reactions provide a distinct advantage for constructing cyclic frameworks under mild conditions.
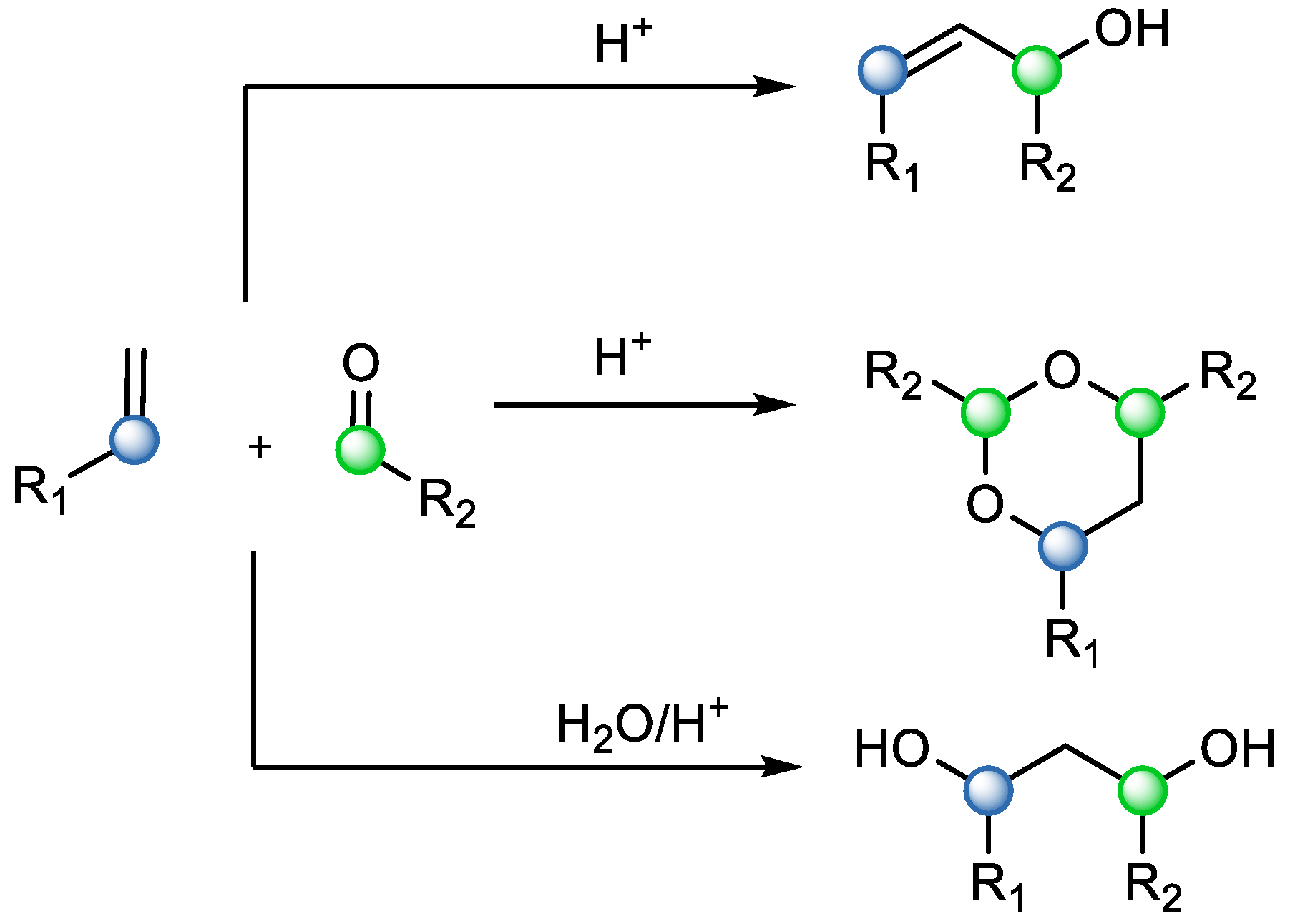
Another option that a synthetic chemist could use is the Prins reaction—the electrophilic addition of a ketone or an aldehyde to an alkene. While this reaction has better atom economy than the previous two, it is not a direct equivalent to carbonyl–olefin metathesis, since the reaction mostly results in 1,3-diols, 1,3-dioxanes, or unsaturated alcohols (
Scheme 6) [
8].
Considering the unique advantages of COM reactions, we will present an overview of the historical development of carbonyl–olefin metathesis methods, and ways of applying them to natural product synthesis with examples and some mechanistic insights. Furthermore, we will discuss their generality, reaction conditions, robustness, selectivity, simplicity, and advantages and disadvantages in general. COM reactions can be categorized based on the type of catalyst or auxiliary used to enable the transformation. That can be either light, metal complexes, organic catalysts, Lewis acids, or Brønsted acids. In the following sections, each of these strategies will be discussed separately but also comparatively. This review also complements others in the field by focusing on natural product utilization and highlights recent advances in carbonyl–olefin metathesis, along with its future prospects [
2,
4,
9,
10,
11,
12].
2. Photochemical Carbonyl–Olefin Metathesis
Photochemical methods can be used for metathesis reactions. They can be applied to both olefin–olefin metathesis and carbonyl–olefin metathesis; the latter will be discussed in this paper. In fact, the first carbonyl–olefin metathesis reactions were based on photochemical conditions. One of the advantages of photochemical processes compared to thermal ones is the ability to obtain thermodynamically less favored products, since they are abe to overcome large energy barriers. These use of these protocols has been well-established in pharmaceutical companies to produce drugs [
13]. These photochemical processes usually require irradiation with UV light, but metathesis reactions using visible light are emerging, replacing short-wavelength mercury vapor lamps with harmless visible-light sources.
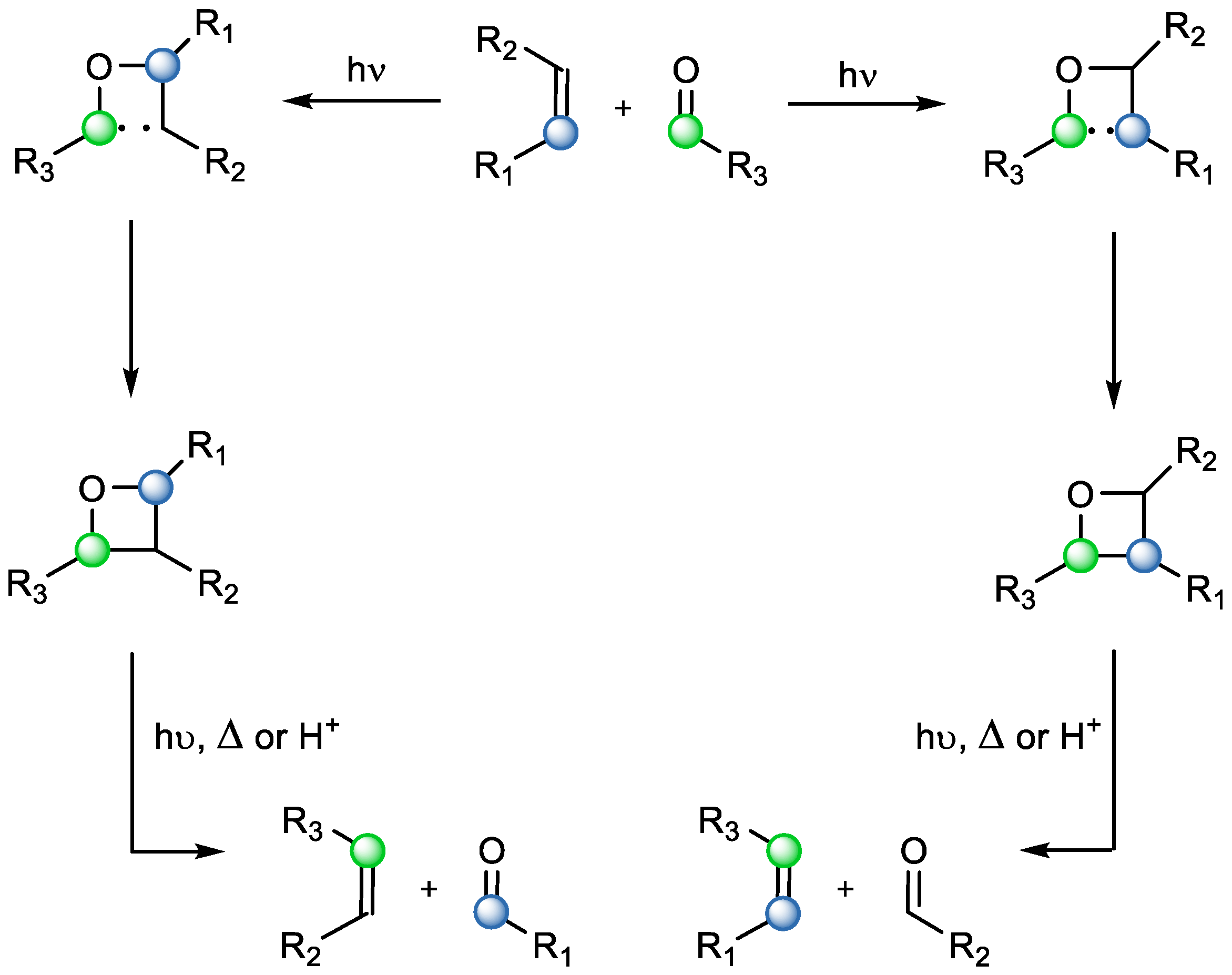
Most photochemical COM reactions involve a [2+2] photochemical cycloaddition followed by fragmentations. The first part is the well-established Paternò–Büchi reaction, which can lead to different regioisomers of the oxetane, depending on the stability of intermediates generated upon light irradiation (
Scheme 7) [
14]. However, different reaction types arise whether fragmentations are based on either heat (pyrolytic), acid catalysis (protolytic), or light (photolytic), which are briefly discussed below.
Metathesis with Paternò–Büchi oxetane formation, followed by pyrolytic fragmentation, is the least used method for the synthesis of natural products. The problem with this method is the high heat required for the fragmentation based on a retrocycloaddition step. Harsh conditions make the reaction scope highly limited and thus not useful for the synthesis of natural products [
13].
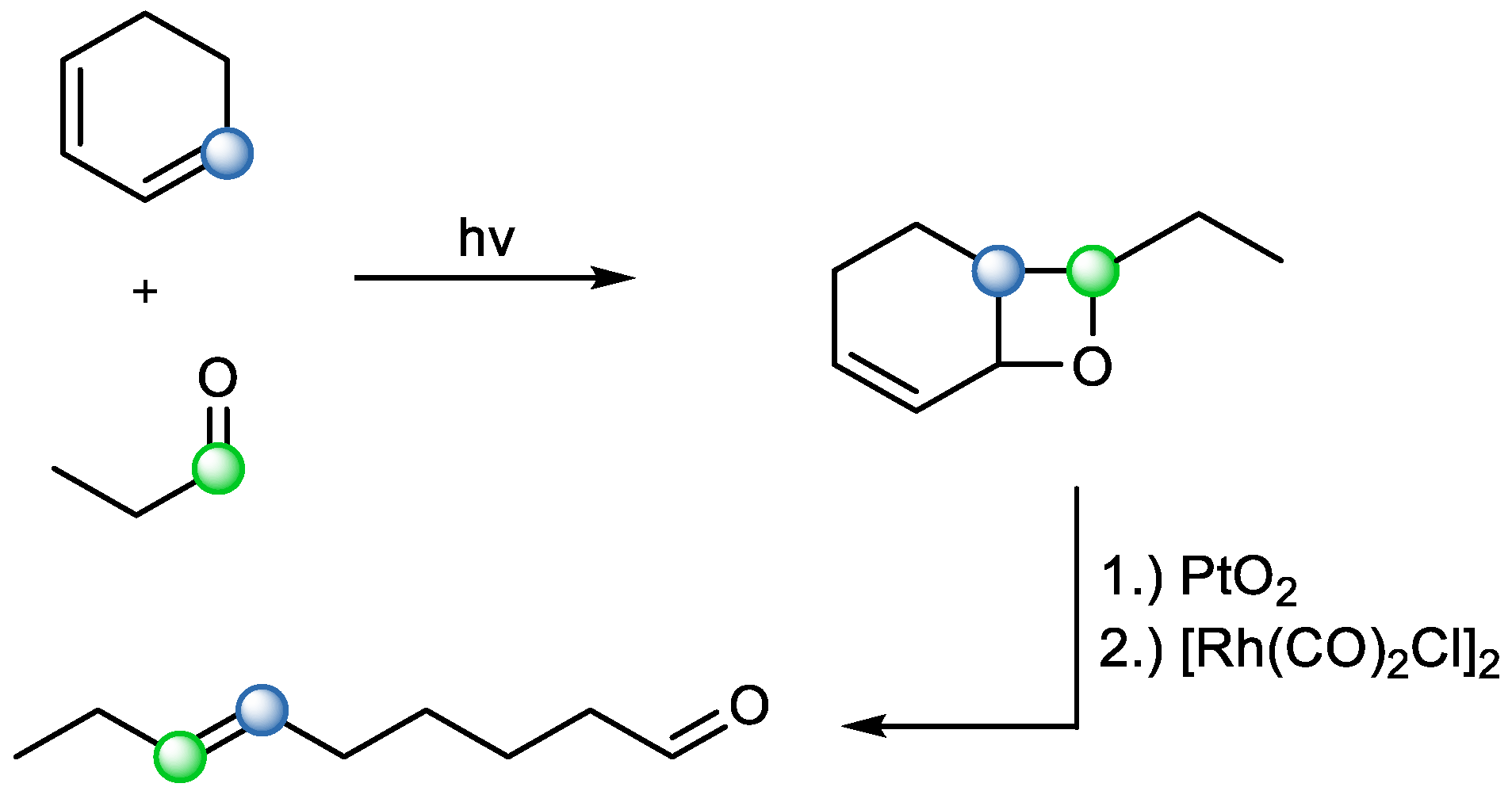
Fragmentation methods based on acid catalysis enable broader use. This is possible using both Brønsted and Lewis acids. The ring opening happens via a carbocation intermediate. This method was used in 1975 for the synthesis of
trans-non-6-en-1ol (
Scheme 8). The main step of the synthesis was based on the photochemical [2+2] cycloaddition of cyclohexa-1,3-diene and propionaldehyde, followed by catalytic hydrogenation, and Lewis acid-catalyzed fragmentation [
13].
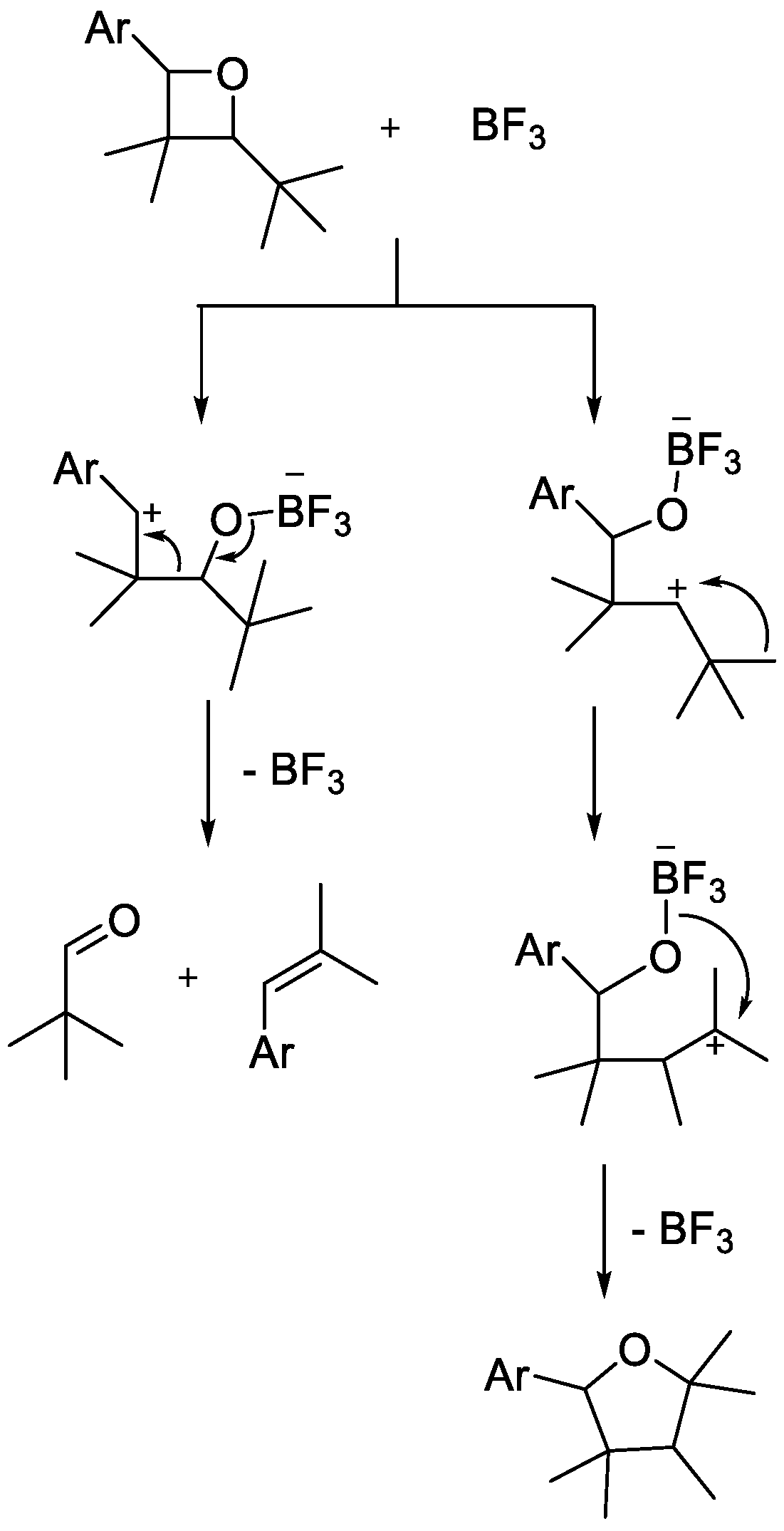
Similar compounds have been prepared using
p-toluenesulfonic acid as a Brønsted acid catalyst for the fragmentation of photochemically prepared oxetane [
15]. However, the fragmentation step does not always lead to the desired metathesis product. Instead of producing a new alkene, ring expansion of an oxetane can occur, which leads to a tetrahydrofuran derivative. The product of fragmentation is predominantly determined by oxetane’s substituents. While electron-rich substituents stabilize the intermediate carbocation and thus tend to favor the ring-opening metathesis reaction, electron-poor substituents destabilize the intermediate cation, and this leads to ring expansion via 1,2-methyl shift (
Scheme 9) [
16].
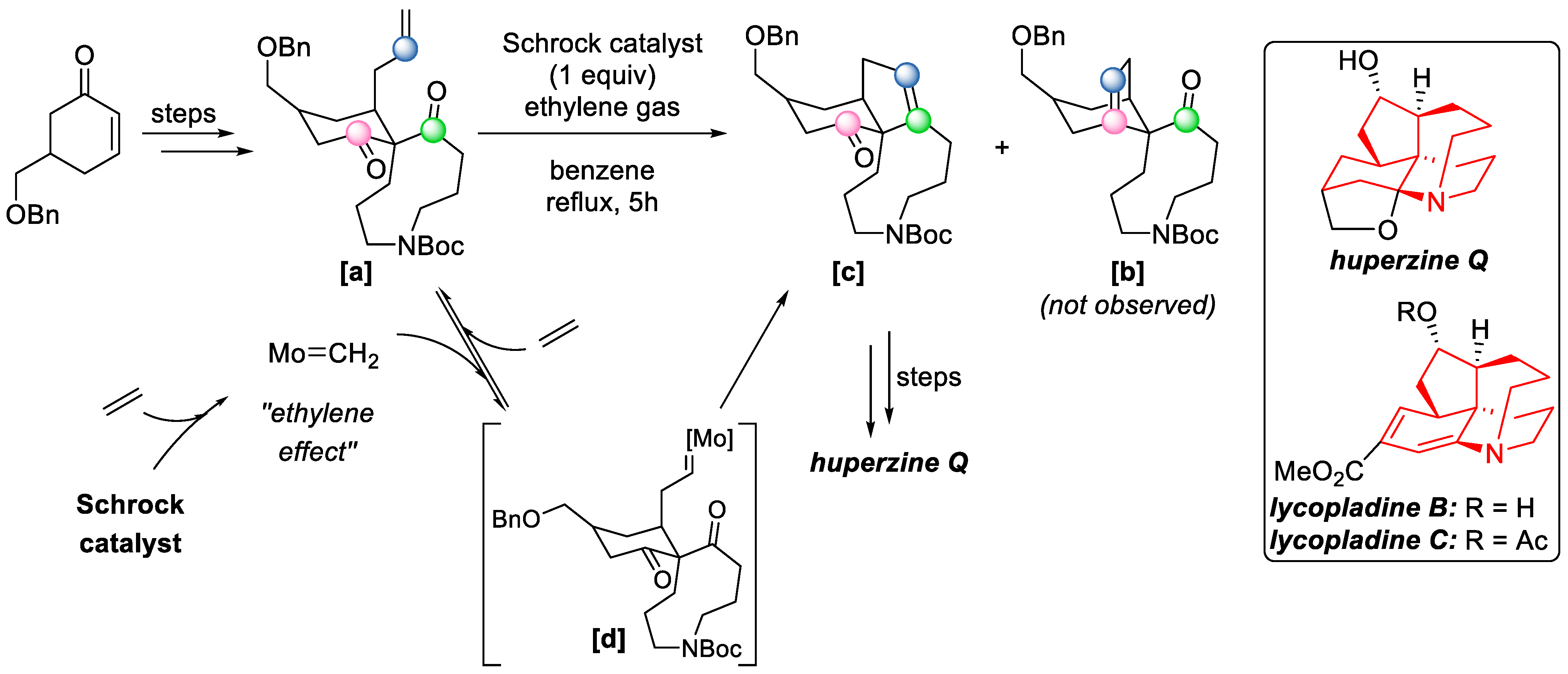
Another possible way of oxetanes, previously prepared via Paternò–Büchi reaction, to undergo retrocycloaddition, is via a photochemical mechanism. This can happen by direct excitation of their own chromophores. After this, both the Paternò–Büchi reaction and the ring-opening step occur photochemically; the reaction cannot always be stopped after the first step. However, there are limitations regarding this synthetic approach. For example, Hong et al. wanted to apply photochemical carbonyl–olefin metathesis to the total synthesis of (−)-Huperzine Q, (+)-Lycopladine B, (+)-Lycopladine C, and (−)-4-epi-Lycopladine D, but the reaction failed (
Scheme 10). They reported that the precursor
[a] decomposed. Thus, they had to resort to metal-catalyzed metathesis methods (Scheme 23) [
17]. Alternatively, ring-opening can be induced by photoinduced electron transfer (either photooxidation or photoreduction) using a photosensitizer like thiapyrylium salt, but these methods have been reported only for the synthesis of small molecules [
13].
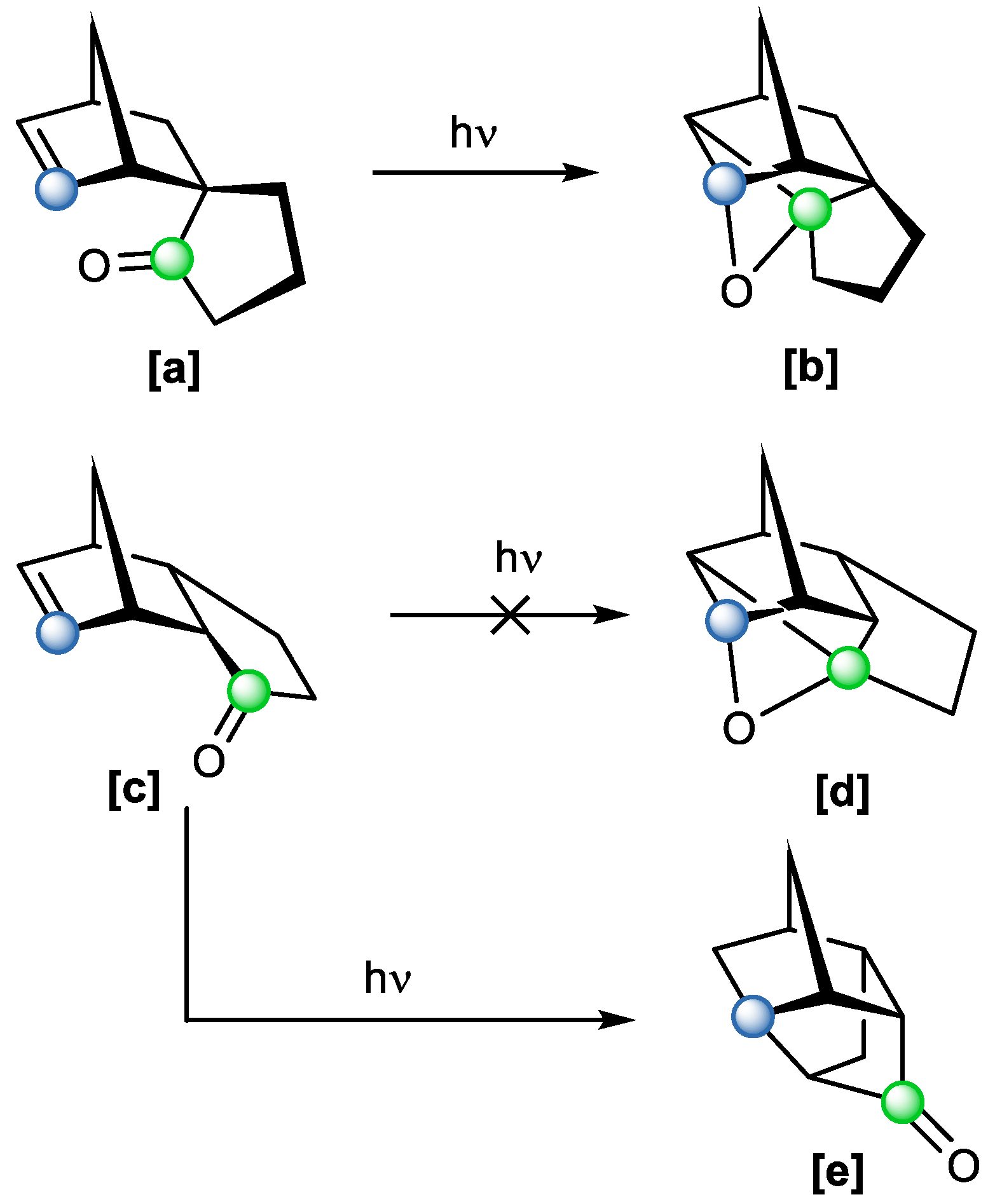
Sauers and coworkers encountered problems when trying to access the Paternò–Büchi product in similar polycyclic compounds with different ring strains [
18]. While the spiro compound with a five-membered ring
[a] gave a desired Paternò–Büchi product with oxetane moiety
[b], this did not happen in the case of a similar compound with a fused five-membered ring
[c] (
Scheme 11). The carbonyl functional group was not involved in the reaction and remained untouched. The formation of a non-expected product
[e] and the subsequent failure of the Paternò–Büchi reaction was rationalized with a high ring strain and the π-π* excited state of the double bond [
18].
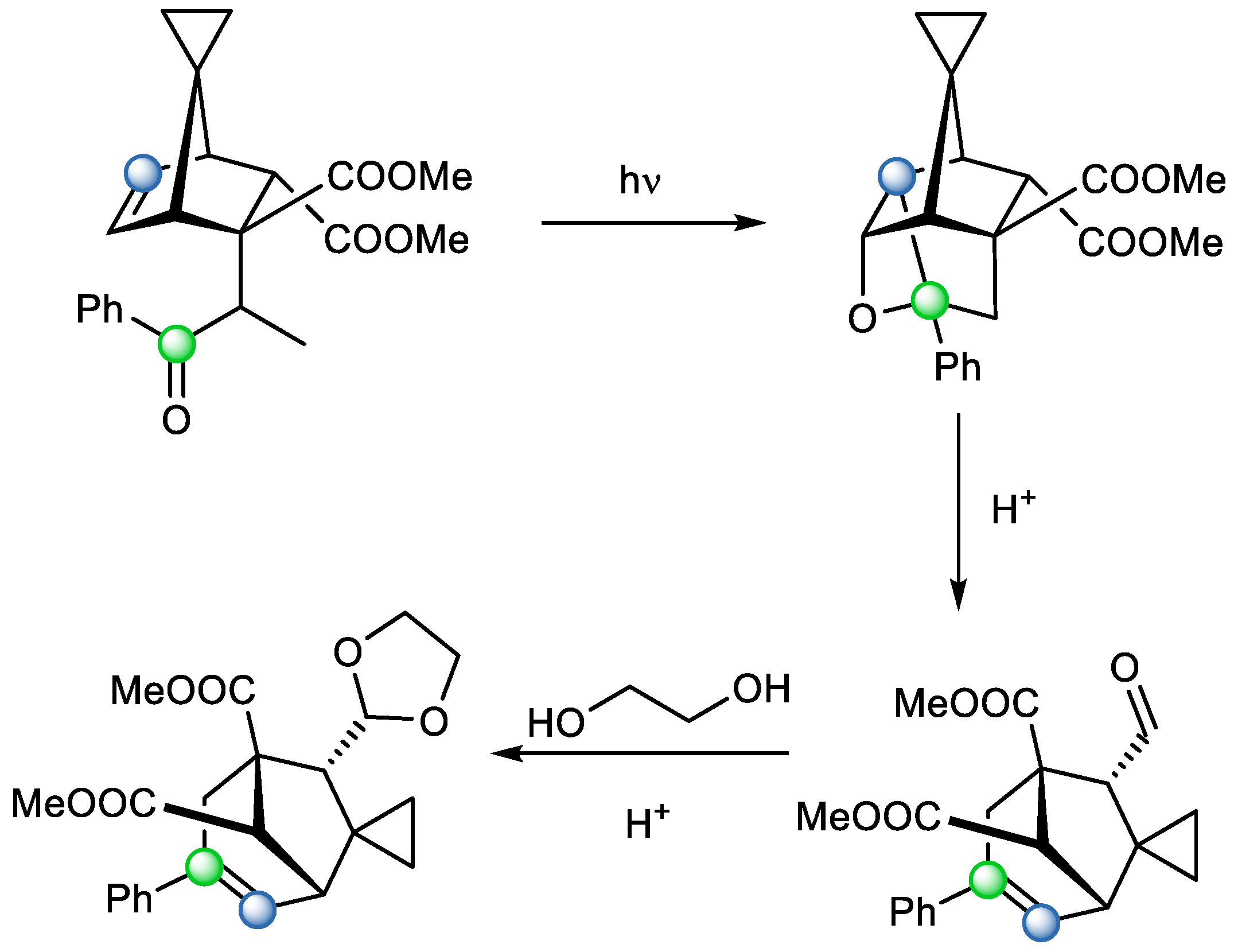
Since substrates that can successfully undergo metathesis reactions based on Paternò–Büchi are quite limited, Valiulin et al. [
19] suggested installing a chromophore on a substrate via Diels–Alder reaction followed by Michael’s addition to make the substrate suitable for further Paternò–Büchi reaction. They proposed introducing benzoyl or heterobenzoyl moieties. This also improved the reactivity of conformationally constrained precursors. They were further fragmented in mildly acidic conditions. However, in certain cases where aldehyde groups were generated, this led to epimerization, which could be an issue for application in natural product synthesis. They were able to partially solve the problem with the transformation of aldehyde to acetal (
Scheme 12).
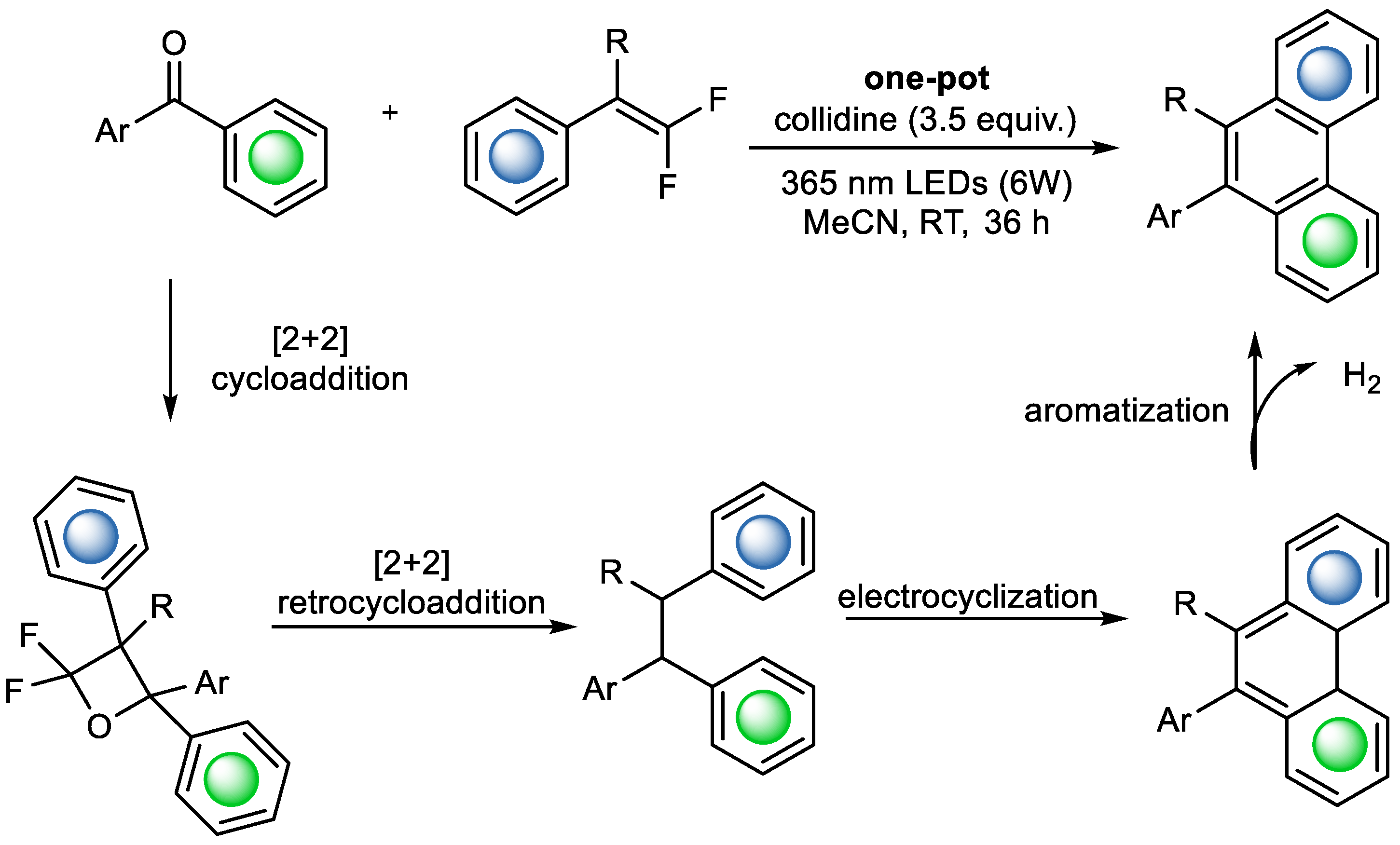
Although most examples of photo-induced COM reactions focus on obtaining a mostly saturated structure, it is also feasible to obtain polyaromatic structures with the method. In 2024, Zhang et al. published a one-pot cascade reaction combining photoinduced [2+2] cyclization, thermally controlled [2+2] retrocyclization, and dehydrogenative cyclization. They used aromatic ketones and aromatic
gem-difluoroalkanes to obtain phenanthrenes, which can be used for drug design (
Scheme 13) [
20].
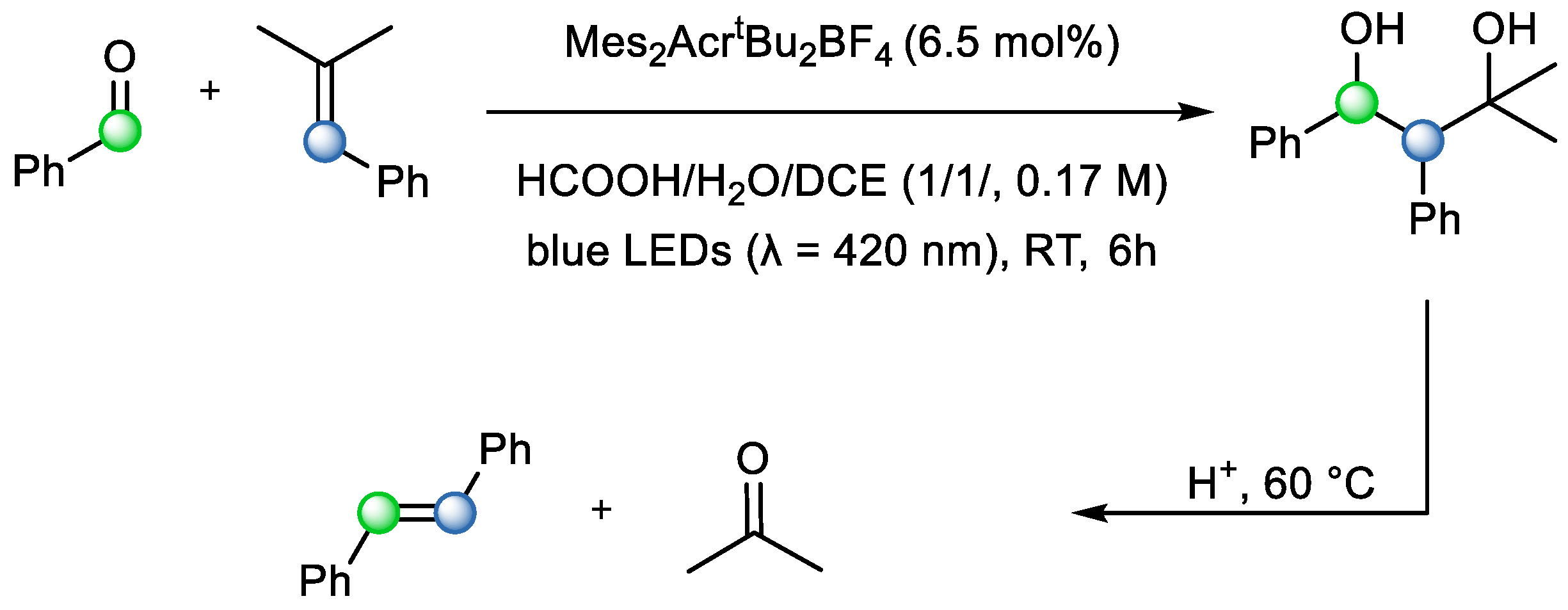
Even though the utilization of photochemical methods in carbonyl–olefin metathesis is usually considered outdated and connected or even equated to Paternò–Büchi [2+2] cycloaddition forming oxetanes, this is not always the case. In 2018, Pitzer et al. presented a method for visible-light mediated carbonyl–olefin cross-metathesis, which is a unique example of an intermolecular reaction that does not result in ring-closing or -opening in the final product (
Scheme 14). The mechanism was rationalized as a hole catalysis reaction, where the formation of an initial radical cation intermediate triggers the remainder of the mechanism. It was used to promote the formation of 1,3-diols from aldehydes and styrenes, which were further fragmented under acidic conditions. The advantage of this approach is synthesis via energetically less demanding 1,3-diols instead of oxetanes. This approach was also more orthogonal, requiring no external additives, and afforded a highly stereoselective reaction [
21]. The concept is based on photoredox catalysis, where carbonyls can be used as radical acceptors, resulting in the formation of benzylic alcohols (diols). They are further fragmented via acid-induced Grob-type fragmentation that predominantly produces
E-stereoisomers (
Scheme 14) [
21].
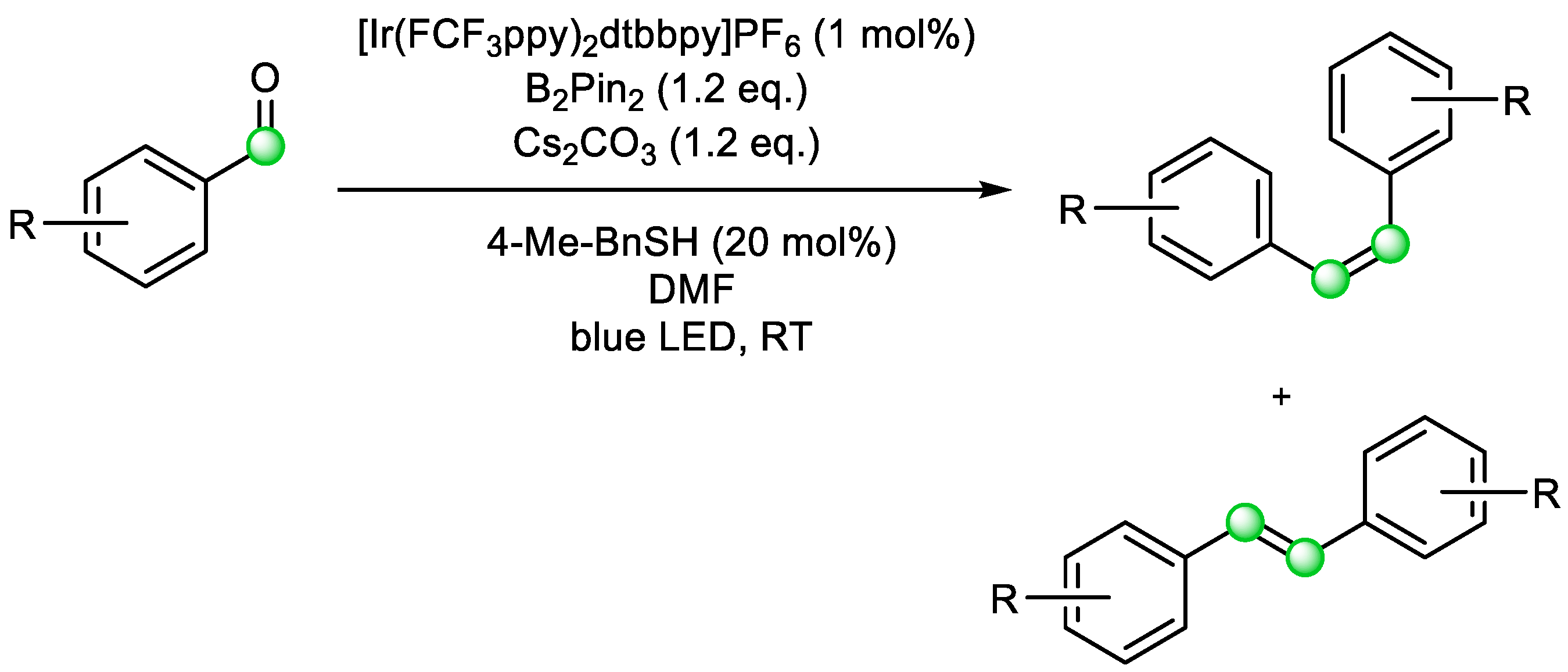
This photoredox approach has enabled different substrates to be employed using the same mechanistic framework. In 2019, Wang et al. expanded this strategy to a ‘carbonyl-carbonyl metathesis’ method (
Scheme 15). They used visible light and an iridium catalyst to obtain an alkene from a pair of aldehydes. The reaction is similar to the McMurry reaction, where the carbonyl groups are activated by a diboronate compound to drive the deoxygenation process. The reaction was designed to use thiol as a co-catalyst. After deprotonation using Cs
2CO
3, the sulfur anion reduces the photoexcited iridium catalyst, followed by SET from the Ir(II) to benzaldehyde. Thiol is further regenerated in a catalytic cycle. The research group has shown both homocoupling and cross-coupling as possible reaction options (
Scheme 14) [
22]. Although not a formal COM reaction, this example illustrates how the rational use of COM reaction mechanisms can inspire the development of new synthetic methods.
3. Metal Alkylidene Mediated Carbonyl–Olefin Metathesis
Compared to the multi-step Paternò–Büchi process, which is often time-consuming and inefficient, the COM reactions mediated by metal alkylidene complexes have shown a great advantage as the metathesis is often performed as a single-step process. As extremely successful reagents for olefin–olefin metathesis reactions, metal alkylidene complexes have also shown considerable promise in carbonyl–olefin metathesis (COM) reactions [
2]. A range of metal-based catalysts with diverse ligand architectures has been developed, enabling broad substrate scope and compatibility with structurally complex molecules [
2]. The following section will discuss notable examples of successful metal alkylidene complexes and their development for COM reactions. Selected applications in the synthesis of structurally complex molecules will also be highlighted to illustrate their synthetic utility.
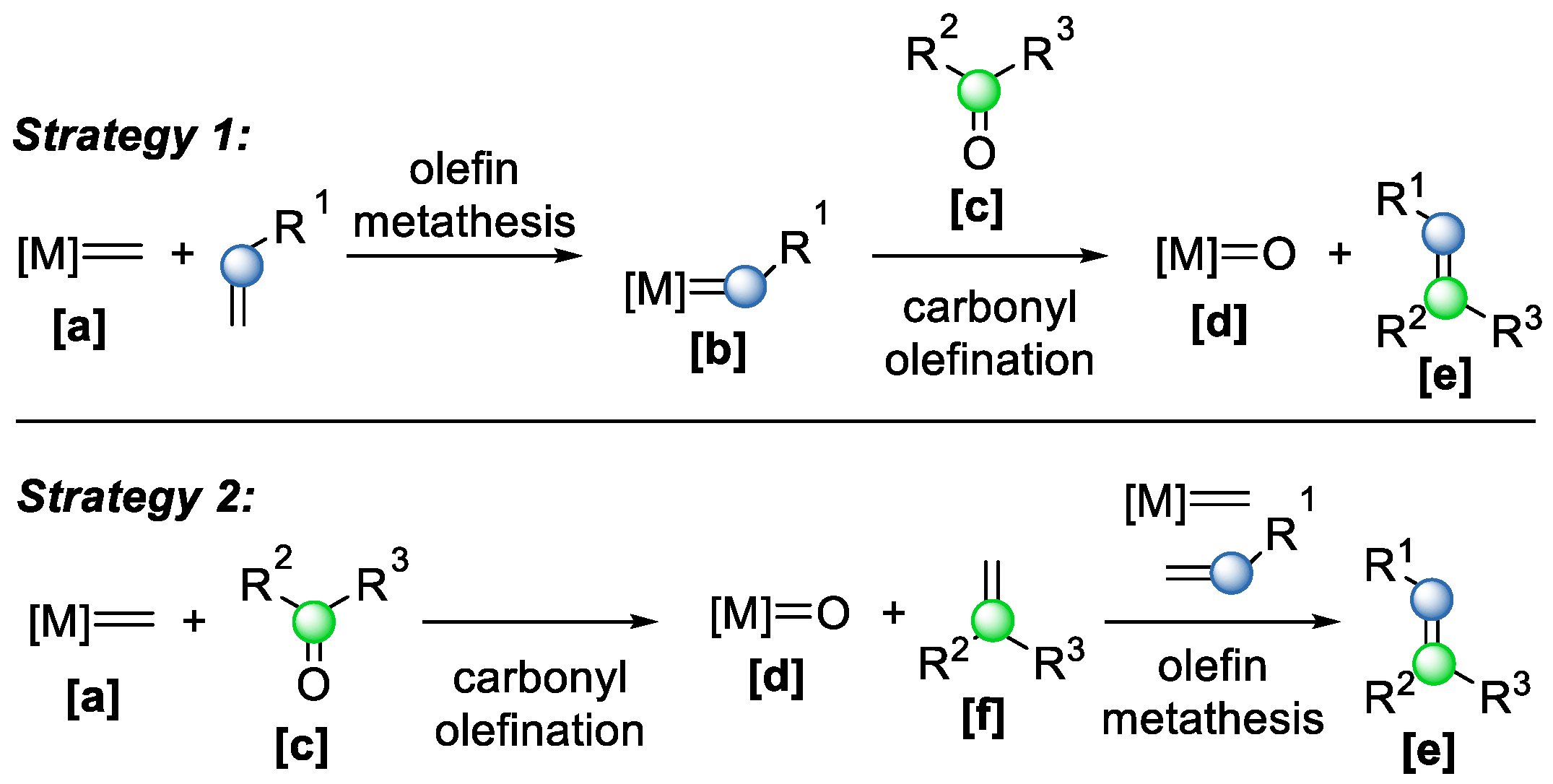
Generally, two main mechanistic strategies are employed in metal alkylidene-mediated COM reactions (
Scheme 16) [
2]. In the first strategy, a metal alkylidene complex
[a] reacts with an olefin to form another alkylidene intermediate
[b], which subsequently undergoes carbonyl olefination with a carbonyl compound
[c], yielding the desired olefin product
[e] and an inactive metal–oxo species
[d]. In the second strategy, the reaction begins with carbonyl olefination between the metal alkylidene and the carbonyl compound, producing an intermediate olefin
[f] and the same metal–oxo byproduct
[d]. This olefin then undergoes olefin–olefin metathesis with a second equivalent of the metal complex to generate the ring-opening metathesis (ROM) product
[e]. In both strategies, however, the formation of the unreactive metal–oxo species
[d] renders the process stoichiometric rather than catalytic, which limits the atom economy and efficiency of the transformation.
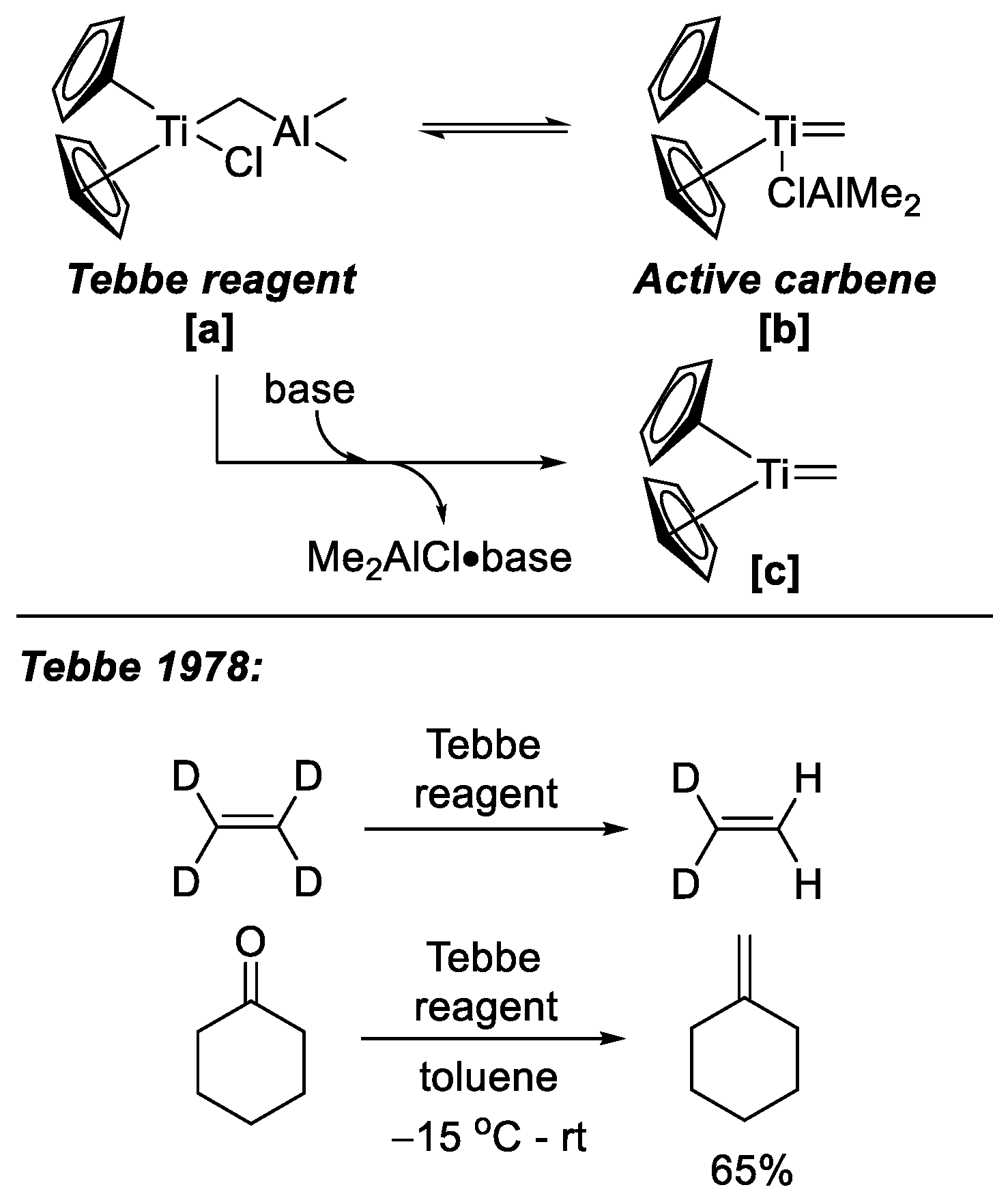
In 1978, Tebbe and coworkers developed a set of methylene-bridged compounds
[a] (
Scheme 17), known as the Tebbe reagent, which showed reactivity upon both regular olefin metathesis and Wittig-like reaction with carbonyl groups [
23]. The Petasis reagent, i.e., a simpler dimethyltitanocene equivalent to the Tebbe reagent, was reported later by Petasis et al., which exhibited a similar reactivity [
24]. In both cases, the reactivity arises from the in situ generation of a titanocene methylidene species
[b], which possesses a metal alkylidene structure analogous to those found in classical metathesis catalysts. The metathesis activity of this species can be significantly enhanced by the addition of a strong Lewis base co-catalyst, which facilitates the formation of the active metathesis reagent [
25]. With mild conditions and promising yield, such compounds have shown great success in several syntheses of different complex molecules.
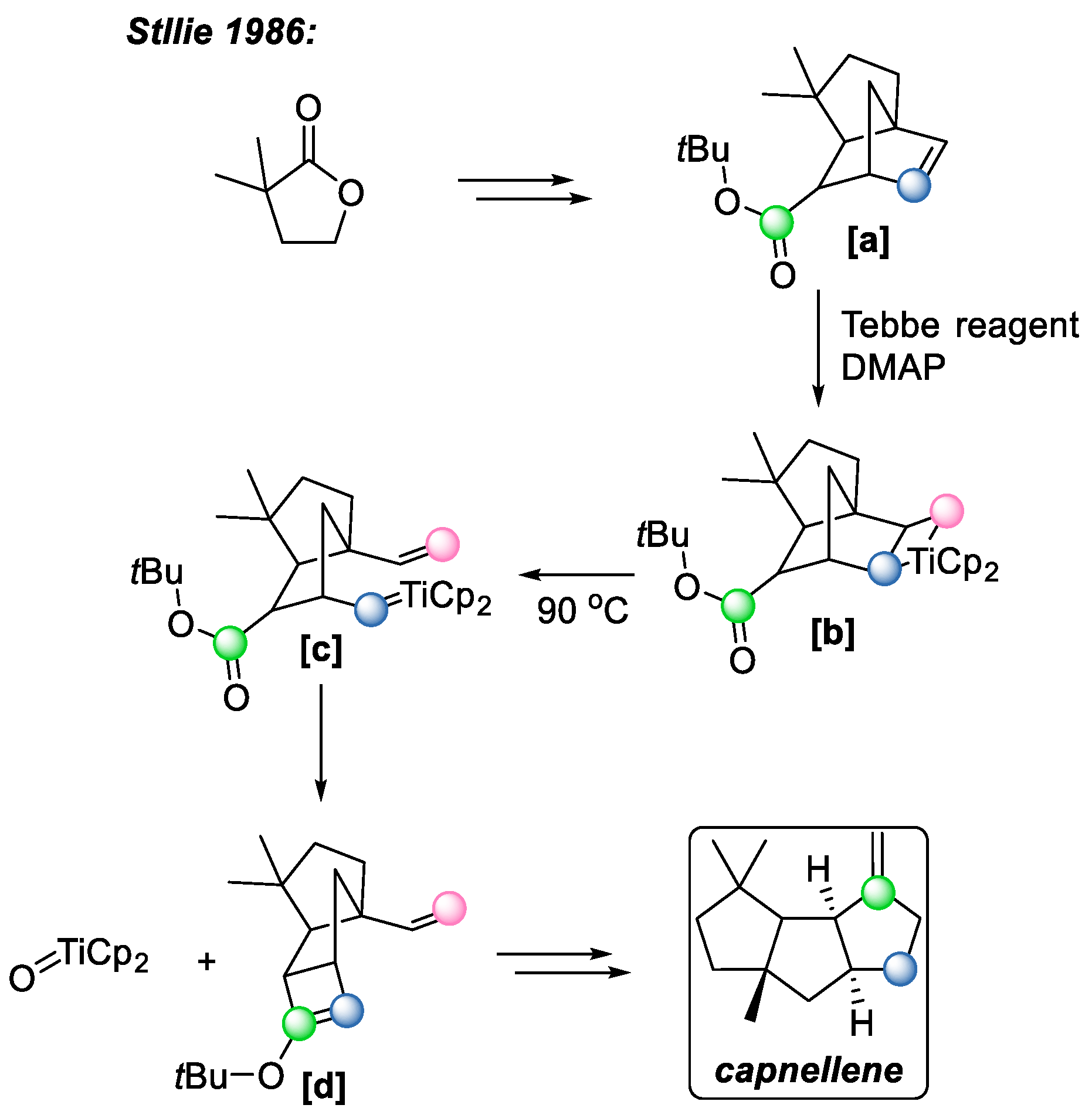
In 1986, Stille et al. achieved the total synthesis of capnellane [
26] (
Scheme 18), which is a natural product that attracts great interest from a synthetic point of view due to its challenging
cis-anti-cis tricyclo-undecane skeletal framework. The key conversion during the synthetic process involved the transformation from bicyclic intermediate
[a] to tricyclic product
[d] via the Tebbe reagent. This transformation mirrors the first mechanistic pathway outlined in
Scheme 16. While transformation from intermediate
[a] to
[c] resembles olefin metathesis, the following carbonyl olefination provides the intramolecular formation of the four-membered ring, and the metal–oxo species. Notably, these transformations occur without altering the stereochemistry of the final product, which is predetermined upon the formation of intermediate
[a].
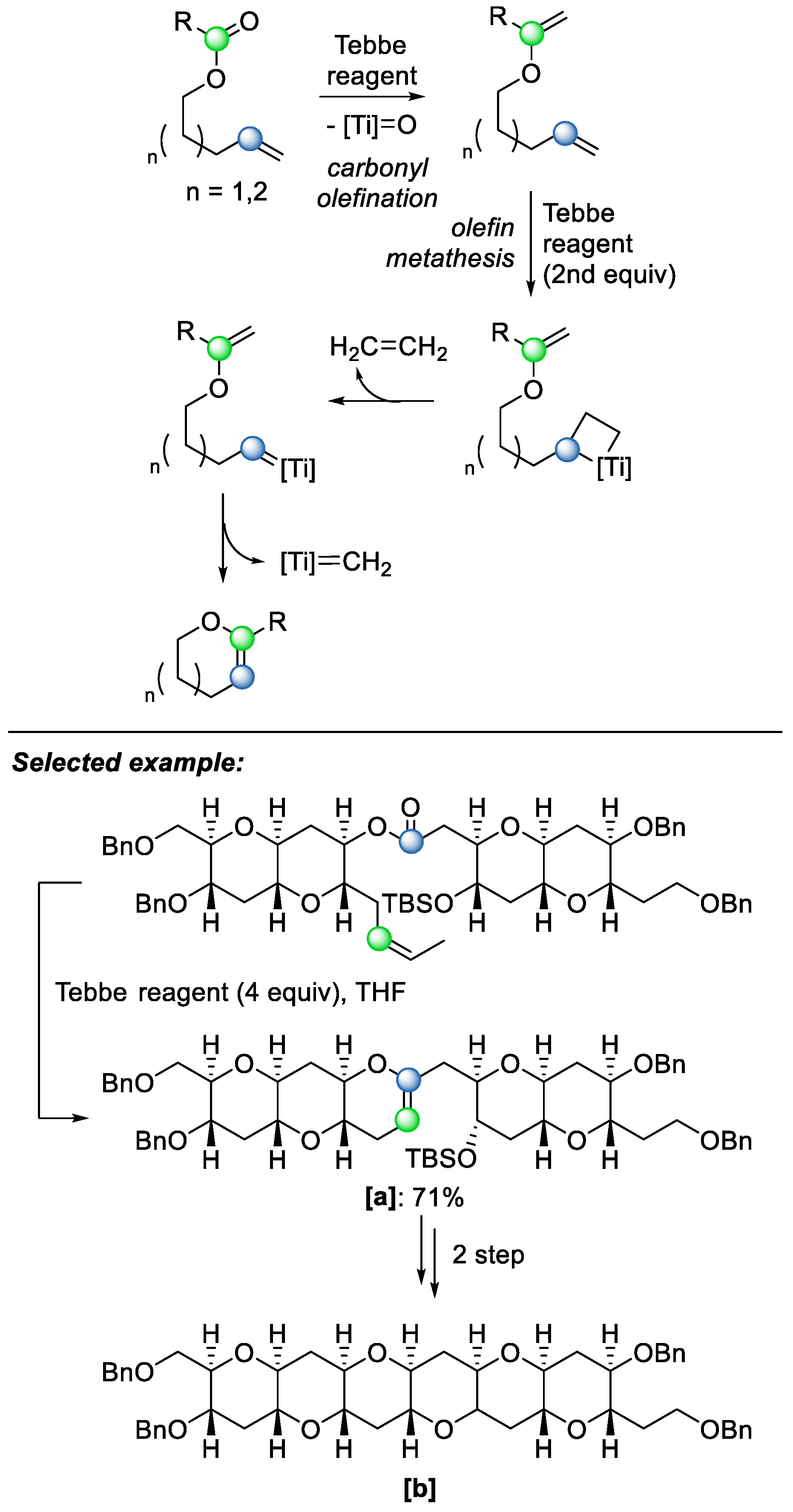
The same reagent can also be used for the second strategy. In 1996, Nicolaou and coworkers exploited this strategy to produce cyclic enol ethers from olefinic esters directly, using an excess equivalence of Tebbe reagent [
27]. The mechanism begins with the olefination of the carbonyl group by the Tebbe reagent, followed by a second equivalent of the Tebbe reagent participating in the reaction, yielding intramolecular olefin–olefin metathesis (
Scheme 19). The reaction scope was investigated by employing the methodology of a variety of substrates, including a hexacyclic skeleton
[b], which is reminiscent of the structures of marine neurotoxins such as brevetoxin B, ciguatoxin, and maitotoxin.
While successful applications of the Tebbe reagent have been developed in various synthetic categories, the Lewis acidity of the Tebbe reagent can often lead to the decomposition of highly substituted targets or substrates which have functionality sensitive to Lewis acid [
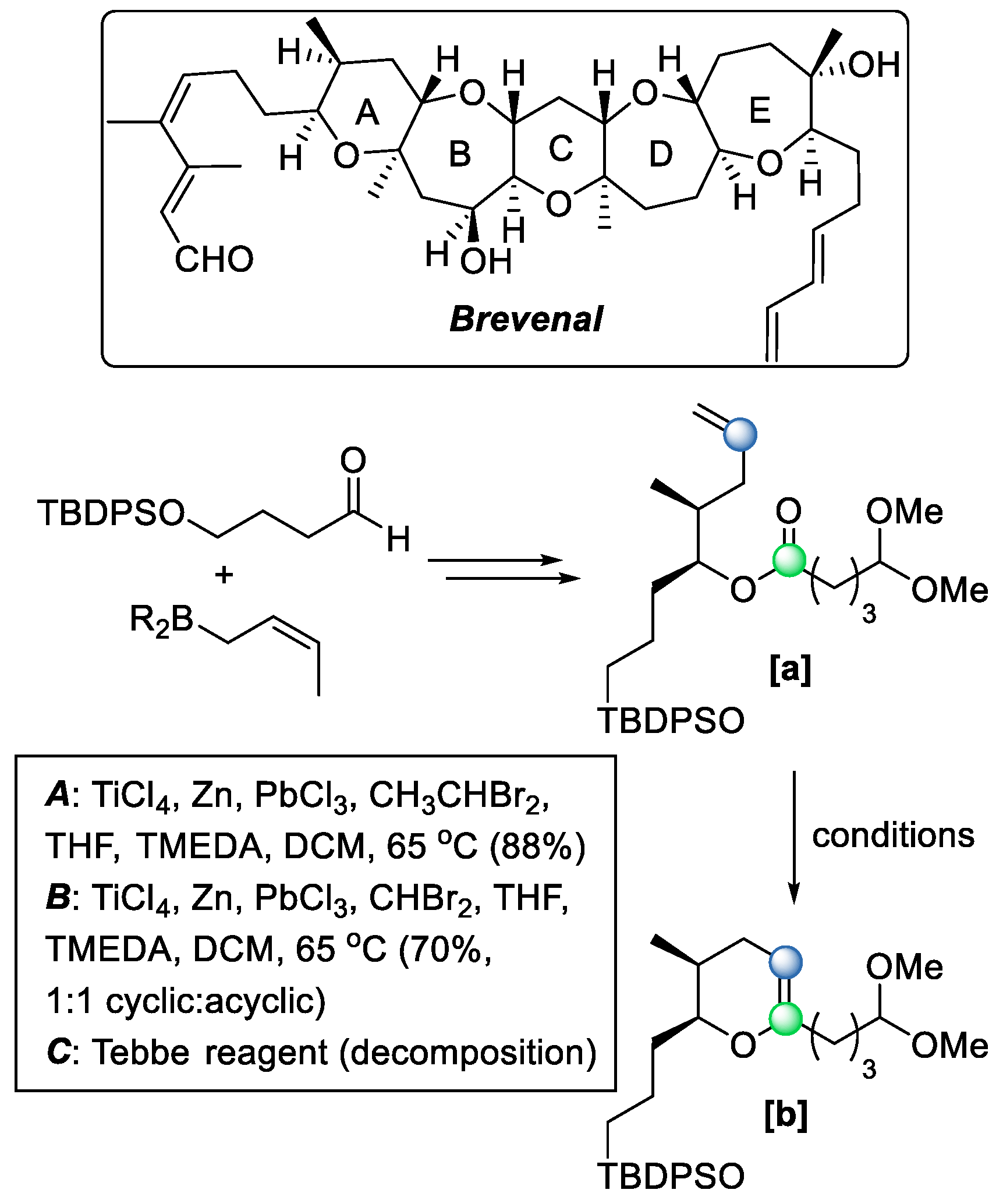
28]. For instance, in 2011, Zhang and coworkers delivered the total synthesis of the marine ladder toxin brevenal [
29] (
Scheme 20), where the conversion of olefinic ester
[a] to enol ether
[b] is required during the building of the A ring (
Scheme 20). However, treating ester
[a] with the Tebbe reagent leads to the decomposition of the starting material (condition
C). In such cases, the Takai–Utimoto reagent, with diminished Lewis acidity, can serve as a useful alternative.
Similar to the expected outcome with the Tebbe reagent, the Takai–Utimoto condition allows an intramolecular COM reaction to form olefinic ester
[a] via the strategy I reaction mechanism, where an olefin metathesis reaction occurs first, followed by a carbonyl olefination reaction forming the cyclic enol ether metathesis product [
28]. In the synthesis of brevenal [
29] (
Scheme 20), the use of the Takai–Utimoto condition successfully afforded ring A via an intramolecular ring-closing reaction in the skeleton (condition
A,
B). A similar strategy was also used for the construction of the C, D, and E rings of Brevenal in advanced stages of the synthesis, affording yields of up to 88%. This clearly showcases the broad scope and mild conditions provided by the Takai–Utimoto conditions.
The Takai–Utimoto reaction condition utilizes a combination of alkyl halide, PbX
2, TiCl
4, and Zn. In 1978, Takai and coworkers first reported that the combination of alkyl halide, TiCl
4 (or AlMe
3), and Zn yields a Wittig-like olefination of carbonyl compounds [
30]. In 1994, they further realized that the addition of PbX
2 accelerates the reaction by transmetallation [
31]. The authors proposed a mechanism (
Scheme 21), explaining the effectiveness of such a combination: two successive rounds of transmetalation from the dihalogenated compound
[a] generate dizinc intermediate
[e]. This intermediate then reacts with TiCl
4 to form a titanium-containing geminal dimetallic compound
[f], which resembles the Tebbe reagent. In situ, this species forms a titanium-based alkylidene
[g], which undergoes the reversible olefination of the carbonyl compound. This strategy has been used extensively in the total synthesis of natural compounds due to its mild condition and broad scope [
32,
33,
34]. Later iterations of the Takai–Utimoto conditions featuring Zr instead of Ti (aka, Takai–Lombardo conditions) have also been employed for the total synthesis of natural products such as (+)-Waixenicin A and Cotylenin A [
35,
36].
Other than the Tebbe reagent and Takai conditions, where the active carbene species is generated in situ, pre-formed metal alkylidene reagents are well known for their excellent performance in promoting COM reactions. For example, the Schrock reagent is a set of molybdenum-based alkylidenes
[a] [
37] which have shown reactivity in Wittig-like carbonyl olefination reactions. In 1993, Fu and Grubbs first realized the potential of such reagents in intramolecular COM reactions (
Scheme 22) [
38]. Via strategy 1 (
Scheme 16), the molybdenum-based carbene
[a] undergoes [2+2] type cyclisation with alkene
[b], giving intermediate
[c], and the new alkylidene
[d] is formed following the retro-[2+2] reaction. The desired cyclized product is formed following a carbonyl olefination reaction through metallaoxetane intermediate
[e]. The preference for an initial olefin metathesis reaction over the carbonyl olefination ensures the ring-closing COM occurs instead of the formation of an acyclic diene.
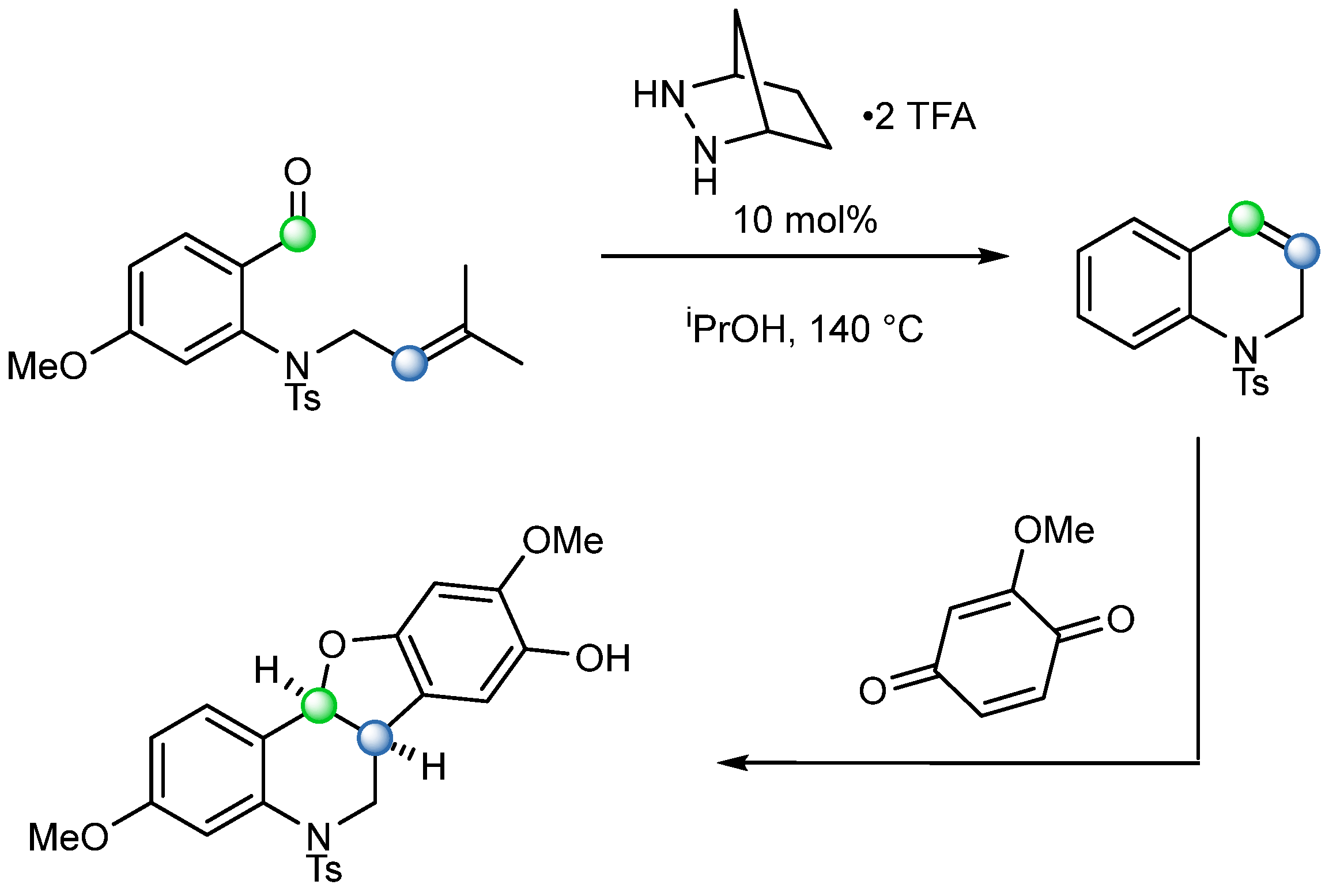
So far, there have been several examples in which Schrock’s reagent was utilized for the synthesis of complex molecules. In 2015, Hong and coworkers finished the total synthesis of (−)-Huperzine Q and (
+)-Lycopladines B and C [
39], a set of structurally unique natural products with a tetracyclic skeleton with a dienamine moiety (
Scheme 23). Schrock catalyst-mediated COM cyclisation was the key step for the construction of the cyclopentanone ring skeleton. The reaction was regiospecific, affording only the desired product
[c], due to the high torsional strain that exists in the disfavored bridged product
[b]. The reaction condition was optimized with the presence of ethylene gas, which presumably increased the catalyst reactivity by means of converting the bulky carbene into the more active species [Mo=CH
2]. The excess of ethylene also helped to prevent the remaining molybdenum alkylidene intermediate
[d] forming side products by prompting recovery of the starting material
[a].
Although the molybdenum-based carbene has shown great success in mediating COM reactions, the extreme sensitivity to air and moisture of such compounds has caused difficulty in storage, and decomposition issues [
40] limit the reaction solvent and the scope of functional groups that are tolerated, thus restricting the adoption of such a method in synthetic methodology. In 1995 and 1999, two generations of ruthenium-based carbene complexes were reported by the group of Grubbs (
Scheme 24) [
40,
41], with improved air and water stability; the reagents had shown great capacity in mediating the olefin–olefin metathesis reaction. In 2016, Chakraborty and co-workers [
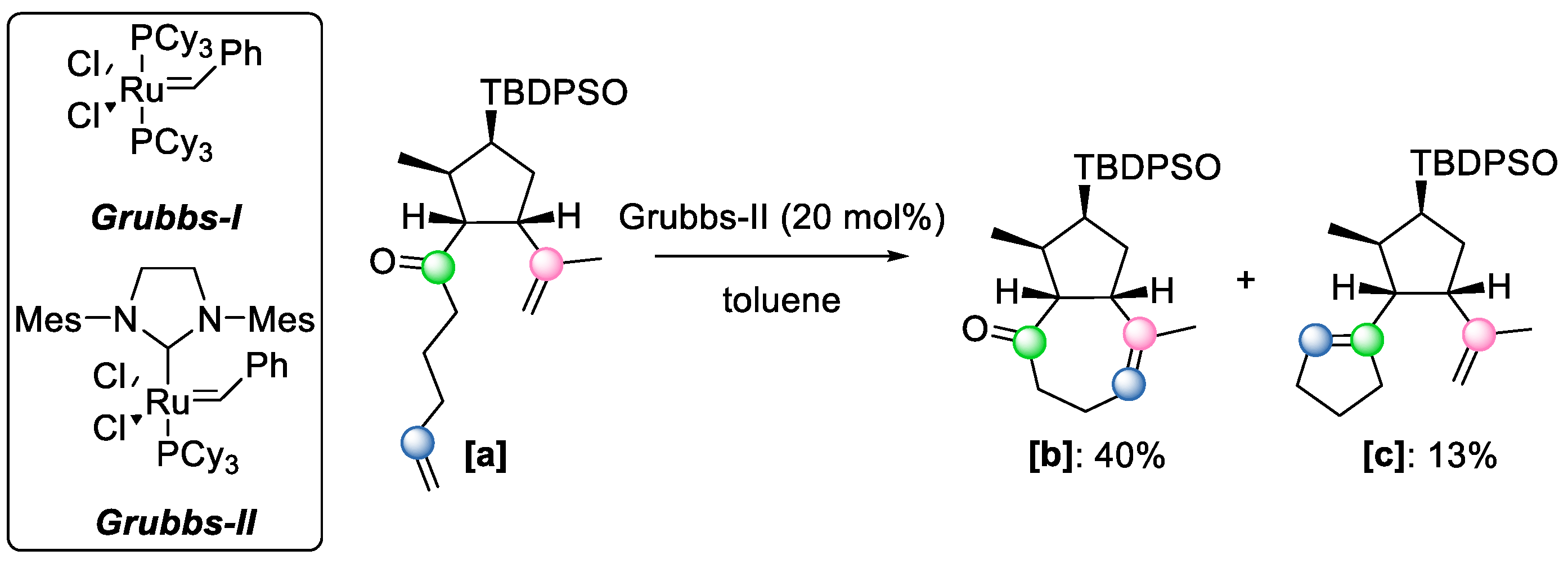
42] observed the formation of COM byproduct
[c] while treating compound
[a] with the Grubbs II reagent for the intramolecular formation of a seven-membered ring
[b] (
Scheme 24), indicating the potential of such a compound in mediating COM reactions. The authors proposed that the mechanism proceeds through strategy 1 (
Scheme 16).
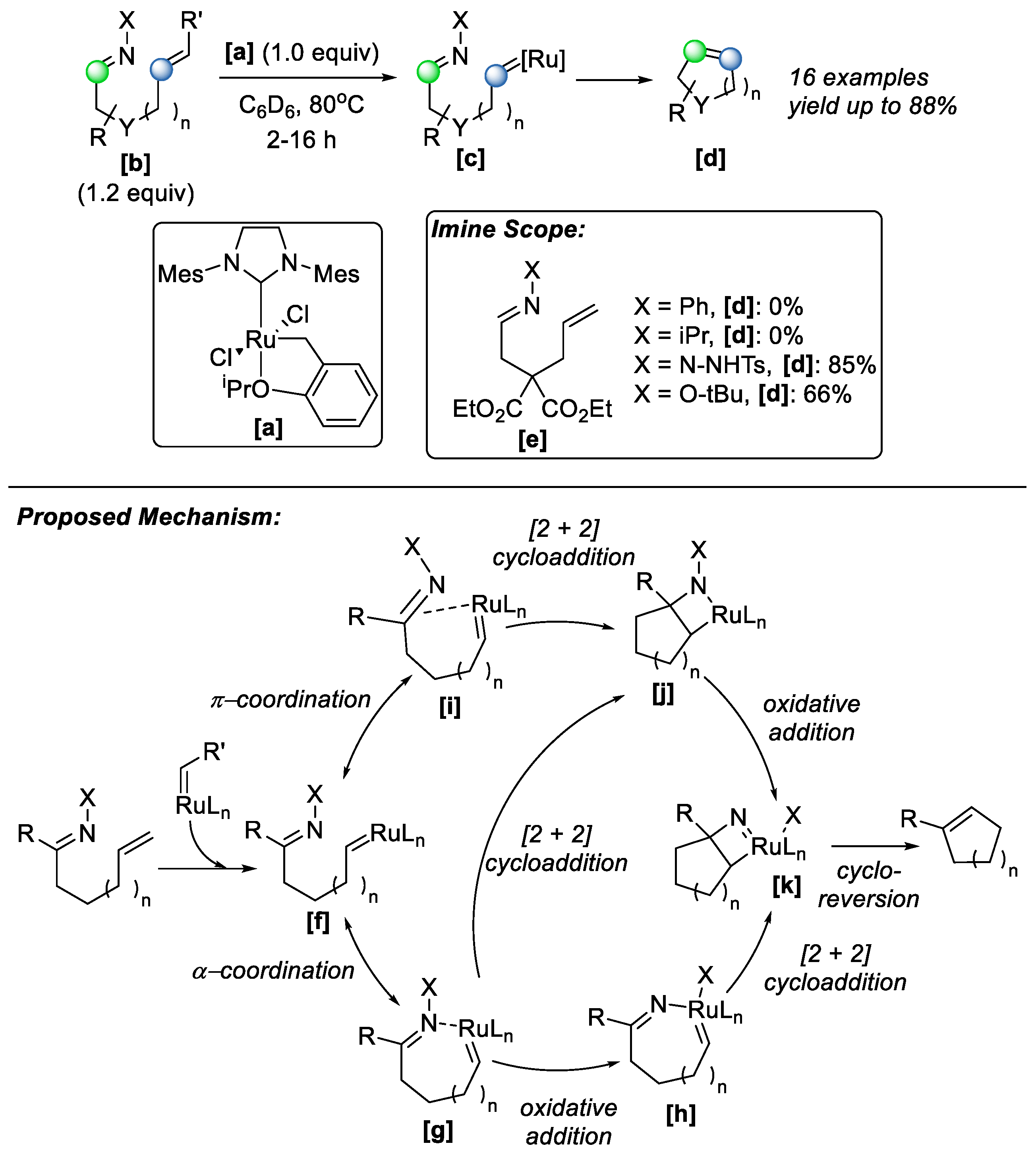
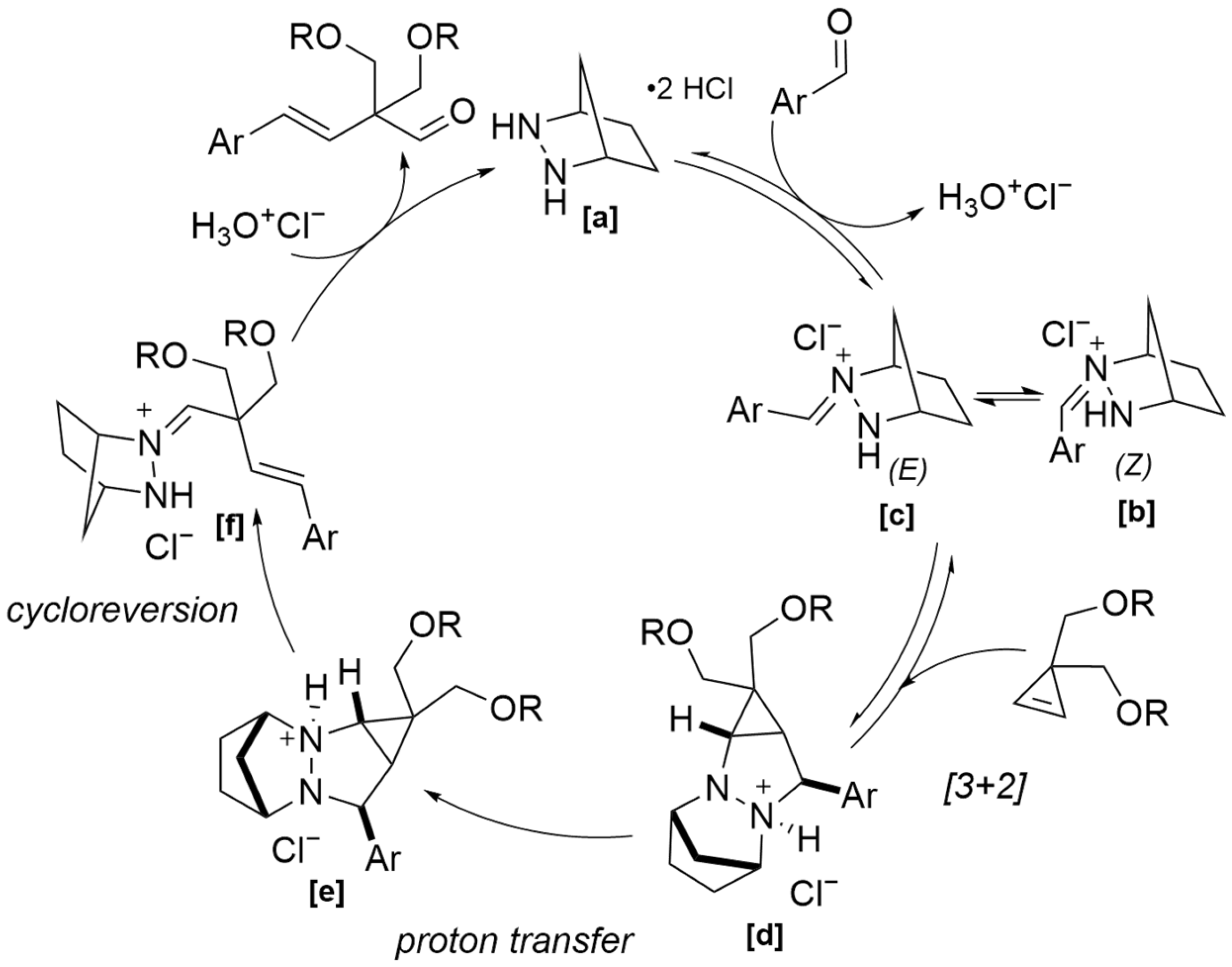
Interestingly, the Grubbs reagent was also successfully exploited in the untapped topic of imine–olefin metathesis. In 2022, Nasrallah and co-workers reported that several commercially available Grubbs II-type ruthenium alkylidene complexes can mediate imine–olefin metathesis in stoichiometric amounts [
43], with alkylidene
[a] being the optimized catalyst (
Scheme 25). The substrate scope was investigated, revealing that electron-rich imines, such as hydrazones and oximes, exhibit enhanced reactivity. This improved performance was attributed to their lower polarity and higher HOMO energy levels, which promoted productive interactions with the metal alkylidene species. An uncommon oxidative step occurs in this reaction, making it distinct from all other metathesis pathways of ruthenium alkylidenes. The Ru–alkylidene intermediate
[f] formed with the substrate undergoes a two-step sequence of [2+2]-cycloaddition/oxidative addition reactions (in any order), yielding aza-ruthenocyclobutane complex
[k], which, upon cycloreversion, generates the metathesis product [
44]. Under optimized conditions, the methodology demonstrated broad functional group tolerance: substrates bearing amides, halides, and Lewis-basic functionalities, including protected amines, were well tolerated. A diverse range of cyclized products was obtained, with yields reaching up to 88%.
As with carbonyl–olefin metathesis (COM), imine–olefin metathesis reactions face a significant challenge: the formation of low-reactivity metal–imido species [M]=NR, which impedes catalytic turnover. However, in 2023, Fan et al. reported a cobalt-catalyzed olefin–imine metathesis reaction [
45] that enables intermolecular metathesis between enaminones
[a] and amidines
[b] (
Scheme 26). Notably, this transformation proceeds via a distinct mechanism in which a metal–alkylidene intermediate is not formed (
Scheme 26). The enaminone
[a] and amidine
[b] undergo an amine-exchange process to generate intermediate
[e].
The active cobalt catalyst [d], produced through halide abstraction from [CoCp*(CO)I2] by AgSbF6, coordinates with intermediate [e], yielding intermediate [f] and facilitating a cyclization step that yields intermediate [g] and regenerates catalyst [d]. A subsequent retro-[2+2] ring-opening of intermediate [g] releases the desired product, with HCN formed as a byproduct. However, the intermediate [e] can also isomerize to the cis-conformation and form a pyrimidine side-product [j], significantly reducing the reaction yield.
While the preceding example demonstrates the feasibility of catalyzing imine–olefin metathesis, the reaction generally proceeds with low yields and involves the release of HCN as a byproduct, posing significant safety concerns. These limitations underscore the fact that current developments in imine–olefin metathesis remain relatively underexplored. Importantly, further advancement in this area could be viewed as complementary to established metathesis methodologies, as Lewis acid-catalyzed carbonyl–olefin metathesis is often restricted to nucleophilic, terminal-inert alkenes. In contrast, the incorporation of functionalized amines in imine substrates offers broader electronic tunability of the C=N π-bond, potentially enabling a wider range of reactivity and more efficient interactions with metal alkylidenes [
43,
45]. Therefore, the continued exploration of imine–olefin metathesis is both needed and promising. Moreover, the application of Grubbs-type catalysts in this context—and in COM reactions more broadly—remains a valuable direction for future research.
5. Lewis Acid-Catalyzed Carbonyl–Olefin Metathesis
The potential of a one-step COM reaction via the in situ formation of an oxetane intermediate was recognized in the 1990s. During this period, several studies reported both inter- and intramolecular COM reactions catalyzed by metal-containing Lewis acids such as SnCl
4 and ZnCl
2 (EPZ-10, clay-supported ZnCl
2) [
47,
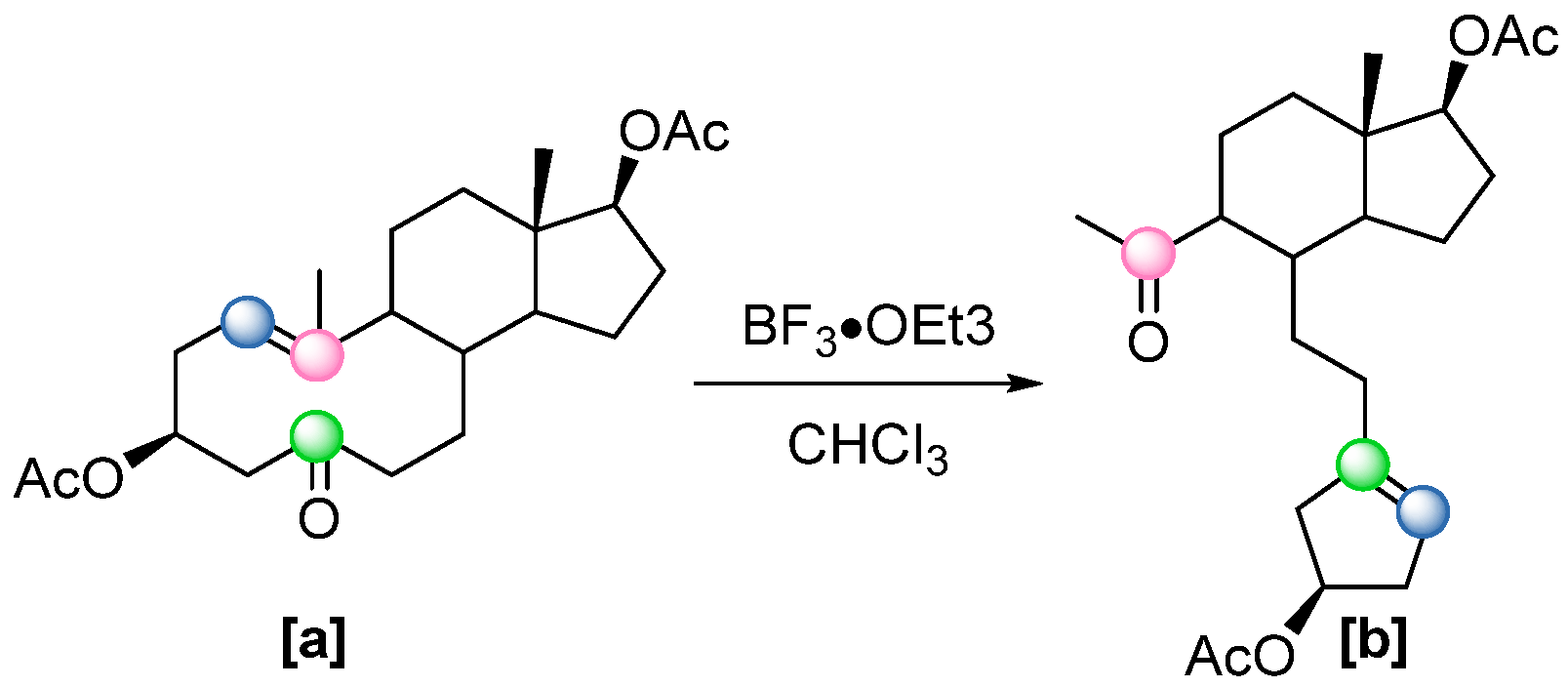
48]. However, these early approaches suffered from significant drawbacks, including low conversion rates and prolonged reaction times. Conversely, two examples of metal-free Lewis acid-catalysed COM reactions were later observed as unexpected side reactions in complex molecule synthesis, mediated by similar boron-based Lewis acids. In 2006, Khripach and coworkers, while investigating the synthesis of a steroid framework, unexpectedly obtained a cyclized product
[b] from precursor
[a] (
Scheme 29) [
49]. This transformation occurred when BF
3·OEt
3 was used to protect the keto group as a dithioketal, giving the cyclized product in a 60% yield. Later, the selectivity of this transformation could be controlled using different Lewis acids (see Scheme 41).
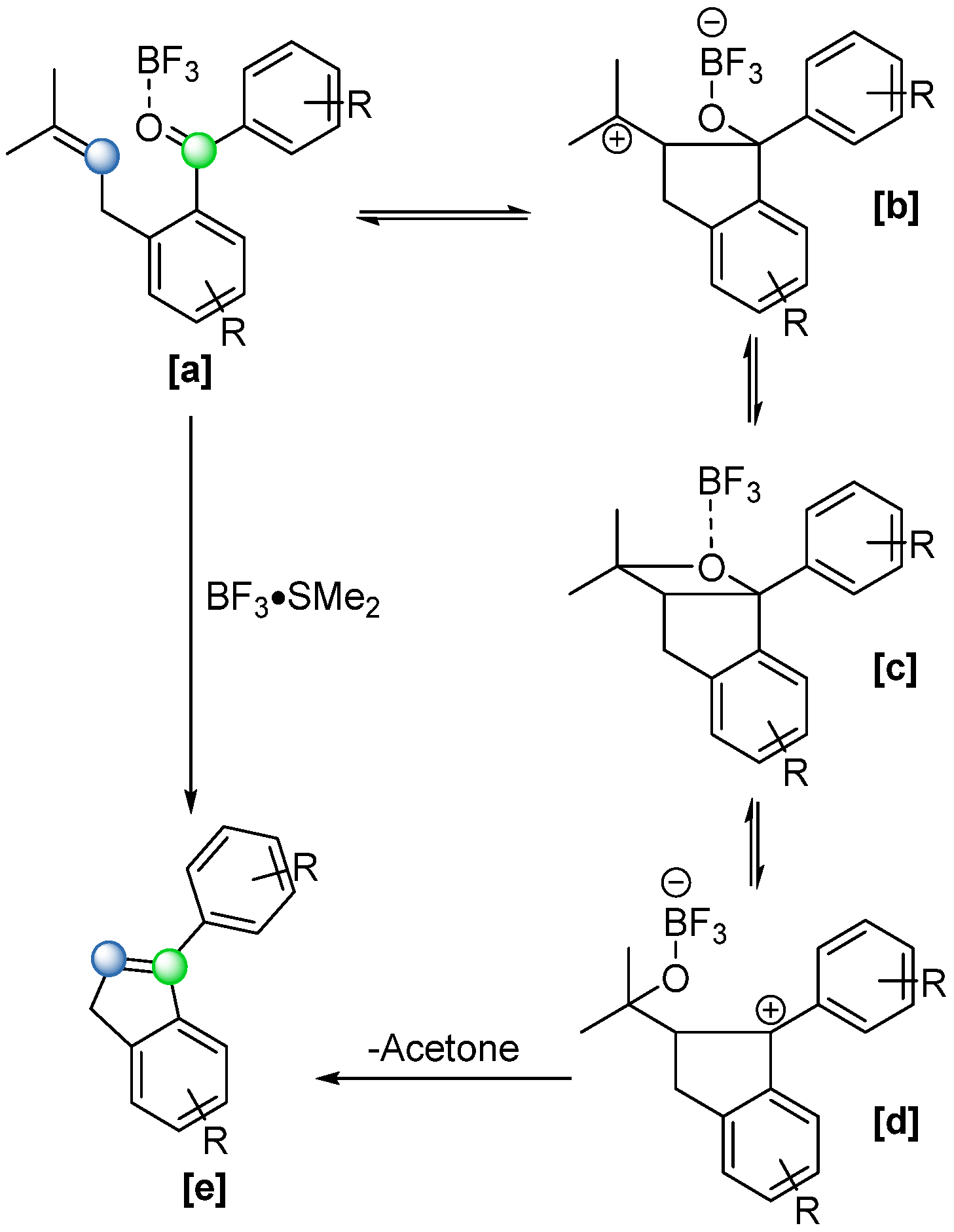
Similarly, in 2010, Slavov and coworkers [
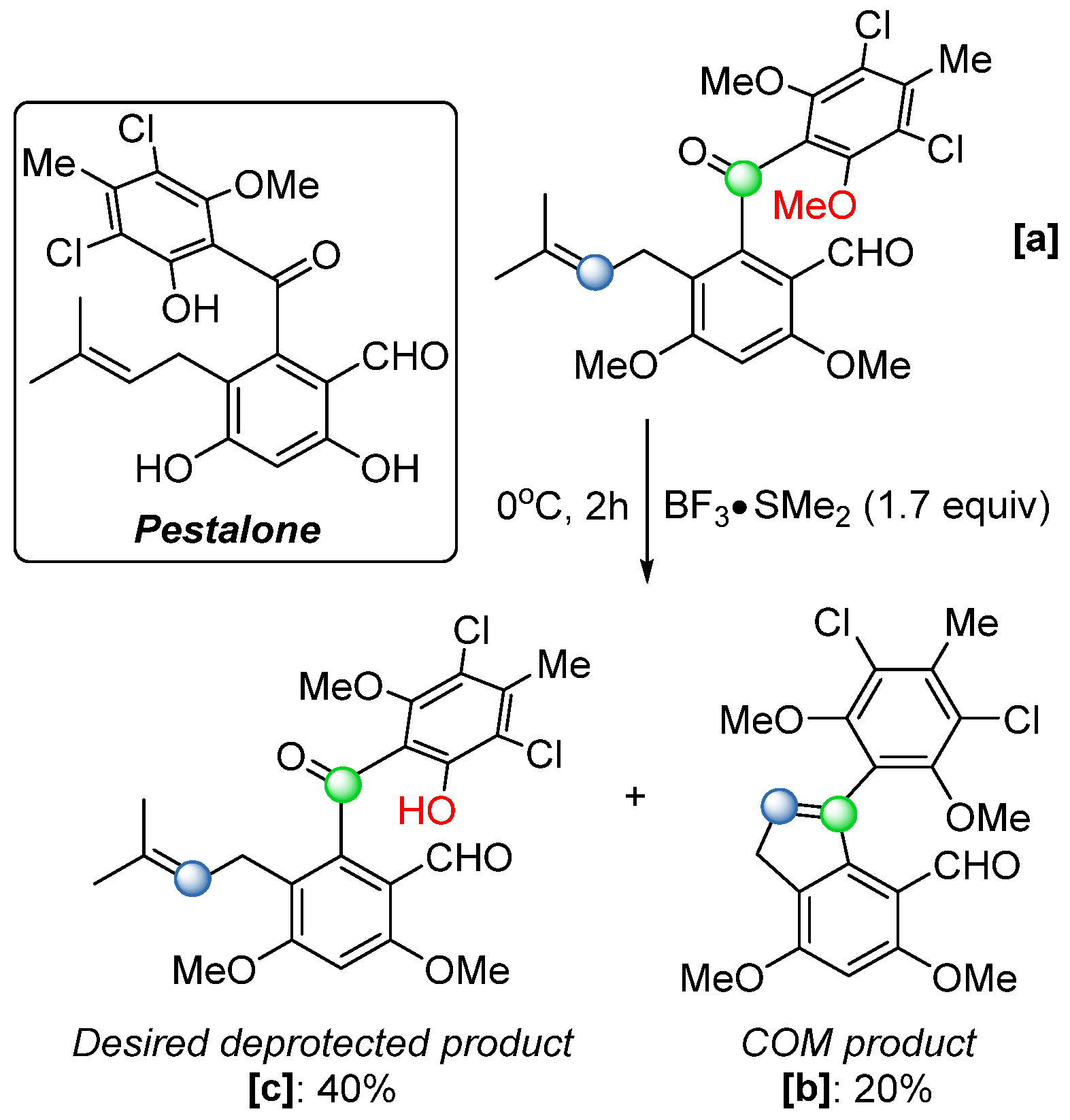
50] observed an analogous intramolecular cyclization during their total synthesis of the marine natural product pestalone (
Scheme 30), a highly substituted benzophenone with potent antibiotic activity. When treating the indene derivative
[a] with BF
3·SMe
2 to remove the O-methyl protecting group, they obtained the cyclized side product
[b] in a 20% yield (
Scheme 30). Building on this discovery, the same research group proposed a reaction mechanism (
Scheme 31). The Lewis acid activates the carbonyl group, increasing its susceptibility to nucleophilic attack by the double bond, giving rise to a carbenium ion intermediate
[b], which subsequently forms an oxetane intermediate
[c]. The final step involves the fragmentation of
[d], driven by the release of acetone. In 2011, the group conducted a comprehensive study to further investigate the reaction mechanism and enhance its efficiency [
51]. By utilizing 2.5 equivalents of BF
3·OEt
3, they successfully improved the reaction yield to 93% and broadened the scope of the optimized conditions to include both inter- and intramolecular reactions.
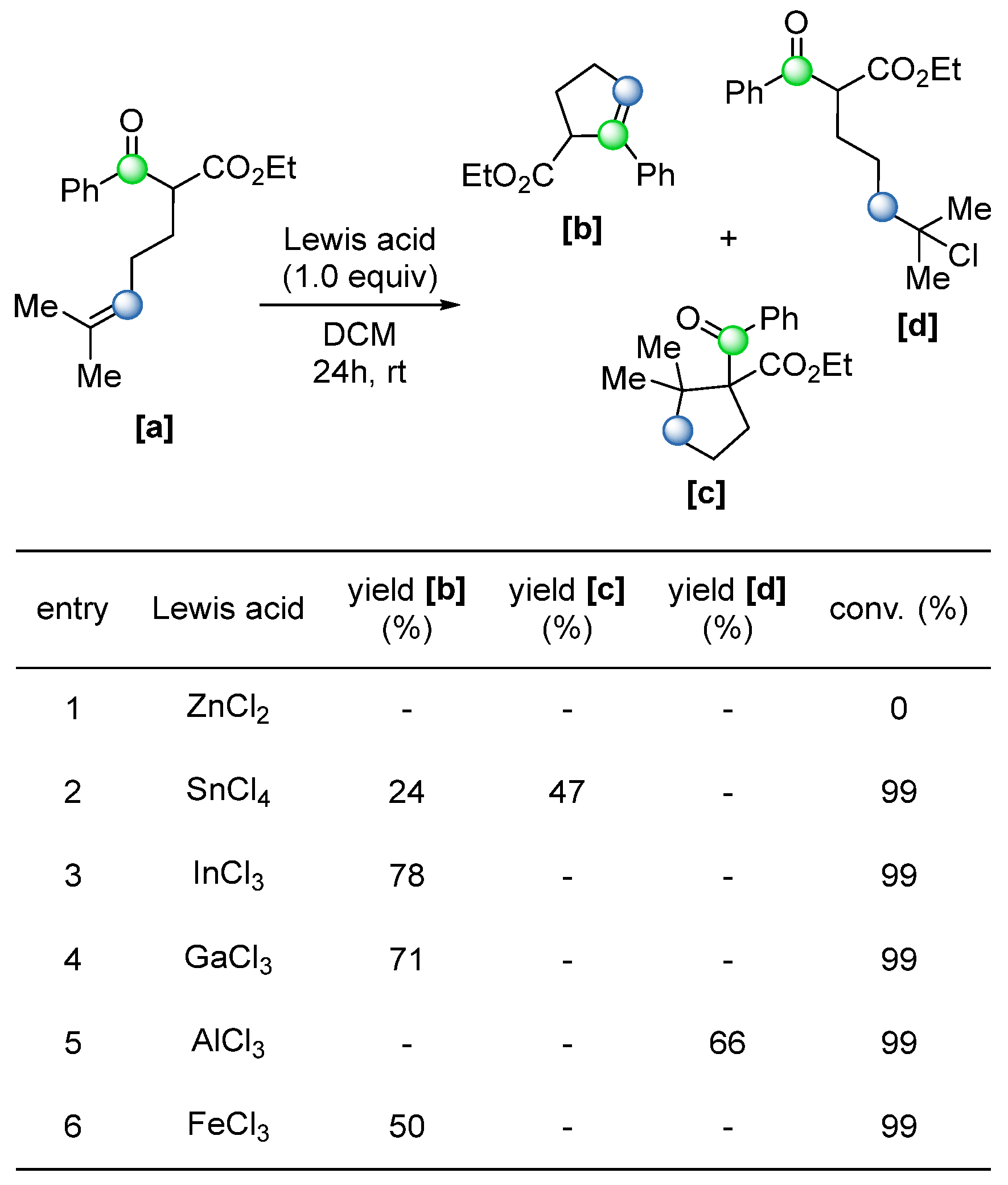
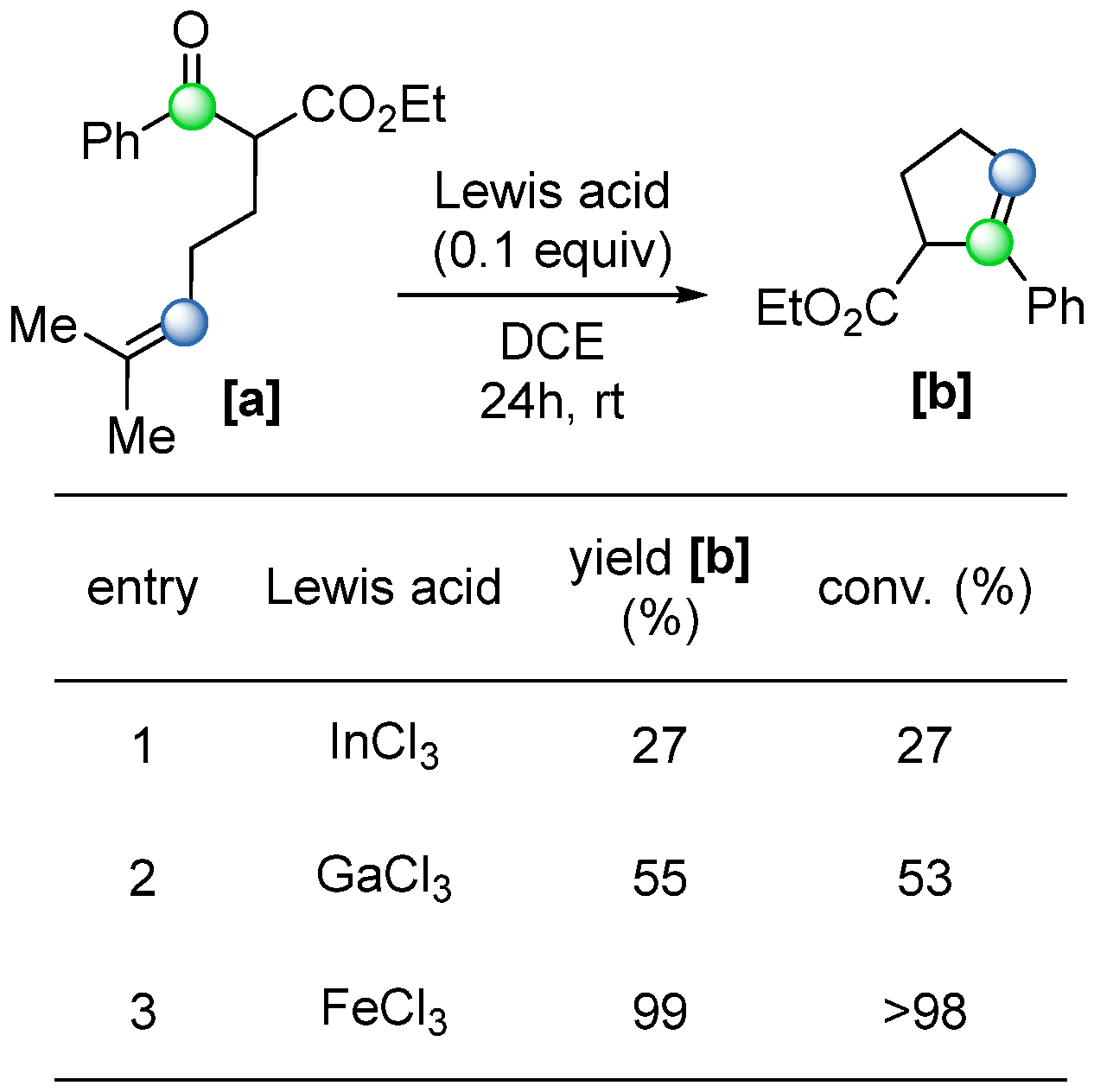
While earlier examples of COM reactions typically required stoichiometric amounts of metal alkylidene mediator or Lewis acid, a significant advancement was reported in 2016 by Ludwig and coworkers, who demonstrated the first catalytic ring-closing COM reaction using a metal-based Lewis acid [
52]. An initial study was conducted with stoichiometric amounts of Lewis acids [
2,
52,
53] (
Scheme 32). The results indicated that several metallic Lewis acids, including FeCl
3, InCl
3, and GaCl
3, both showed satisfying activity towards the COM reaction, yielding COM product
[b], while strong Lewis acids such as AlCl
3 gave the addition product
[d], and weak Lewis acids such as ZnCl
2 did not show any reactivity. Further tests were carried out with a catalytic amount of Lewis acid (10 mol%), where FeCl
3 showed privileged reactivity with a yield of 99% (
Scheme 33). After further optimization, 5 mol% FeCl
3 was identified as the optimal condition.
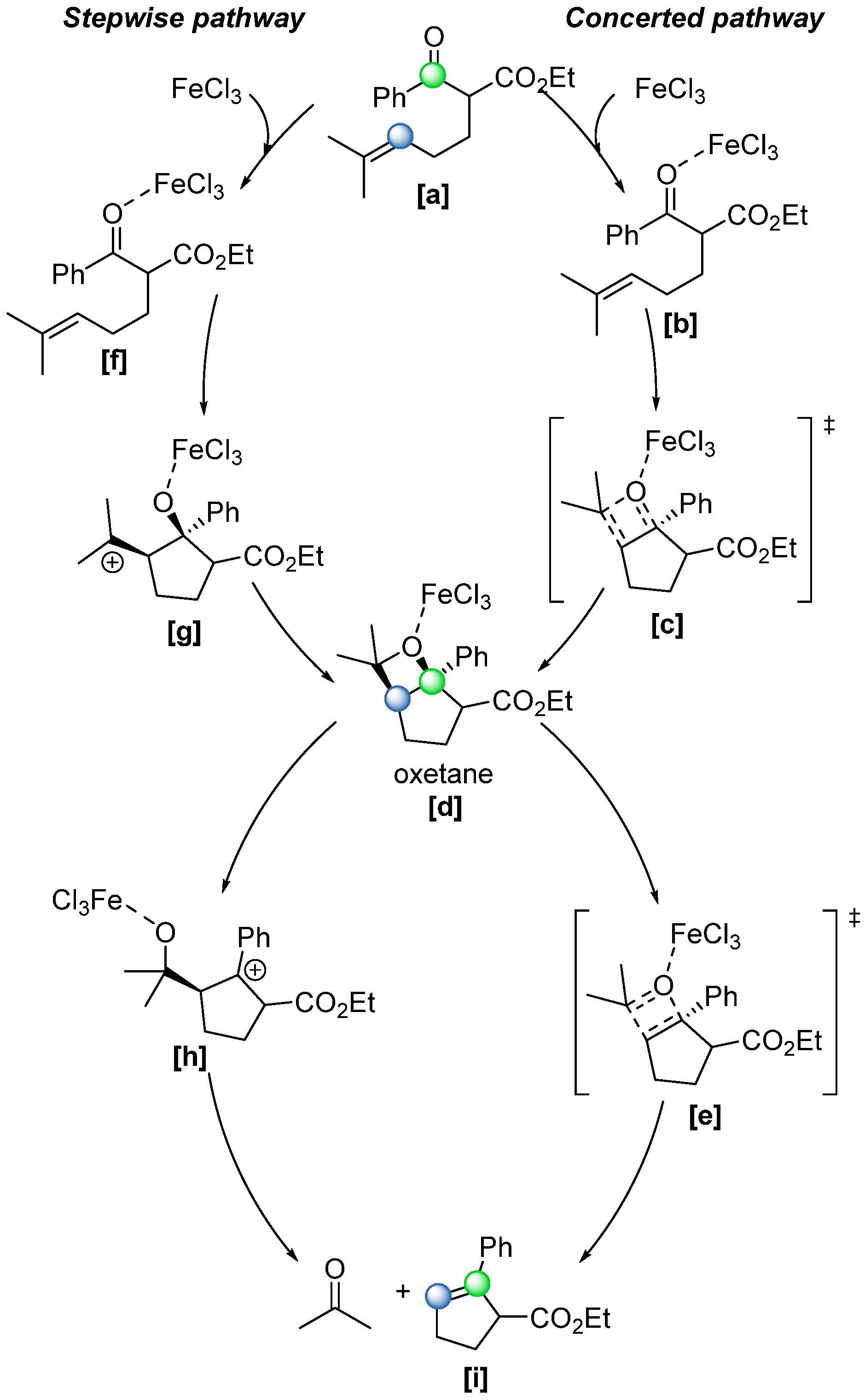
A detailed mechanistic investigation of the FeCl
3-catalysed COM reaction was conducted by the same group [
53]. They proposed two possible reaction pathways (
Scheme 34): a concerted mechanism involving the simultaneous formation and fragmentation of the oxetane ring
[d], and a stepwise mechanism in which the oxetane
[d] forms first and subsequently undergoes ring-opening via carbocation intermediates
[g] and
[h] (
Scheme 34). Interestingly, computational studies revealed that the lowest energy pathway for oxetane formation proceeds through a concerted, asynchronous [2+2] cycloaddition. This finding was further supported by potential energy surface (PES) analyses and carbocation-trapping experiments, which failed to detect any products arising from the intermolecular addition of nucleophiles to carbocation. Conversely, the fragmentation of the oxetane ring can be concerted or stepwise depending on the nature of the C–C double-bond formed. For example, for
[a] in
Scheme 34, a stable carbocation can be formed via C-O heterolysis.
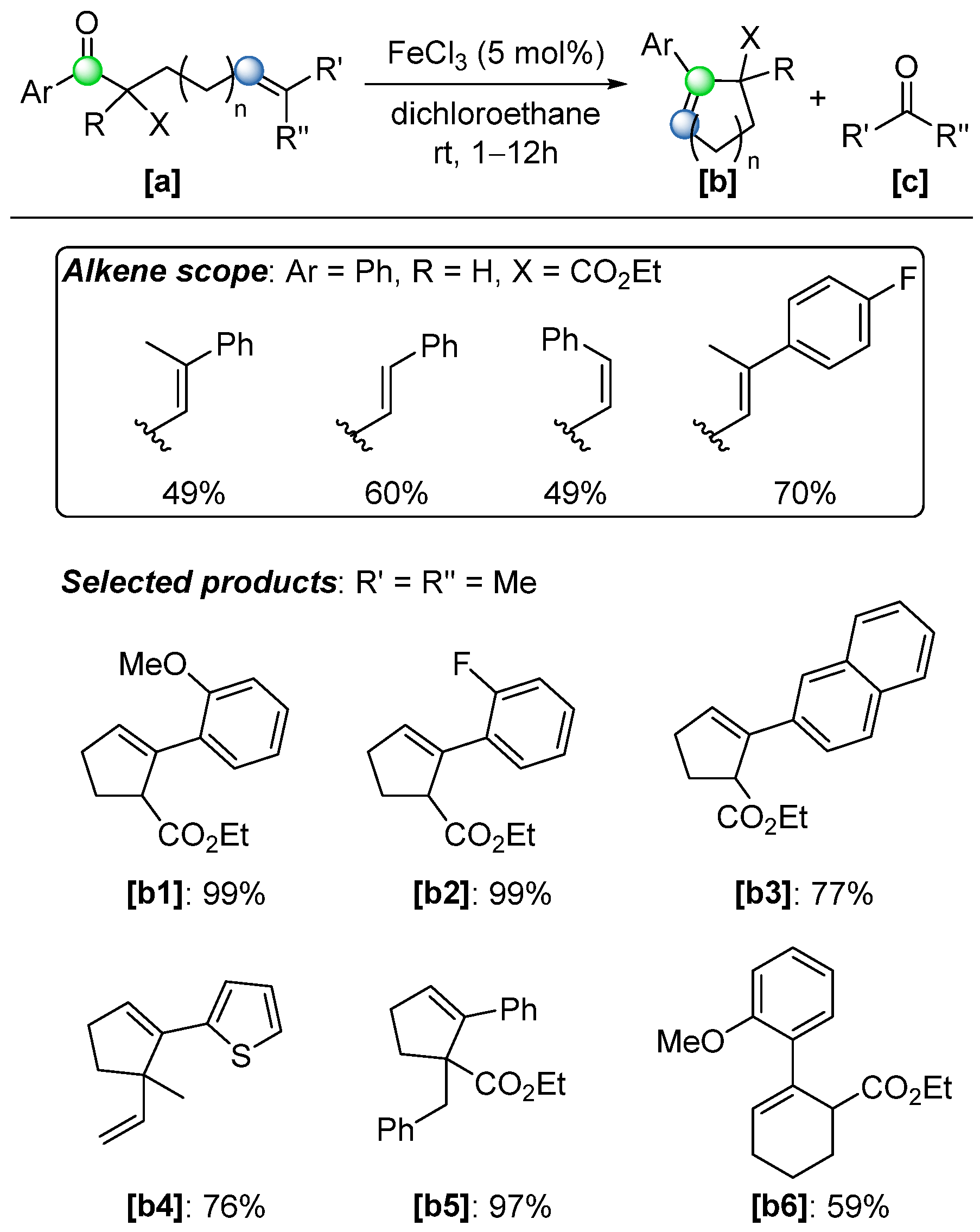
Substrate scope studies confirmed the broad applicability and efficiency of the optimized conditions (
Scheme 35). Using starting material
[a], the reaction tolerated a wide range of substituents, including both electron-withdrawing and electron-donating groups on the aromatic ring (
[b1],
[b2]), as well as modifications to the alkene and β-position of the carbon chain (
[b3],
[b4],
[b5]). This versatility enabled the synthesis of five- and six-membered ring products with structure
[b6], achieving yields up to 99% (
Scheme 35). However, the catalyzed COM reaction relied exclusively on aryl ketone substrates, with aliphatic ketone analogues largely excluded. This limitation arose from the inherently weaker coordination of aliphatic ketones to Lewis acids, and the critical role played by the aromatic ring in stabilizing key transition states
[c] and
[e] (
Scheme 34) through electron density redistribution [
53,
54].
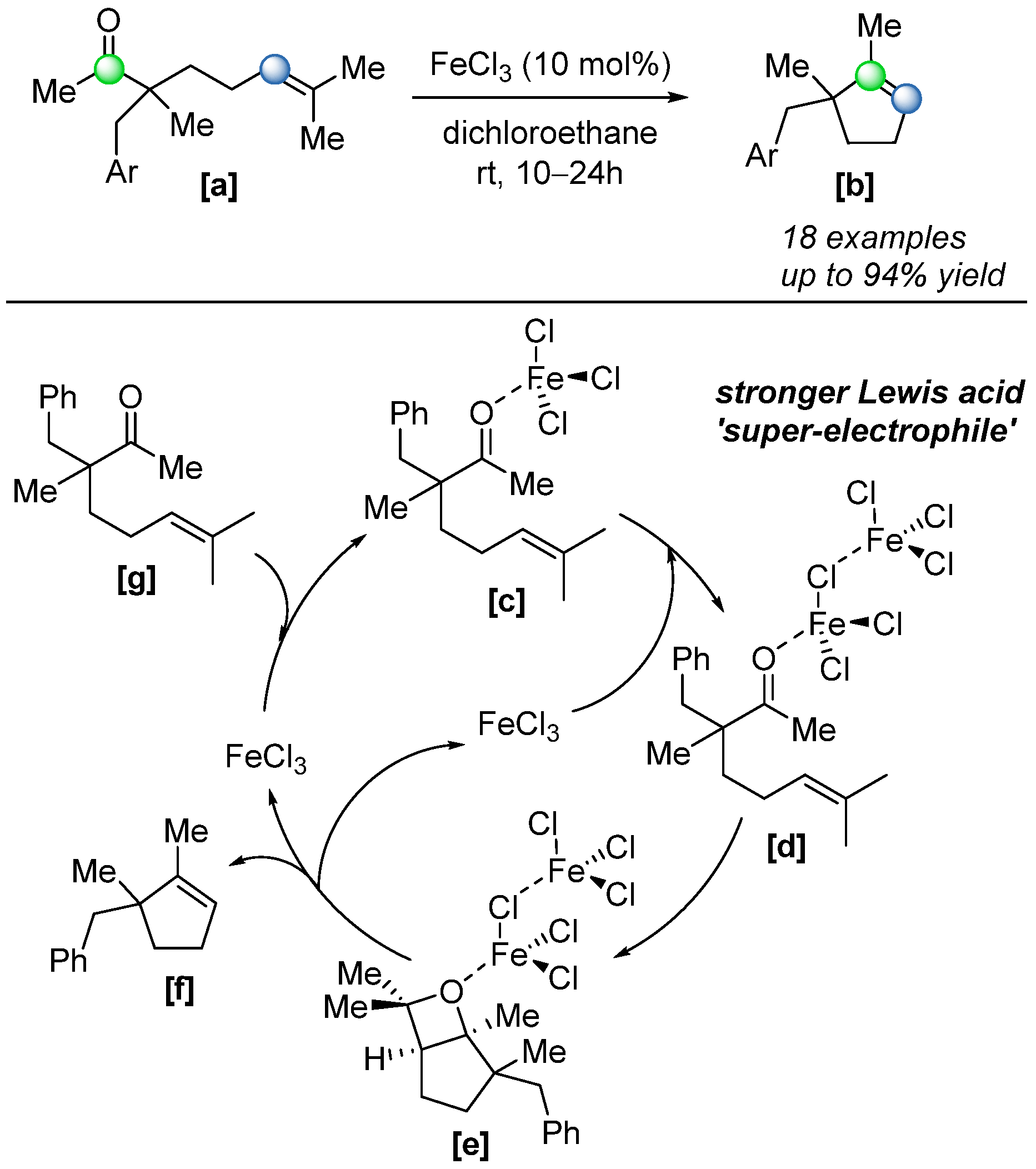
In 2019, Albright and co-workers successfully extended the reaction scope to aliphatic ketones [
54] with the optimized condition of a 10 mol% FeCl
3 catalyst; a range of five-membered rings was formed via intramolecular COM reaction, with yields up to 99% (
Scheme 36). Notably, mechanistic investigations revealed that the reaction was not catalyzed by monomeric FeCl
3, but rather by a homobimetallic, singly bridged iron(III) dimer
[d] formed in situ. This dimer exhibits significantly enhanced Lewis acidity, leading to increased substrate polarization and more effective activation (
Scheme 36).
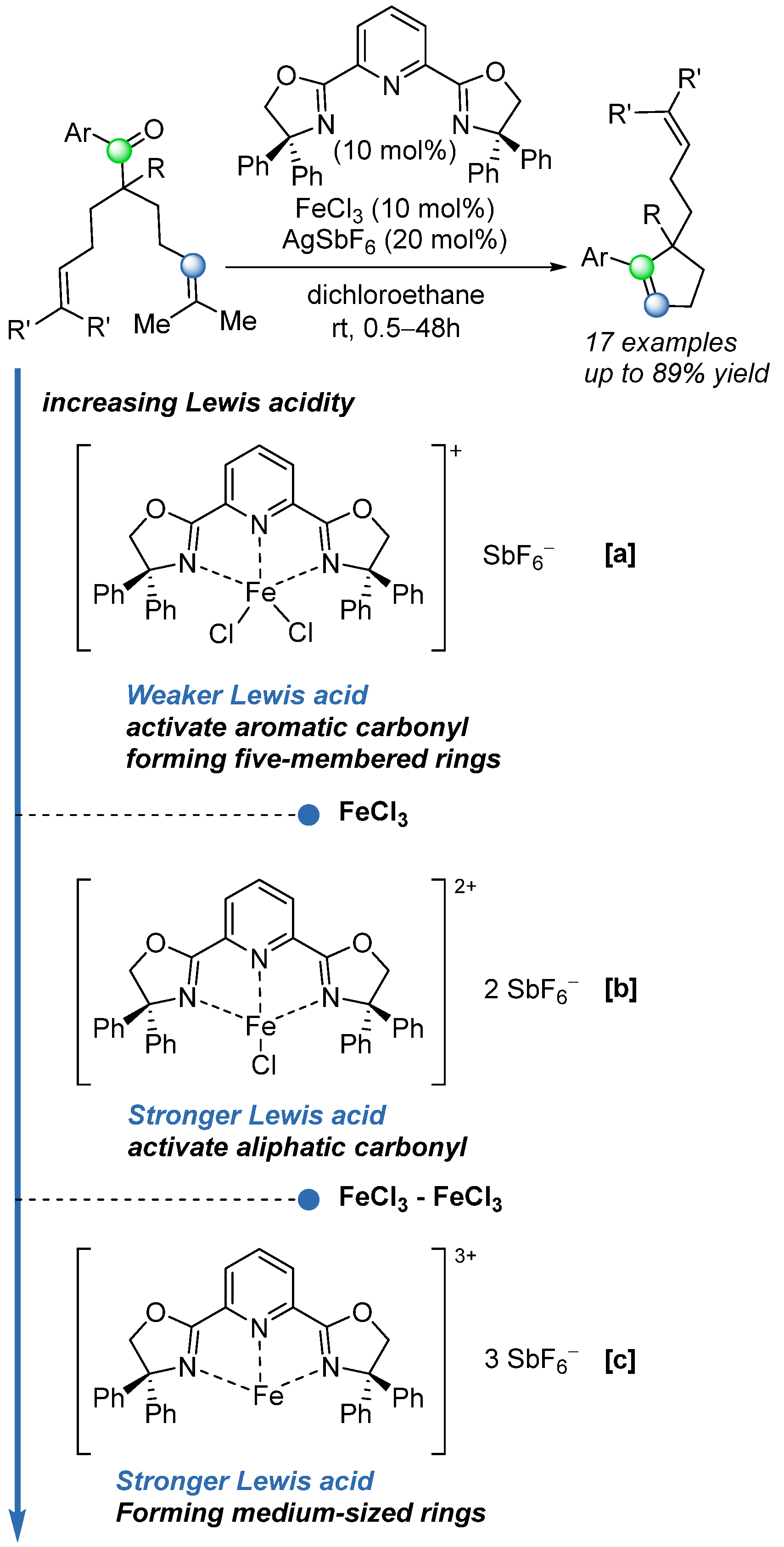
To further extend the reaction scope, in 2024, the same group [
55] reported the development of a bis(oxazoline) ligand (
Scheme 37) that complexes with iron to enable tunable Lewis acidity. By sequentially abstracting chloride ligands from the iron complex, the system allows access to catalysts that are both weaker and stronger than FeCl
3 (
Scheme 37). With the system
[b], a range of COM reactions were successfully carried out on sterically and electronically challenging substrates that had previously proven resistant to metathesis. Despite this advancement, the proposed catalytic systems did not afford enantioenriched products, highlighting the need for the development of asymmetric variants of this transformation.
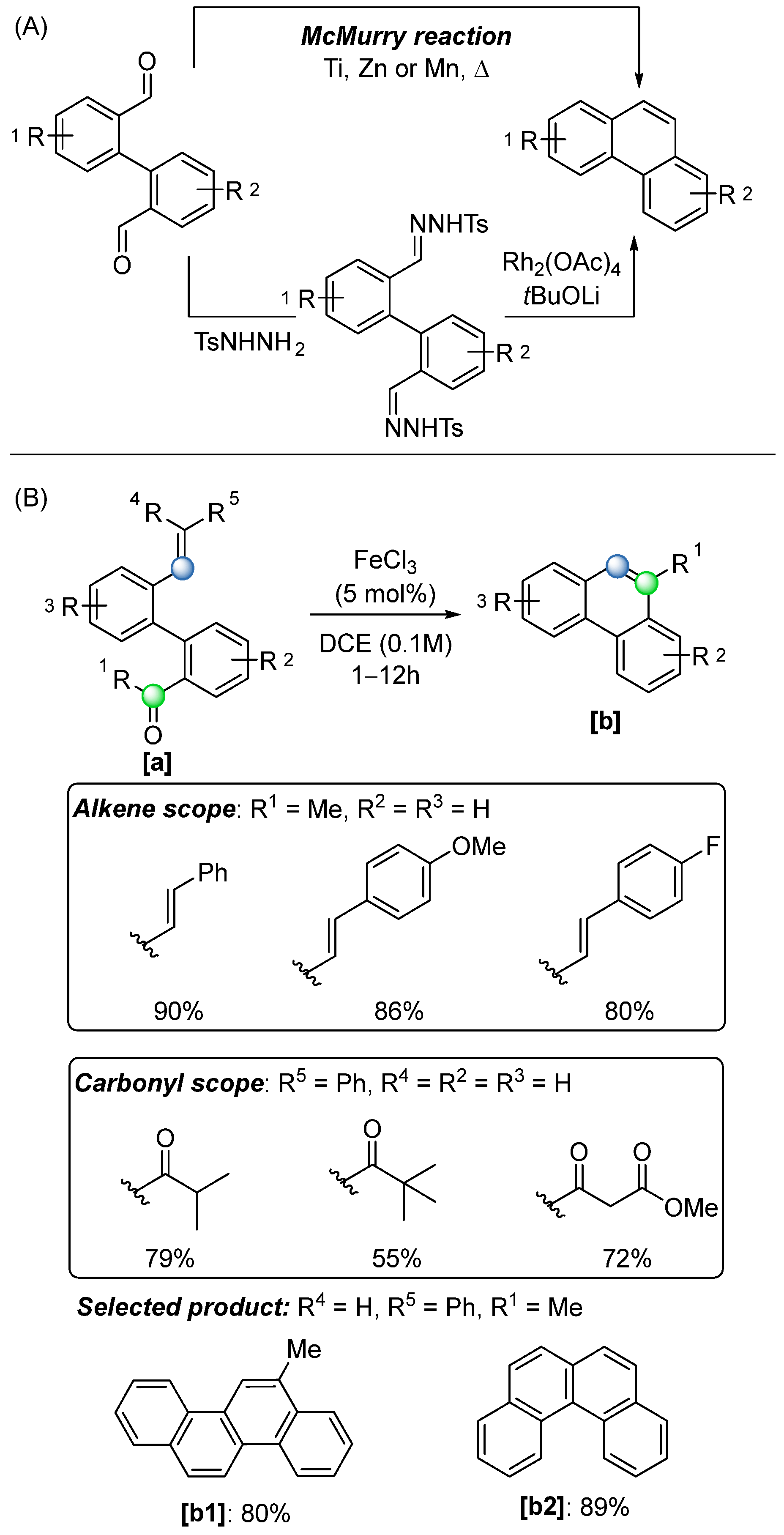
In 2017, they reported the use of FeCl
3-catalysed COM reactions for the construction of polycyclic aromatic compounds [
56] (
Scheme 38B)—an important class of structural motifs in organic chemistry with wide-ranging applications in both materials science and natural product synthesis. Traditional approaches to synthesizing polycyclic aromatic compounds include the classic McMurry reaction, which typically requires stoichiometric amounts of metallic reagents and harsh conditions. More modern methods, including rhodium-catalyzed protocols, often rely on expensive and less readily available metal catalysts (
Scheme 38A). In contrast, the FeCl
3-catalysed approach offers significant advantages, such as operational simplicity, mild conditions, and use of an inexpensive, accessible catalyst. Furthermore, this method demonstrates good to excellent yields across a broad substrate scope, accommodating various substitutions on the aromatic ring, the olefin, and the carbonyl group (
Scheme 38B).
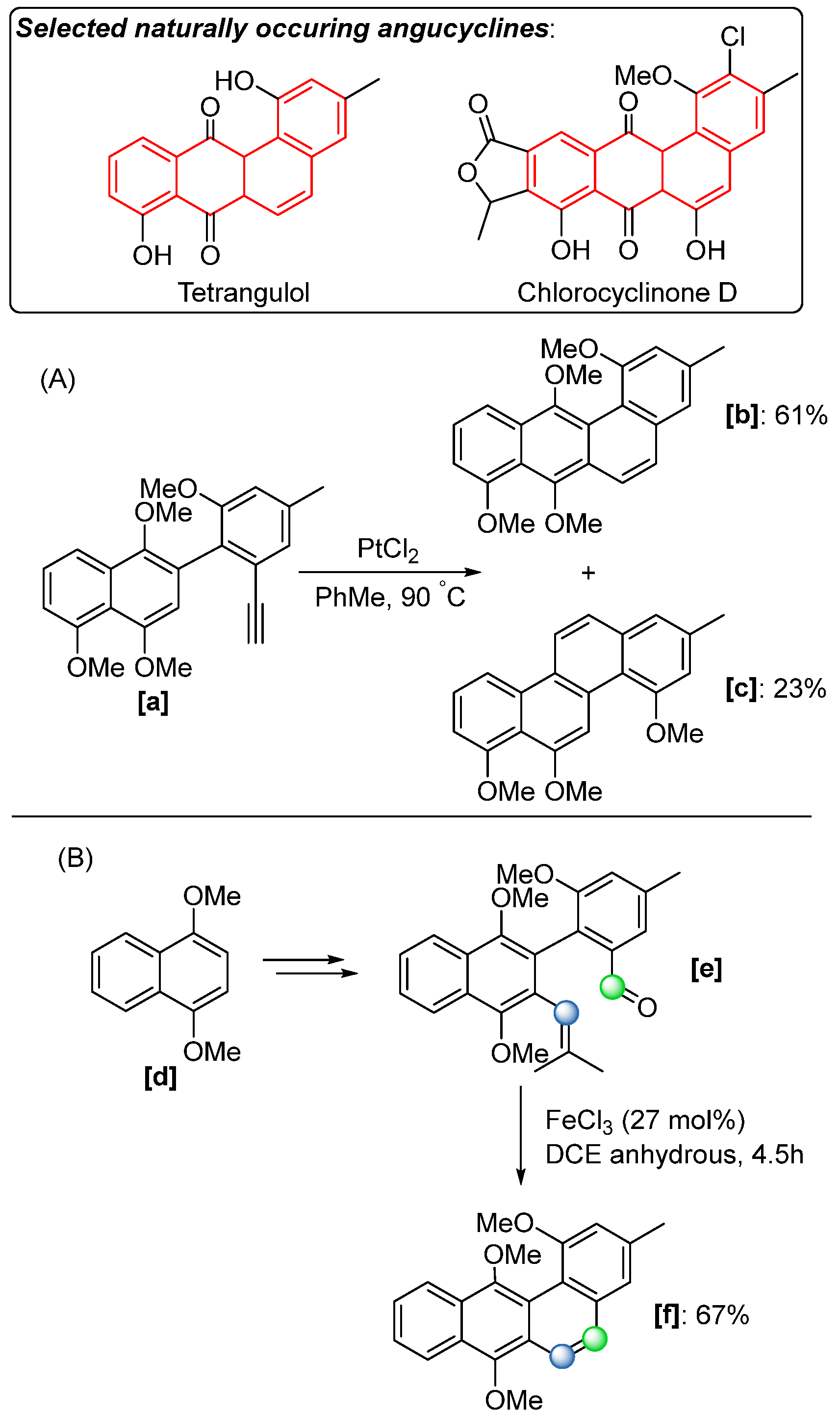
In 2022, Jagot and coworkers applied the same ring-closing COM strategy to construct the carbon skeleton of angucycline (
Scheme 39) [
57], which is a class of naturally occurring quinones known for their diverse biological activities, including anticancer and antibacterial properties. The COM reaction was employed at a late stage of the synthesis (
Scheme 39B), where the precursor
[e], bearing the requisite carbonyl group and a trisubstituted alkene, while treated with a catalytic amount of FeCl
3 (27 mol%), successfully yielded the desired anthracene derivative
[f] in a 67% yield. Compared to traditional approaches for synthesizing the angucycline core (
Scheme 39A), such as the cyclization of naphthyl phenylacetylene, which typically requires expensive transition metal catalysts like platinum(II) chloride and often results in undesired by-products
[c], the FeCl
3-catalysed method offers several key advantages: high chemoselectivity, milder reaction conditions, and the use of a cost-effective catalyst, making it a more practical and sustainable alternative for complex polyaromatic synthesis.
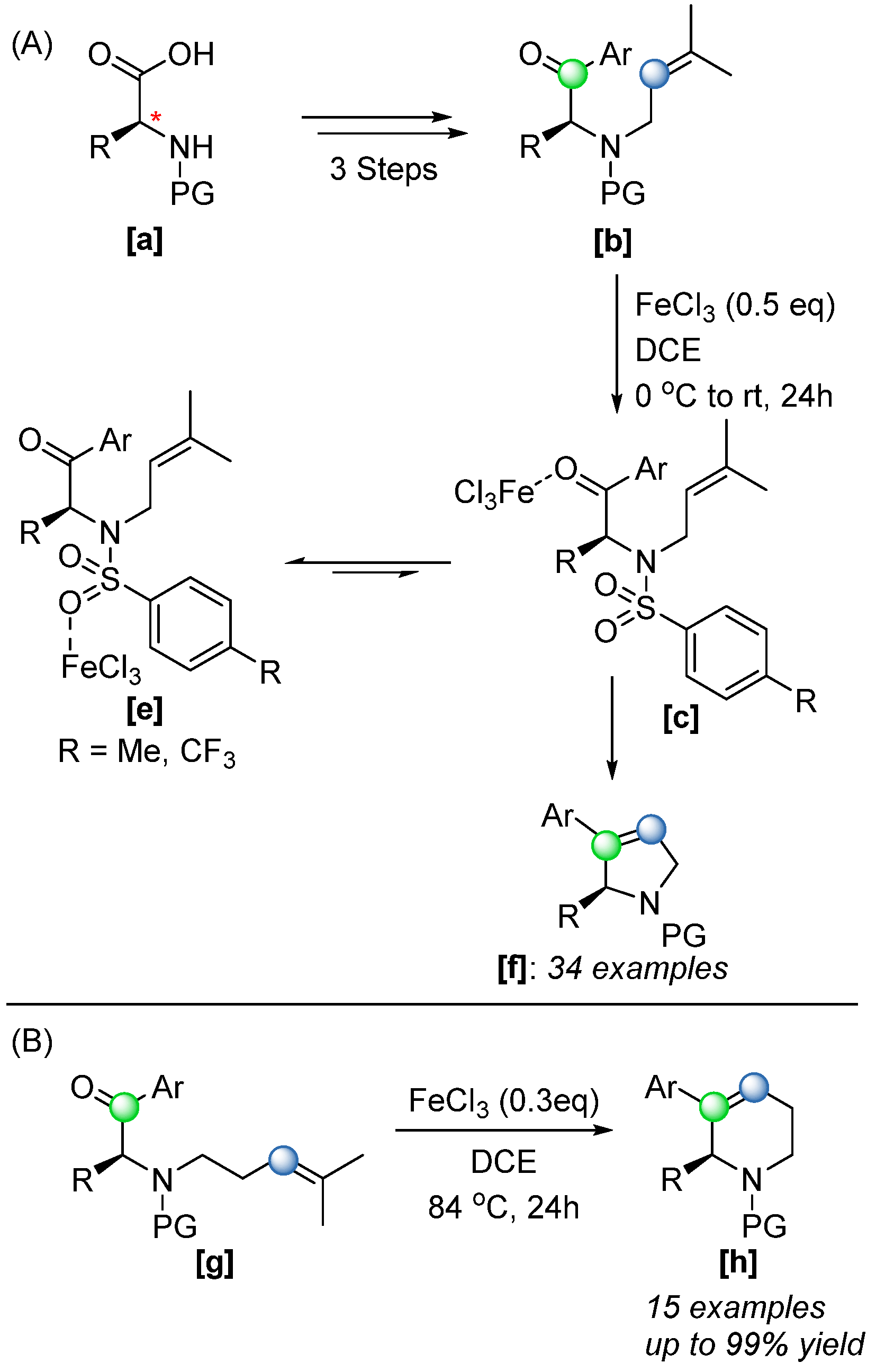
In 2018, the Schindler group further expanded the scope of Fe(III)-catalyzed COM reactions by applying the methodology to the synthesis of dihydropyrroles [
58], a type of structural motif commonly found in biologically active natural products and synthetic pharmaceuticals. The strategy (
Scheme 40A) utilized a commercially available chiral pool amino acid
[a], which enables distinct variations of both the α-amino and aryl ketone substituents to access a range of secondary amine derivatives
[b]. Upon treatment with FeCl
3, the desired cyclized products were obtained in excellent yield up to 99% with high enantioselectivity (98%
ee). Relatively high equivalents of FeCl
3 were required in this system due to the presence of additional Lewis basic sites, which can coordinate to the catalyst. However, improved yields were achieved by employing more electron-withdrawing protecting groups, more specifically by replacing a tosyl group with a 4-trifluoro-methylbenzenesulfonyl group which reduced the amine’s binding affinity, thereby enhancing catalytic efficiency. This approach was further extended in 2020 to the synthesis of six-membered tetrahydropyridines (
Scheme 40B) [
59], demonstrating broader applicability and scalability, including successful gram-scale reactions.
Overall, this Fe(III)-catalyzed methodology offers significant advantages over existing approaches for synthesizing nitrogen-containing heterocycles. Conventional methods often rely on precious metal catalysts, expensive chiral ligands, extended reaction times, or suffer from limited substrate scope [
58,
59]. In contrast, the FeCl
3-based strategy is operationally simple, cost-effective, and exhibits high selectivity. Additionally, the reaction produces only non-competitive byproducts, and final deprotection steps are generally straightforward and have high yield (up to 98%).
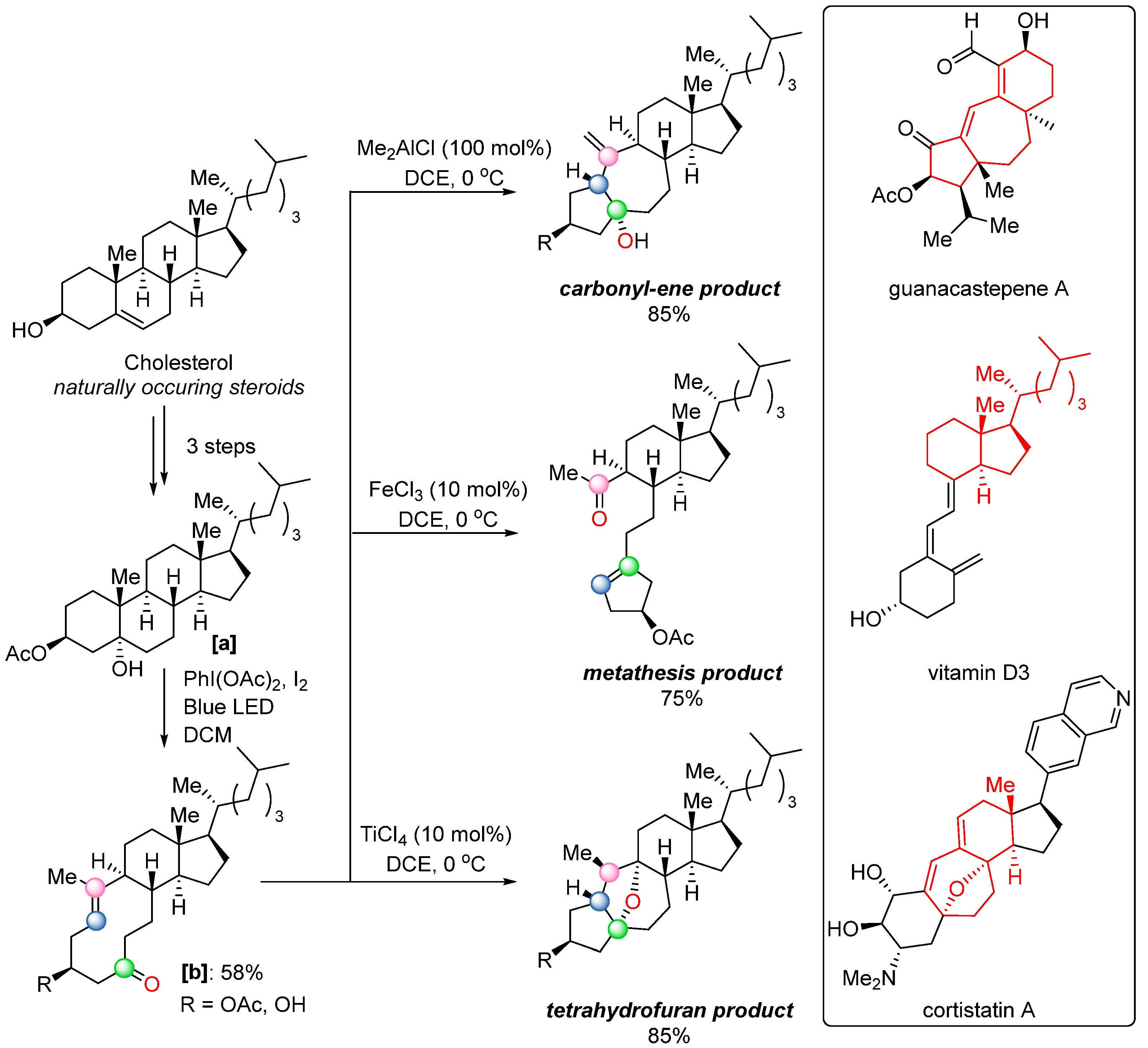
In 2019, the Schindler group reported another notable advancement: a transannular Lewis acid-catalyzed COM reaction (
Scheme 41) [
60] with a functionalized cyclopentenone system
[b], which could be synthesized in a short sequence from naturally occurring steroids. Remarkably, when this system was treated with different Lewis acids, three distinct products were observed—a carbonyl-ene product, a metathesis product, and a tetrahydrofuran product. Through the careful optimization of reaction conditions using various Lewis acids, the formation of each product could be selectively controlled. Intriguingly, all three products structurally resemble biologically significant natural compounds, including guanacastepene A, vitamin D
3, and cortistatin A (
Scheme 41). To rationalize these outcomes, a detailed mechanism was proposed based on both computational and experimental studies (
Scheme 42). Upon activation by a Lewis acid, two competing, reversible pathways can occur: (i) a carbonyl-ene reaction, yielding the kinetic product
[f]; (ii) a concerted asynchronous [2+2] cycloaddition, forming an oxetane intermediate
[c], which can diverge into two distinct pathways. In one, it undergoes fragmentation via elimination, forming a cycloheptene intermediate
[e] that rearranges through intramolecular nucleophilic attack of a tertiary alcohol on the alkene to yield the tetrahydrofuran product
[g]. In the other, the oxetane undergoes a concerted, asynchronous ring opening to produce the carbonyl–olefin metathesis product
[d].
From the examples discussed above, FeCl
3 has proven to stand out as a highly effective Lewis acid catalyst for carbonyl–olefin metathesis, owing to its accessible solution-phase speciation, both as a monomer and in its dimeric form, as well as the efficient interaction between the catalyst and the substrate [
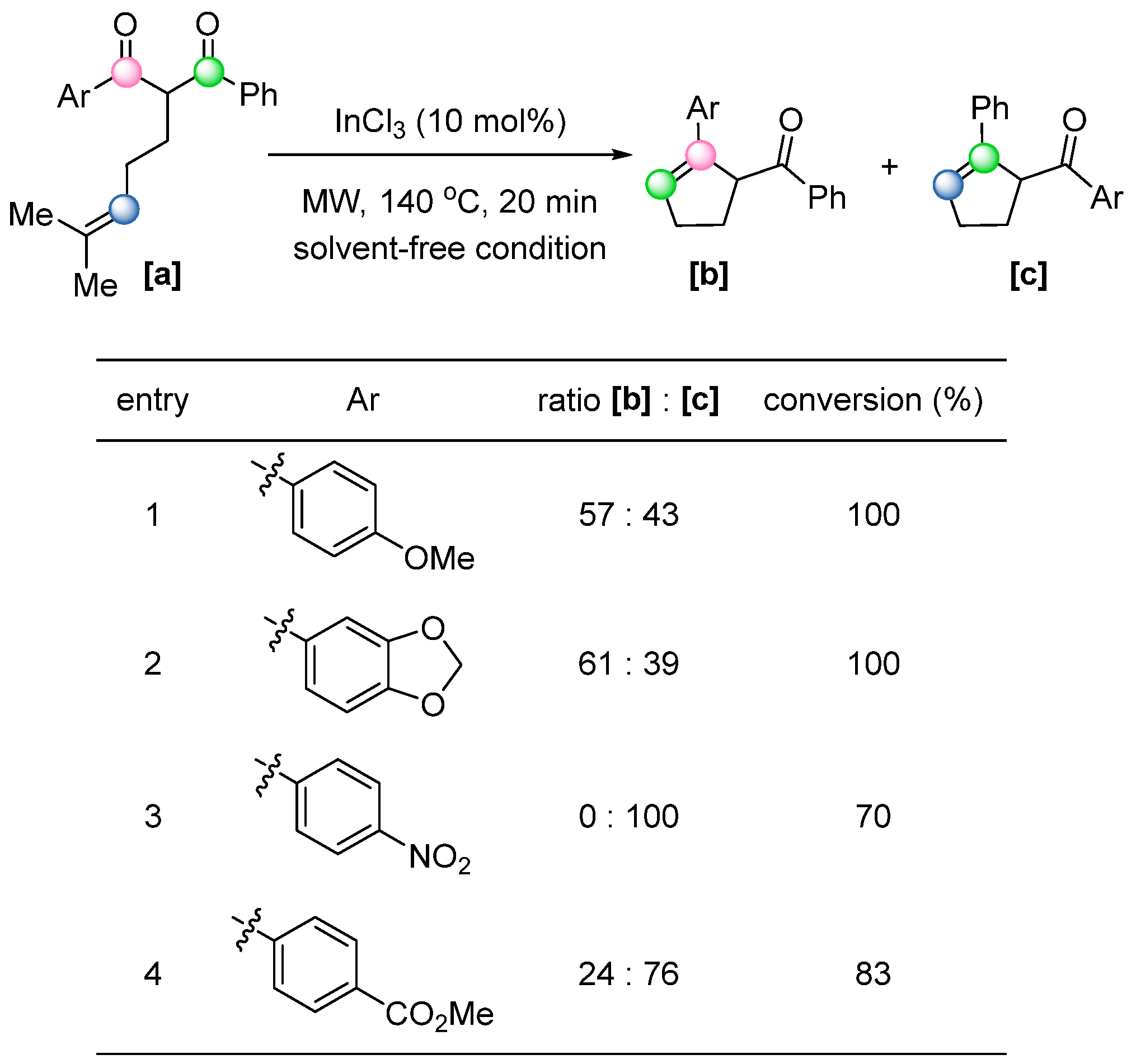
61]. Besides iron, the usage of other metal Lewis acids in COM reactions was also explored. In 2023, Pizzio and co-workers reported a microwave-assisted, InCl
3-catalyzed COM reaction in solvent-free conditions [
62]. The study investigated the reaction scope and regioselectivity using a series of electronically varied 2-(4-methylpent-3-en-1-yl)-1,3-diaryl-1,3-diketones (
Scheme 43).
Substrates bearing electron-withdrawing groups predominantly yielded regioisomer [c], while electron-donating groups favored the formation of alternative regioisomer [b]. This result can be explained by the reaction mechanism, wherein a [2+2] cycloaddition follows selective carbonyl activation by InCl3, which preferentially coordinates to the carbonyl group with the highest electron density. Notably, the reaction proceeds efficiently not only in biodegradable solvents but also under solvent-free conditions, underscoring its potential for sustainable synthesis.
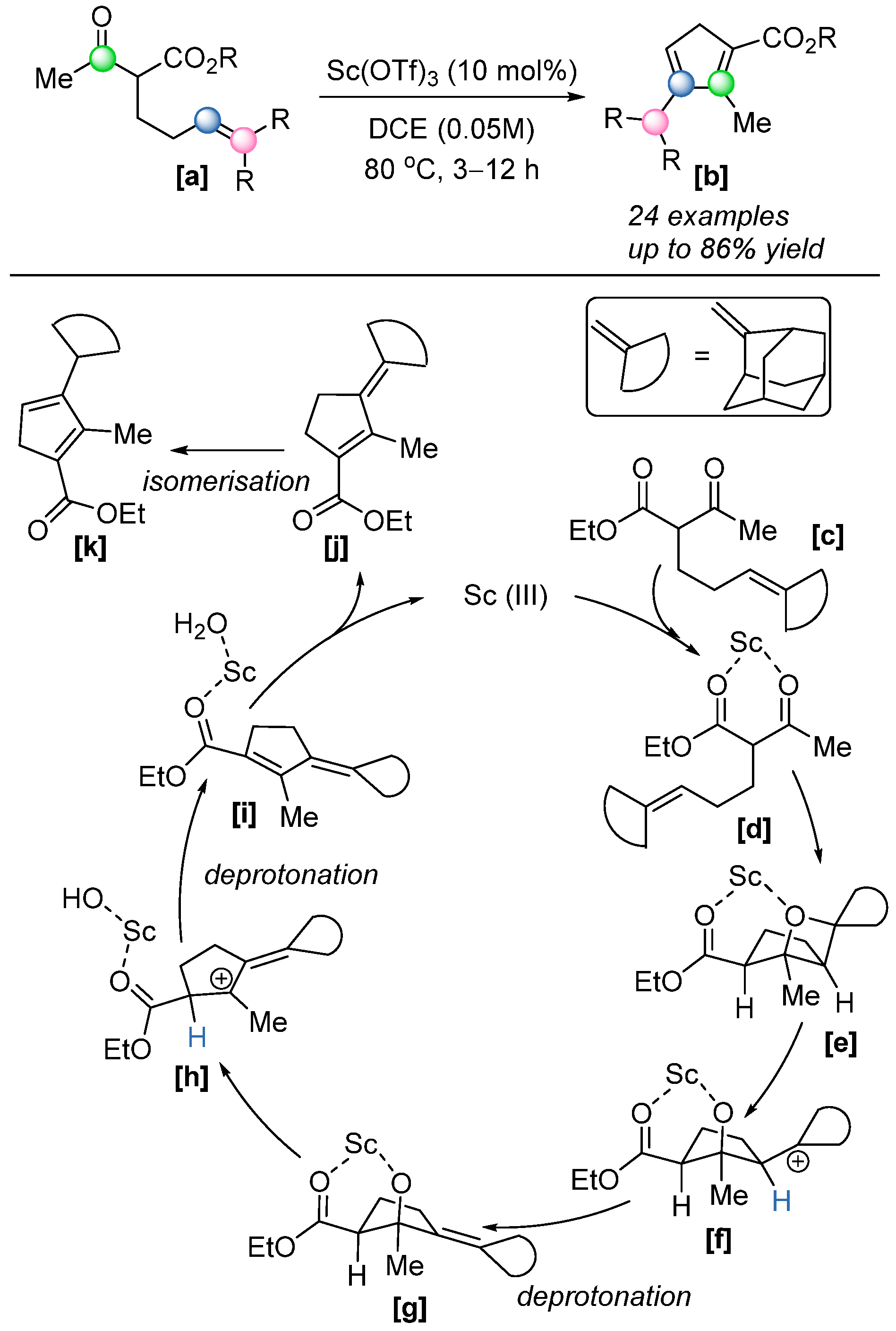
In the same year, McAtee et al. reported a novel strategy for the synthesis of functionalized cyclopentadienes from precursor
[a] [
63] (
Scheme 44). Through a combination of experimental and computational studies, the authors proposed a distinctive reaction mechanism: after the formation of an oxetane intermediate
[e], which undergoes ring fragmentation to generate a carbocation species
[f], stabilized by a six-membered scandacycle. The subsequent deprotonation of a neighboring proton by an external triflate yields an alkoxide intermediate
[g], which then undergoes ring opening to form another carbocation
[h]. A second deprotonation step furnishes the conjugated diene
[i]. Upon the release of the Lewis acid, intermediate
[j] undergoes isomerization to afford the thermodynamically favored cyclopentadiene product
[k]. This methodology presents a valuable alternative to traditional approaches for synthesizing functionalized cyclopentadienes, particularly in overcoming challenges associated with the selective incorporation of electron-withdrawing groups. Such substituents often increase the acidity of the methylene moiety, leading to undesired polymerization or 1,5-hydrogen atom migration. The proposed strategy offers a controlled and efficient route to access these sensitive structures.
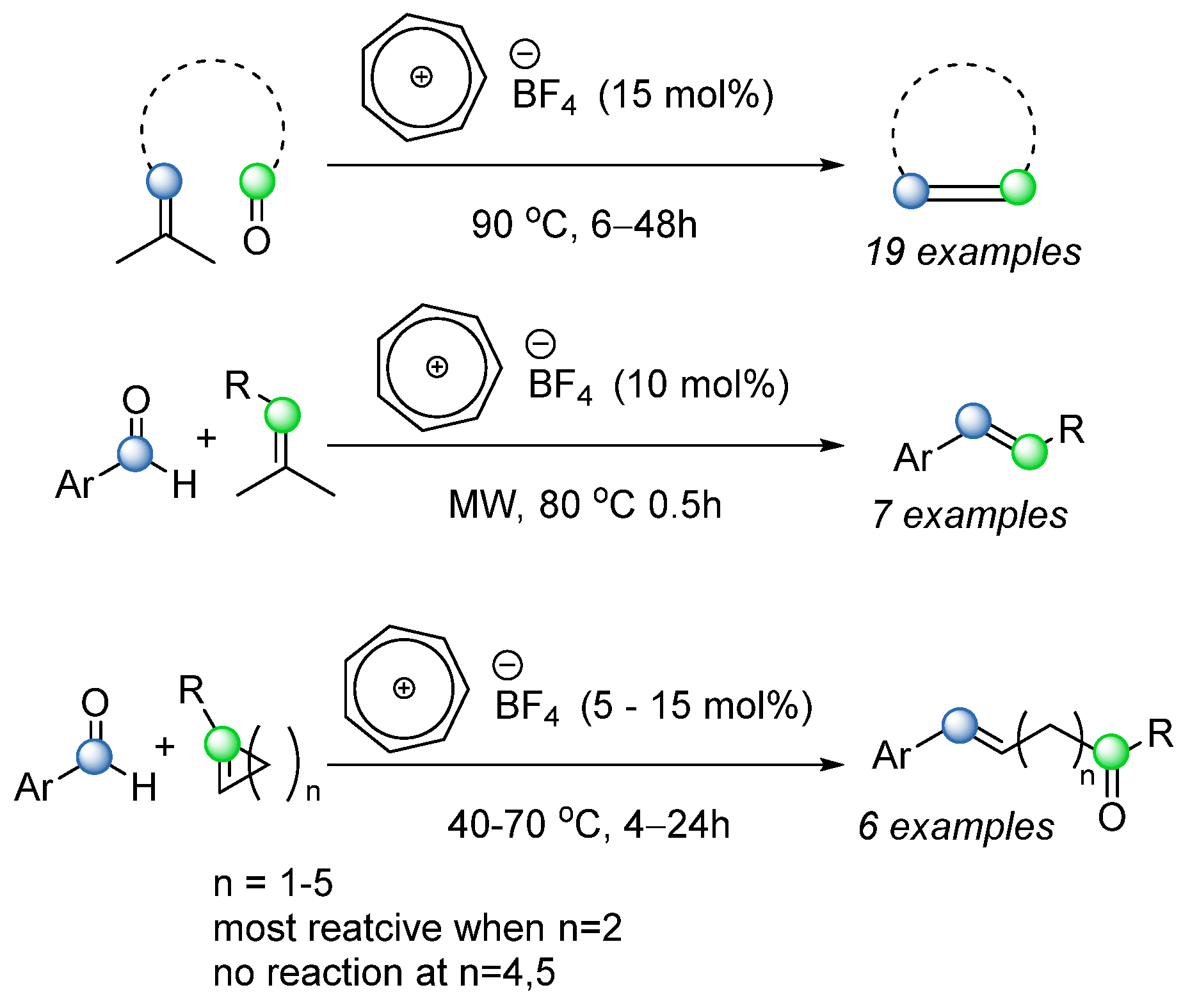
While a lot of exciting and interesting reactions have been developed, most of the reactions show limited scope, where only intramolecular ring closure is observed. This could be explained due to the fact that the intermolecular reaction is generally more demanding entropically. In 2018, Tran and coworkers published a study that indicated that a catalytic amount of tropylium tetrafluoroborate [
64] can promote a COM reaction as a Lewis acid; the reaction worked well intramolecularly on a broad scope of compounds, with moderate to good yield of up to 93%. The intermolecular reaction was tested between a set of aromatic aldehydes and isopropylidene-bearing olefin substrates, with the optimized condition of 10 mol% tropylium catalyst in dichloromethane at 80 °C for 0.5 h under pressurized microwave irradiation. These reactions afforded a promising yield of up to 70% of the desired product, mostly as the
trans isomer. The intermolecular ring-opening reaction was also scoped, where alkene rings from three-membered rings to seven-membered rings were tested, and where four-membered rings showed the highest activity (
Scheme 45).
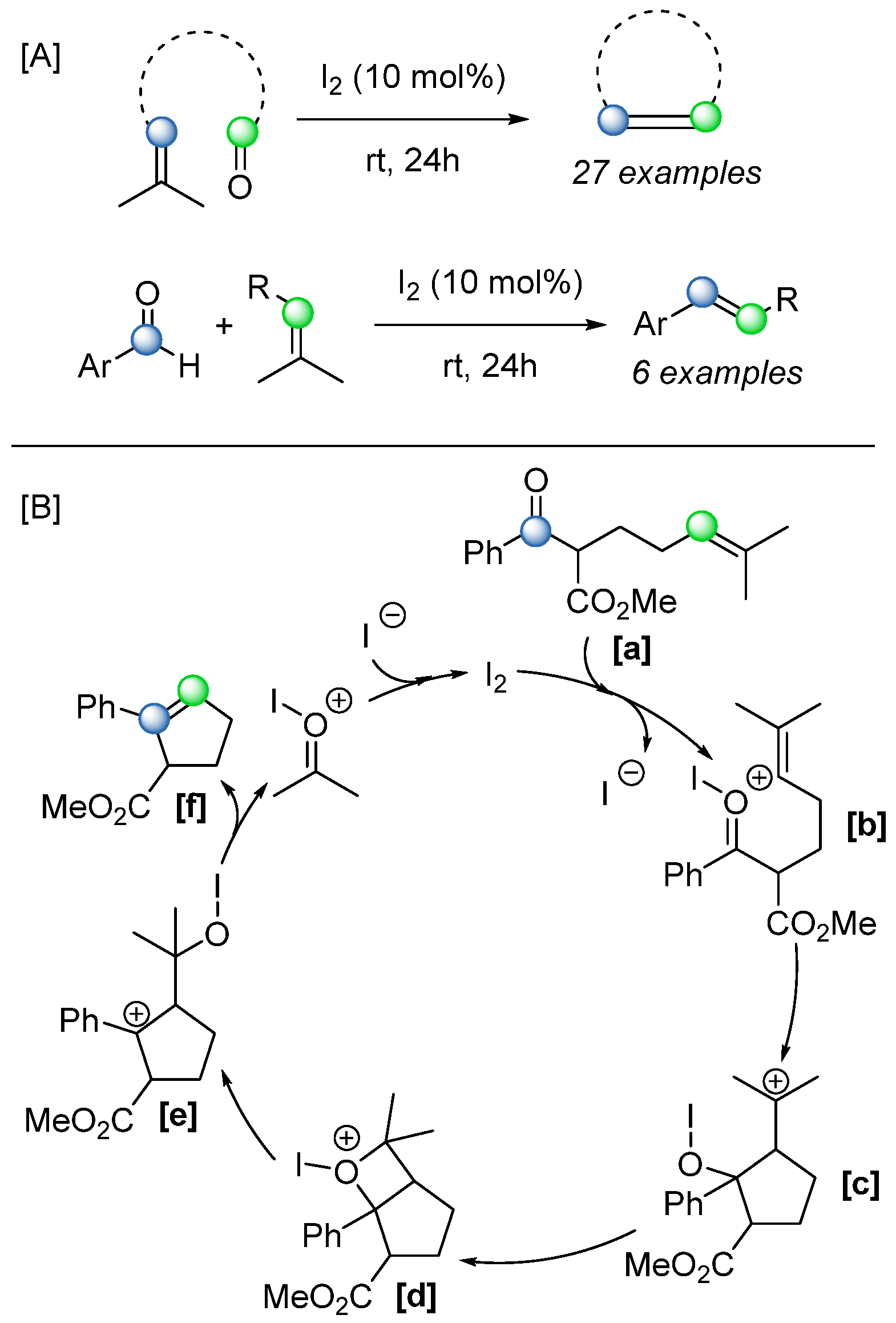
The same group reported in 2019 the use of molecular iodine as an alternative Lewis acid catalyst for COM reactions [
65]. In this system, the iodonium ion serves to activate the carbonyl group, enhancing its susceptibility to nucleophilic attack by the olefin. Based on both computational and experimental evidence, a detailed mechanism was proposed (
Scheme 46). This approach was applied successfully to a wide range of intramolecular substrates and, notably, also demonstrated feasibility for selected intermolecular reactions, with yields reaching up to 50%. The use of iodine as a cheap and accessible catalyst also gives this methodology an extra advantage.
To date, Lewis acids have emerged as some of the most successful and extensively developed catalysts for COM reactions, with demonstrated applicability in both inter- and intramolecular transformations. However, their use in the synthesis of structurally complex molecules remains relatively limited. This can be attributed to the inherent sensitivity of Lewis acids toward Lewis-basic functional groups, as well as their potential to induce decomposition in highly functionalized substrates. Additionally, competing carbonyl–ene side reactions often reduce product yields and generate undesired byproducts, complicating purification and lowering overall efficiency. These challenges highlight the need for continued methodological development to establish more general and robust approaches for COM reactions, particularly for molecules with complex structures.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}