
Recent Advances in Carbon-Centered Radical-Initiated Olefin Transformation Chemistry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
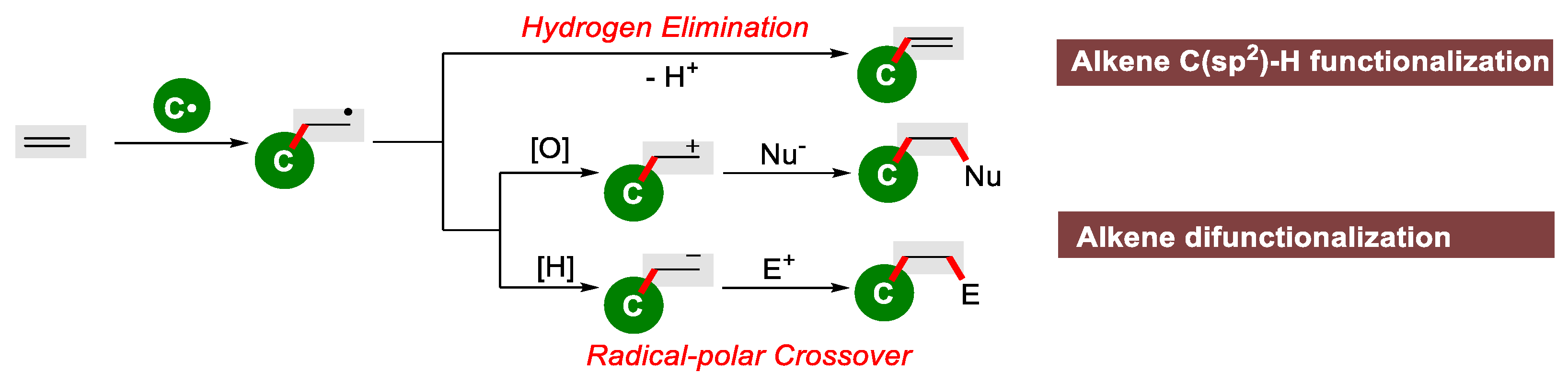
2. Carbon-Centered Radical-Initiated Olefin Transformation
2.1. Alkene Heck-Type Alkylation
2.1.1. Alkene Heck-Type C(sp2)-H Alkylation
2.1.2. Alkene Heck-Type Decarboxylative Alkylation
2.2. Alkene Hydroalkylation
2.2.1. C(sp3)-H Functionalization-Enabled Alkene Hydroalkylation
2.2.2. C-X Functionalization-Enabled Alkene Hydroalkylation
2.3. Alkene Difunctionalization
2.3.1. C-H Functionalization-Enabled Alkene Dicarbofunctionalization
2.3.2. C-H Functionalization-Enabled Alkene Alkyl-Heterofunctionalization
2.3.3. C-X Functionalization-Enabled Alkene Dicarbofunctionalization
2.3.4. C-X Functionalization-Enabled Alkene Carbo-Heterofunctionalization
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, F.-H.; Guo, X.; Zeng, X.; Wang, Z. Asymmetric 1,4-Functionalization of 1,3-Enynes via Dual Photoredox and Chromium Catalysis. Nat. Commun. 2022, 13, 5036. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Radical Aryl Migration Reactions and Synthetic Applications. Chem. Soc. Rev. 2015, 44, 5220–5245. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, Z.; Feng, T.; Zhu, C. Radical-Mediated Rearrangements: Past, Present, and Future. Chem. Soc. Rev. 2021, 50, 11577–11613. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhu, C. Radical-Mediated Remote Functional Group Migration. Acc. Chem. Res. 2020, 53, 1620–1636. [Google Scholar] [CrossRef]
- Beckwith, A.L.; Crich, D.; Duggan, P.J.; Yao, Q. Chemistry of β-(Acyloxy) Alkyl and β-(Phosphatoxy) Alkyl Radicals and Related Species: Radical and Radical Ionic Migrations and Fragmentations of Carbon−Oxygen Bonds. Chem. Rev. 1997, 97, 3273–3312. [Google Scholar] [CrossRef]
- Tang, X.; Studer, A. Alkene 1, 2-difunctionalization by Radical Alkenyl Migration. Angew. Chem. Int. Ed. 2018, 57, 814–817. [Google Scholar] [CrossRef]
- Li, L.; Li, Z.-L.; Gu, Q.-S.; Wang, N.; Liu, X.-Y. A Remote C–C Bond Cleavage–Enabled Skeletal Reorganization: Access to Medium-/Large-Sized Cyclic Alkenes. Sci. Adv. 2017, 3, e1701487. [Google Scholar] [CrossRef]
- Kyne, S.H.; Lefèvre, G.; Ollivier, C.; Petit, M.; Cladera, V.-A.R.; Fensterbank, L. Iron and cobalt catalysis: New perspectives in synthetic radical chemistry. Chem. Soc. Rev. 2020, 49, 8501. [Google Scholar] [CrossRef]
- Liu, Y.; You, T.; Wang, H.-X.; Tang, Z.; Zhou, C.-Y.; Chen, C.-M. Iron- and cobalt-catalyzed C(sp3)–H bond functionalization reactions and their application in organic synth esis. Chem. Soc. Rev. 2020, 49, 5310. [Google Scholar] [CrossRef]
- Lu, H.; Lang, K.; Jiang, H.; Wojtas, L.; Zhang, X.P. Intramolecular 1,5-C(sp3)–H Radical Amination via Co(II)-Based Metalloradical Catalysis for Five-Membered Cyclic Sulfamides. Chem. Sci. 2016, 7, 6934–6939. [Google Scholar] [CrossRef]
- Nobile, E.; Castanheiro, T.; Besset, T. Radical-Promoted Distal C−H Functionalization of C(sp3) Centers with Fluorinated Moieties. Angew. Chem. Int. Ed. 2021, 60, 12170–12191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Studer, A. Chemistry with N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem. 2019, 1, 38–49. [Google Scholar] [CrossRef]
- Stateman, L.M.; Nakafuku, K.M.; Nagib, D.A. Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [Google Scholar] [PubMed]
- Yang, C.-J.; Zhang, C.; Gu, Q.-S.; Fang, J.-H.; Su, X.-L.; Ye, L.; Sun, Y.; Tian, Y.; Li, Z.-L.; Liu, X.-Y. Cu-Catalysed Intramolecular Radical Enantioconvergent Tertiary β-C(sp3)–H Amination of Racemic Ketones. Nat. Catal. 2020, 3, 539–546. [Google Scholar] [CrossRef]
- Zhong, L.-J.; Lv, G.-F.; Ouyang, X.-H.; Li, Y.; Li, J.-H. Copper-Catalyzed Fluoroamide-Directed Remote Benzylic C–H Olefination: Facile Access to Internal Alkenes. Org. Chem. Front. 2022, 9, 4309–4315. [Google Scholar] [CrossRef]
- Liu, J.; Hong, Y.; Liu, Y.-L.; Tan, J.-Y.; Liu, H.-M.; Dai, G.-L.; Chen, S.-L.; Liu, T.; Li, J.-H.; Tang, S. Nickel-Catalyzed Radical Heck-Type C(sp3)–C(sp2) Coupling Cascades Enabled by Bromoalkane-Directed 1,4-Aryl Shift: Access to Olefinated Arylalanines. Org. Lett. 2022, 24, 8192–8196. [Google Scholar] [CrossRef]
- Li, J.; Liu, Z.; Wu, S.; Chen, Y. Acyl Radical Smiles Rearrangement To Construct Hydroxybenzophenones by Photoredox Catalysis. Org. Lett. 2019, 21, 2077–2080. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Chen, W.; Liu, Z.; Tian, J.; Li, J.; Ma, J.; Cheng, X.; Li, G. Building Congested Ketone: Substituted Hantzsch Ester and Nitrile as Alkylation Reagents in Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 12312–12315. [Google Scholar] [CrossRef]
- Phelan, J.P.; Lang, S.B.; Sim, J.; Berritt, S.; Peat, A.J.; Billings, K.; Fan, L.; Molander, G.A. Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc. 2019, 141, 3723–3732. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Li, Z.-H.; Liu, J.-Q.; Wang, X.-S. Copper-Catalyzed Synthesis of Dibenzo [b, f] Imidazo [1, 2-d][1, 4] Oxazepine Derivatives via a Double Ullmann Coupling Reaction. Synthesis 2019, 51, 1662–1668. [Google Scholar]
- Zhao, R.; Chang, D.; Shi, L. Recent Advances in Cyclic Diacyl Peroxides: Reactivity and Selectivity Enhancement Brought by the Cyclic Structure. Synthesis 2017, 49, 3357–3365. [Google Scholar] [CrossRef]
- Zhang, Z.; Rogers, C.R.; Weiss, E.A. Energy Transfer from CdS QDs to a Photogenerated Pd Complex Enhances the Rate and Selectivity of a Pd-Photocatalyzed Heck Reaction. J. Am. Chem. Soc. 2020, 142, 495–501. [Google Scholar] [CrossRef]
- Cao, H.; Kuang, Y.; Shi, X.; Wong, K.L.; Tan, B.B.; Kwan, J.M.C.; Liu, X.; Wu, J. Photoinduced Site-Selective Alkenylation of Alkanes and Aldehydes with Aryl Alkenes. Nat. Commun. 2020, 11, 1956. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, Z.-L.; Ouyang, X.-H.; Song, R.-J.; Li, J.-H. Copper-catalyzed oxidative decarboxylative alkylation of cinnamic acids with 4-alkyl-1,4-dihydropyridines†. Chem. Commun. 2020, 56, 14055–14058. [Google Scholar] [CrossRef] [PubMed]
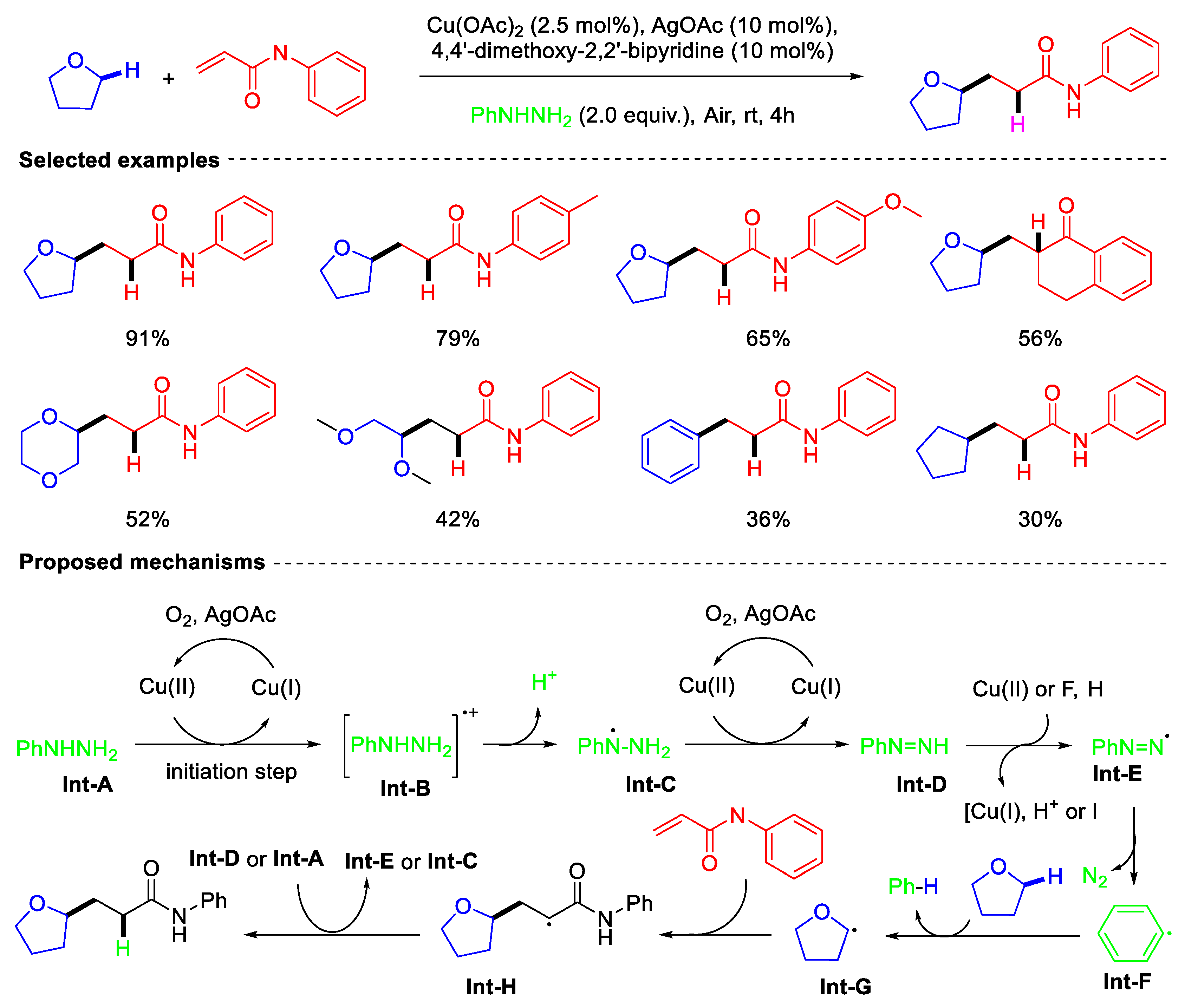
- Bai, P.; Zhang, X.; Su, Q.; Xiao, T.; Jiang, Y.; Zhang, G.; Xie, Y.; Qin, G. Cu-Catalyzed Formal Addition of Aliphatic C(sp3)−H Bonds to Alkenes via Phenyl Radical Involved Intermolecular HAT. Adv. Synth. Catal. 2024, 19, 4042–4048. [Google Scholar] [CrossRef]
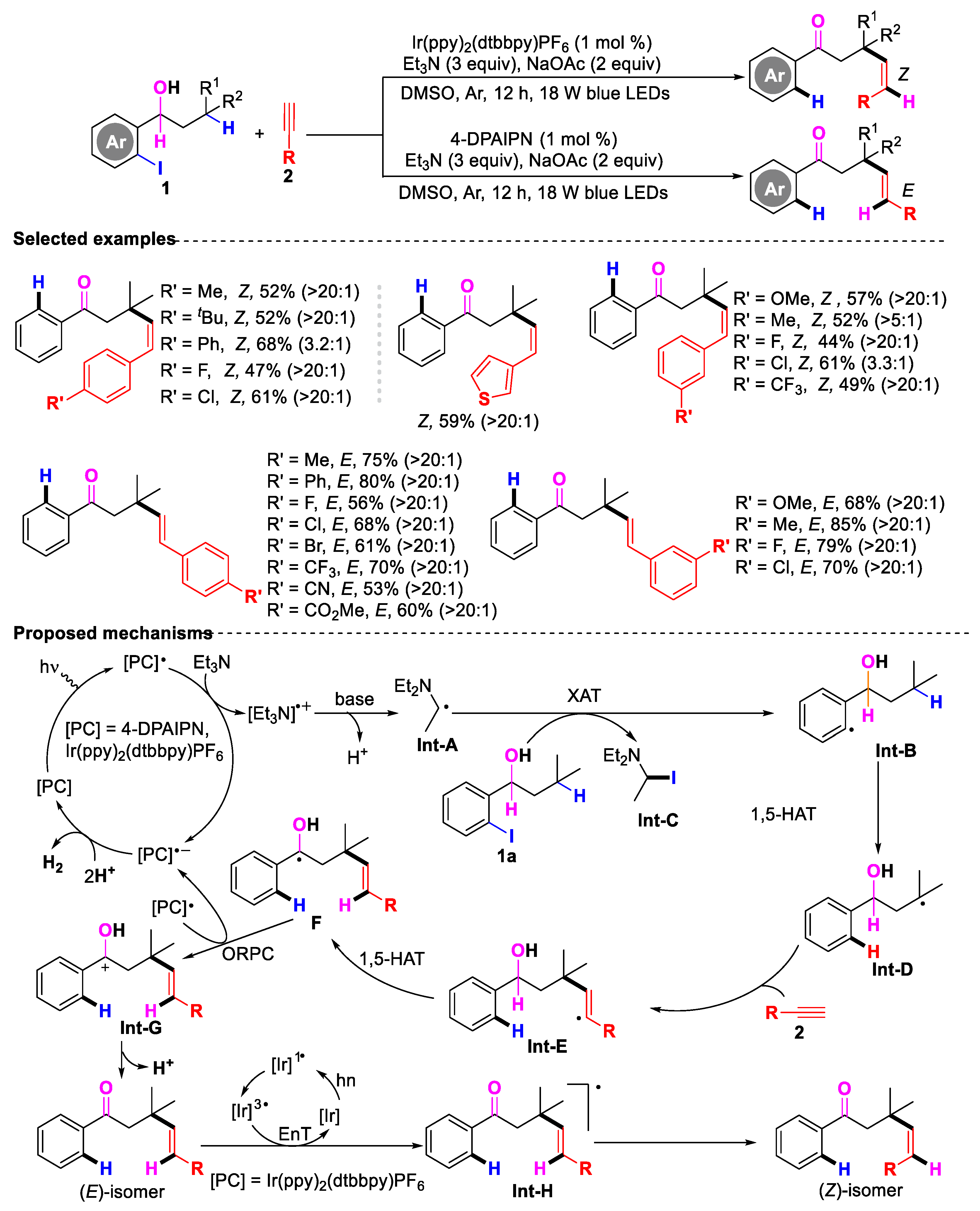
- Zeng, L.; Zhang, Y.; Hu, M.; He, D.-L.; Ouyang, X.-H.; Li, J.-H. Divergent Synthesis of (E)- and (Z)-Alkenones via Photoredox C(sp3)−H Alkenylation−Dehydrogenation of o-Iodoarylalkanols with Alkynes. Org. Lett. 2024, 26, 10096–10101. [Google Scholar] [CrossRef]
- Li, Y.; Nie, W.; Chang, Z.; Wang, J.-W.; Lu, X.; Fu, Y. Cobalt-Catalysed Enantioselective C(sp3)–C(sp3) Coupling. Nat. Catal. 2021, 4, 901–991. [Google Scholar] [CrossRef]
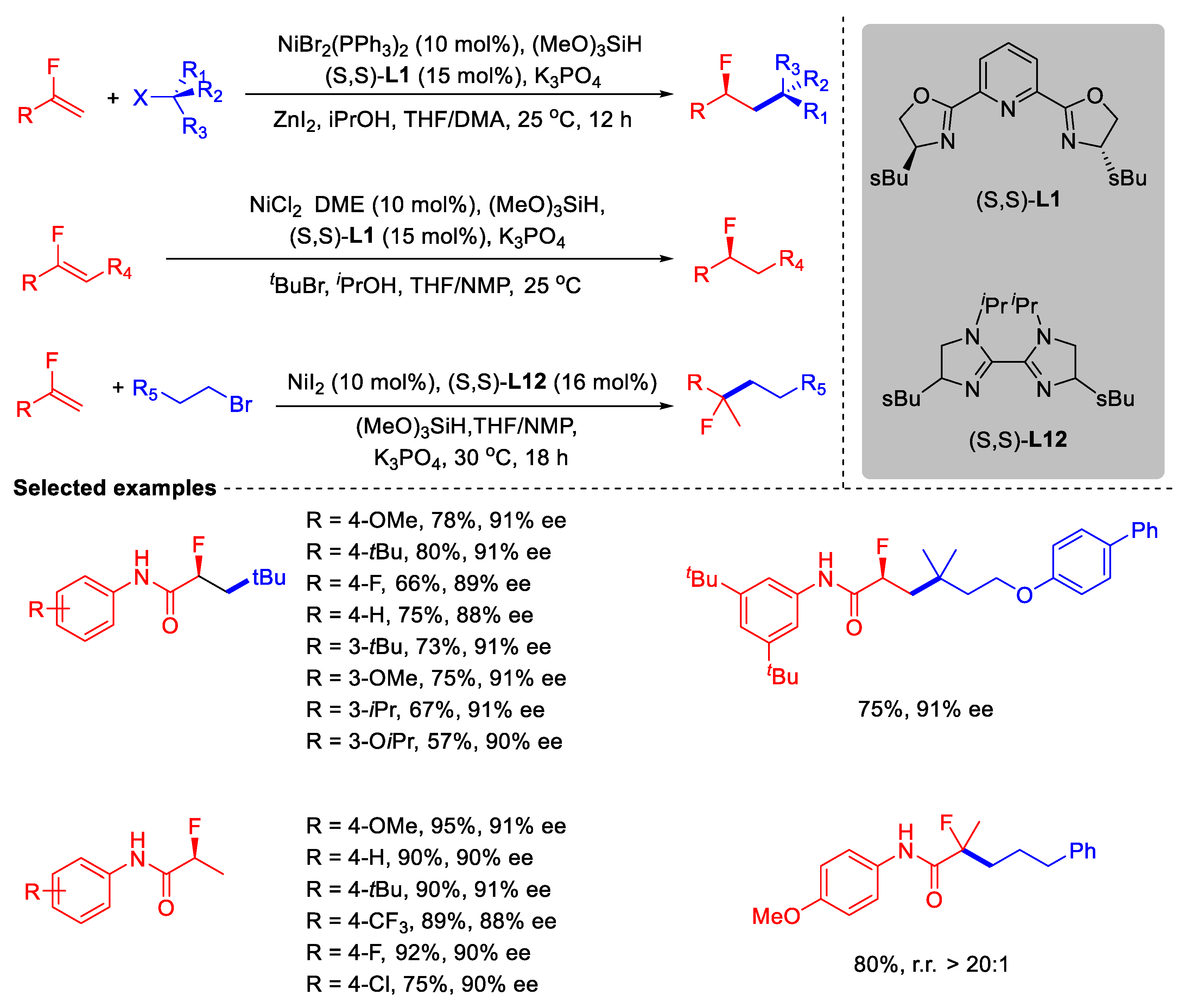
- Chen, F.; Zhang, Q.; Li, Y.; Yu, Z.-X.; Chu, L. Selective Hydrofunctionalization of Alkenyl Fluorides Enabled by Nickel-Catalyzed Hydrogen Atoms and Group Transfer: Reaction Development and Mechanistic Study. J. Am. Chem. Soc. 2024, 146, 11418–11431. [Google Scholar] [CrossRef]
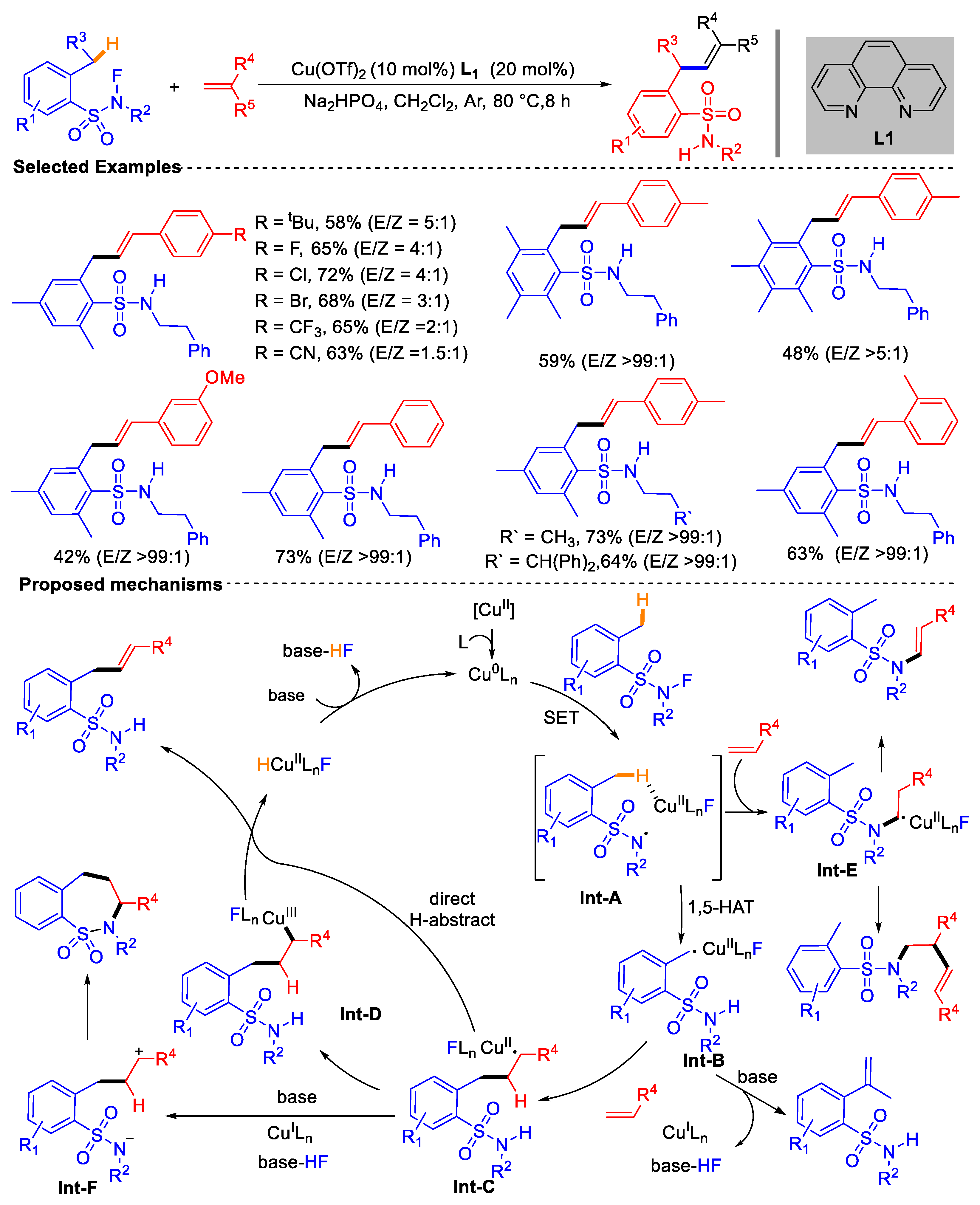
- Zhong, L.-J.; Xiong, Z.-Q.; Ouyang, X.-H.; Li, Y.; Song, R.-J.; Sun, Q.; Lu, X.; Li, J.-H. Intermolecular 1,2-Difunctionalization of Alkenes Enabled by Fluoroamide-Directed Remote Benzyl C(sp3)–H Functionalization. J. Am. Chem. Soc. 2022, 144, 339–348. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Zhang, Z.; Niu, Z.-J.; Shi, W.-Y.; Gong, X.-P.; Jiao, R.-Q.; Gao, M.-H.; Liu, X.-Y.; Liang, Y.-M. Copper-Catalyzed Hydrogen Atom Transfer and Aryl Migration Strategy for the Arylalkylation of Activated Alkenes. Org. Lett. 2022, 24, 4338–4343. [Google Scholar] [CrossRef] [PubMed]
- Rui, R.; Wang, B.; Xu, X.-J.; Zhang, Z.; Chen, X.; Liu, X.-Y. Cu-Catalyzed Alkylarylation of Alkenes via N-Directed Remote C(sp3)–H Functionalization. Org. Chem. Front. 2024, 11, 1484–1489. [Google Scholar] [CrossRef]
- Qin, J.-H.; Luo, M.-J.; An, D.-L.; Li, J.-H. Electrochemical 1,2-Diarylation of Alkenes Enabled by Direct Dual C–H Functionalizations of Electron-Rich Aromatic Hydrocarbons. Angew. Chem. Int. Ed. 2021, 60, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
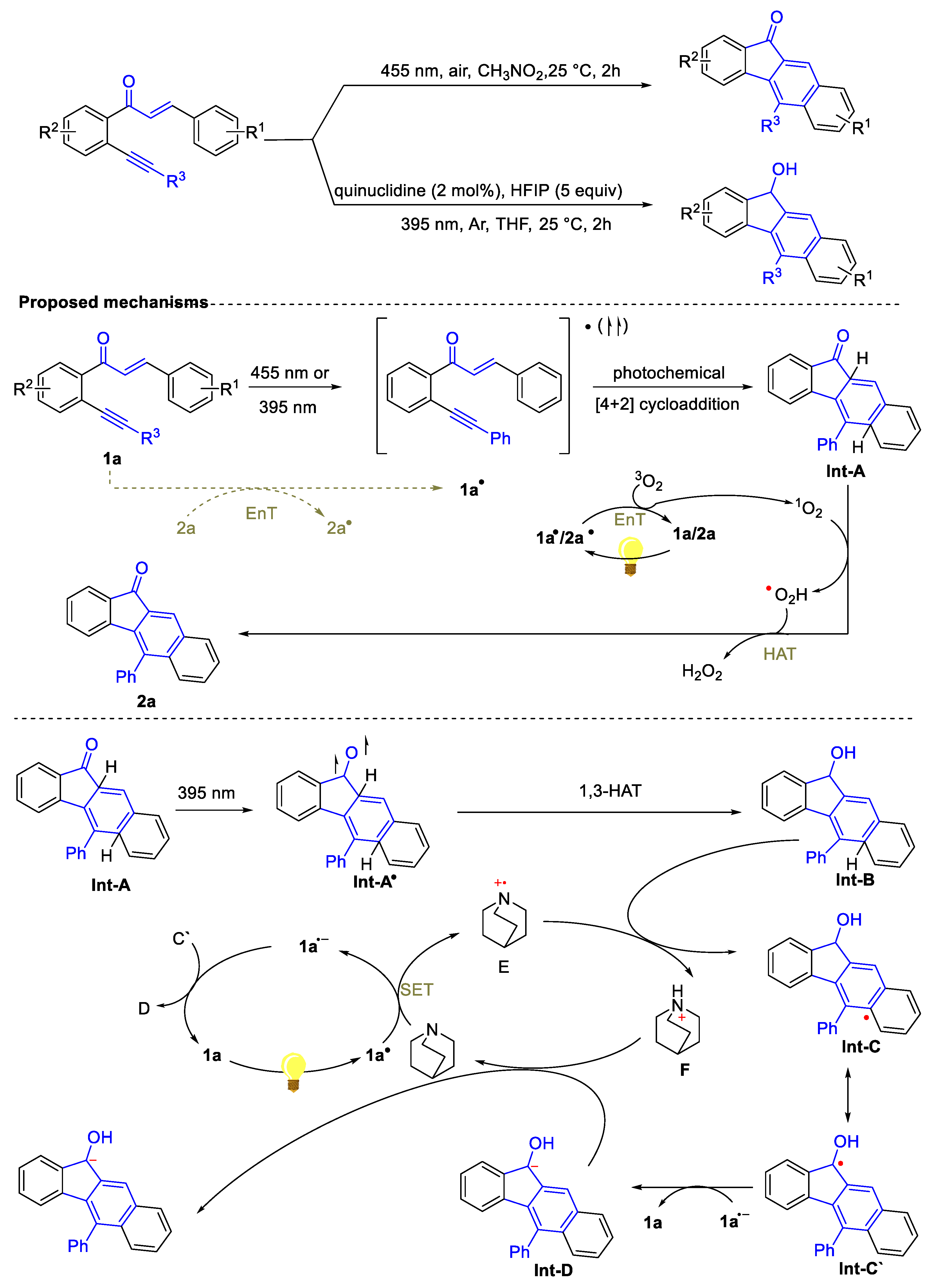
- Shi, J.-F.; Sun, Q.-Y.; Gao, Y.-L.; Liang, D.-Q. Self-photocatalysis with multiple activities: Divergent synthesis of benzo[b]fluorenones and benzo[b]fluorenols from enone-ynes†. Org. Chem. Front. 2025, 12, 2352–2361. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.-L.; Song, R.-J.; Li, J.-H. 1, 2-Alkylarylation of Activated Alkenes with Dual C–H Bonds of Arenes and Alkyl Halides toward Polyhalo-Substituted Oxindoles. Org. Chem. Front. 2014, 1, 1289–1294. [Google Scholar] [CrossRef]
- Lu, M.-Z.; Loh, T.-P. Iron-Catalyzed Cascade Carbochloromethylation of Activated Alkenes: Highly Efficient Access to Chloro-Containing Oxindoles. Org. Lett. 2014, 16, 4698–4701. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Yin, X.-P.; Zhou, J. Internally Reuse Waste: Catalytic Asymmetric One-Pot Strecker Reaction of Fluoroalkyl Ketones, Anilines and TMSCN by Sequential Catalysis. Chin. J. Chem. 2018, 36, 321–328. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Gao, Y.; Fang, H.; Tang, G.; Zhao, Y. Cascade Arylalkylation of Activated Alkenes: Synthesis of Chloro-and Cyano-Containing Oxindoles. J. Org. Chem. 2015, 80, 2621–2626. [Google Scholar] [CrossRef]
- Pan, C.; Gao, D.; Yang, Z.; Wu, C.; Yu, J.-T. Metal-Free Radical Cascade Chloromethylation of Unactivated Alkenes: Synthesis of Polychloro-Substituted Indolines. Org. Biomol. Chem. 2018, 16, 5752–5755. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.-L.; Song, R.-J.; Li, J.-H. Visible-Light-Facilitated 5-Exo-Trig Cyclization of 1,6-Dienes with Alkyl Chlorides: Selective Scission of the C(sp3)–H Bond in Alkyl Chlorides. Eur. J. Org. Chem. 2014, 2014, 1177–1181. [Google Scholar] [CrossRef]
- Liu, Y.; Song, R.-J.; Luo, S.; Li, J.-H. Visible-Light-Promoted Tandem Annulation of N-(o-Ethynylaryl) Acrylamides with CH2Cl2. Org. Lett. 2018, 20, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, J.; Chu, L. Solvent-Tuned Chemoselective Carboazidation and Diazidation of Alkenes via Iron Catalysis. Org. Chem. Front. 2019, 6, 512–516. [Google Scholar] [CrossRef]
- Li, W.-Y.; Wu, C.-S.; Wang, Z.; Luo, Y. Fe-Catalyzed Three-Component Carboazidation of Alkenes with Alkanes and Trimethylsilyl Azide. Chem. Comm. 2018, 54, 11013–11016. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liang, Y.-Y.; Ouyang, X.-H.; Xiao, Y.-T.; Qin, J.-H.; Song, R.-J.; Li, J.-H. Three-Component Photoredox 1,2-Alkylamination of Styrenes with Alkanes and Nitrogen Nucleophiles via C(sp3)–H Bond Cleavage. Org. Chem. Front. 2021, 8, 7009–7014. [Google Scholar] [CrossRef]
- Liang, W.; Jiang, K.; Du, F.; Yang, J.; Shuai, L.; Ouyang, Q.; Chen, Y.-C.; Wei, Y. Iron-Catalyzed, Iminyl Radical-Triggered Cascade 1, 5-Hydrogen Atom Transfer/(5+ 2) or (5+ 1) Annulation: Oxime as a Five-Atom Assembling Unit. Angew. Chem. Int. Ed. 2020, 59, 19222–19228. [Google Scholar] [CrossRef]
- Jiang, K.; Li, S.-J.; Liu, Q.-P.; Yu, N.; Li, Y.-L.; Zhou, Y.-Q.; He, K.-C.; Lin, J.; Zheng, T.-Y.; Lang, J. Iminyl Radical-Triggered Relay Annulation for the Construction of Bridged Aza-Tetracycles Bearing Four Contiguous Stereogenic Centers. Chem. Sci. 2022, 13, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
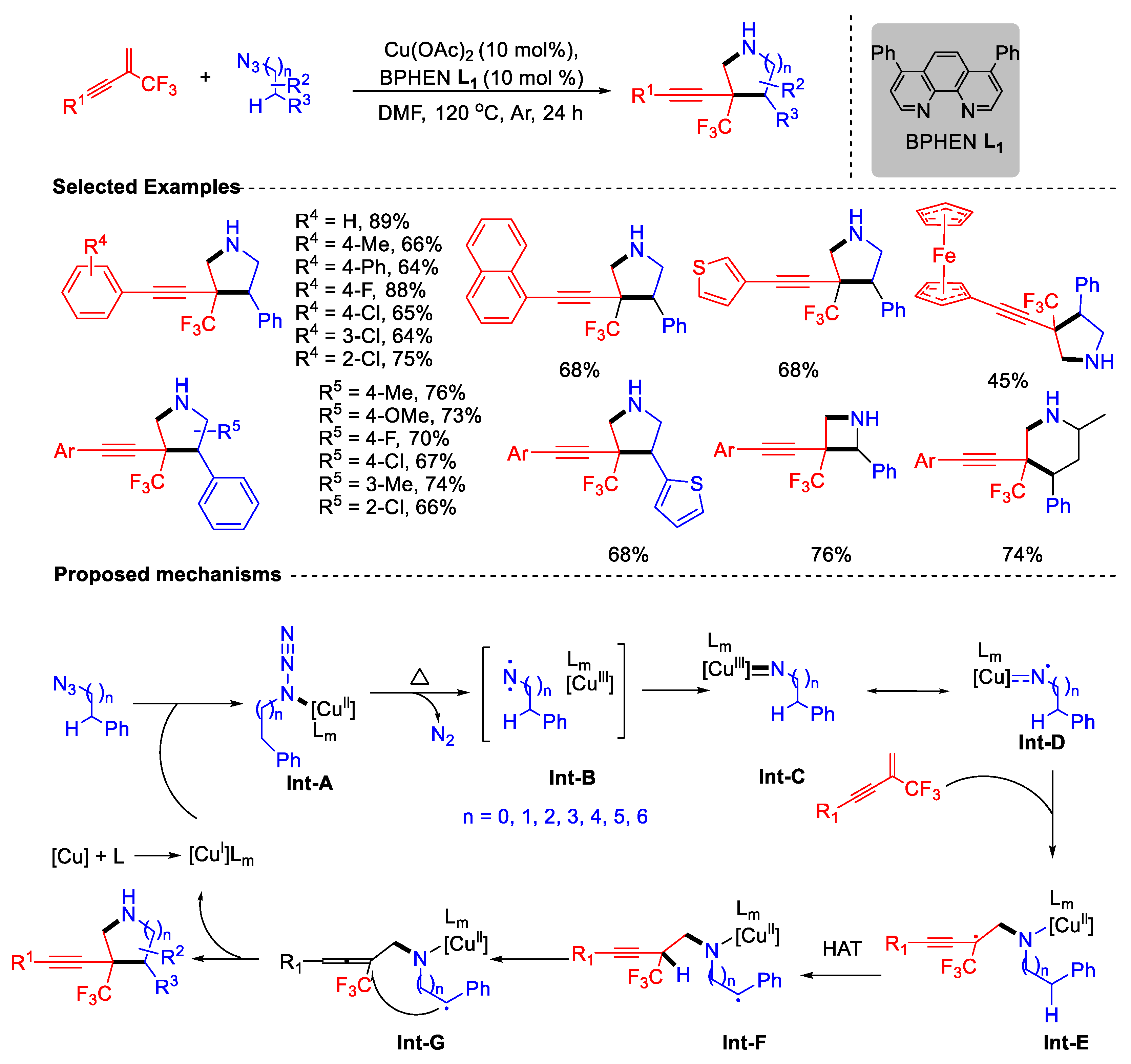
- Yang, Y.-Z.; Xue, Q.; Xiong, Z.-Q.; Li, Y.; Ouyang, X.-H.; Hu, M.; Li, J.-H. Divergent [2 + n] Heteroannulation of β-CF3-1,3-Enynes with Alkyl Azides via Hydrogen Atom Transfer and Radical Substitution. Org. Lett. 2024, 26, 889–894. [Google Scholar] [CrossRef]
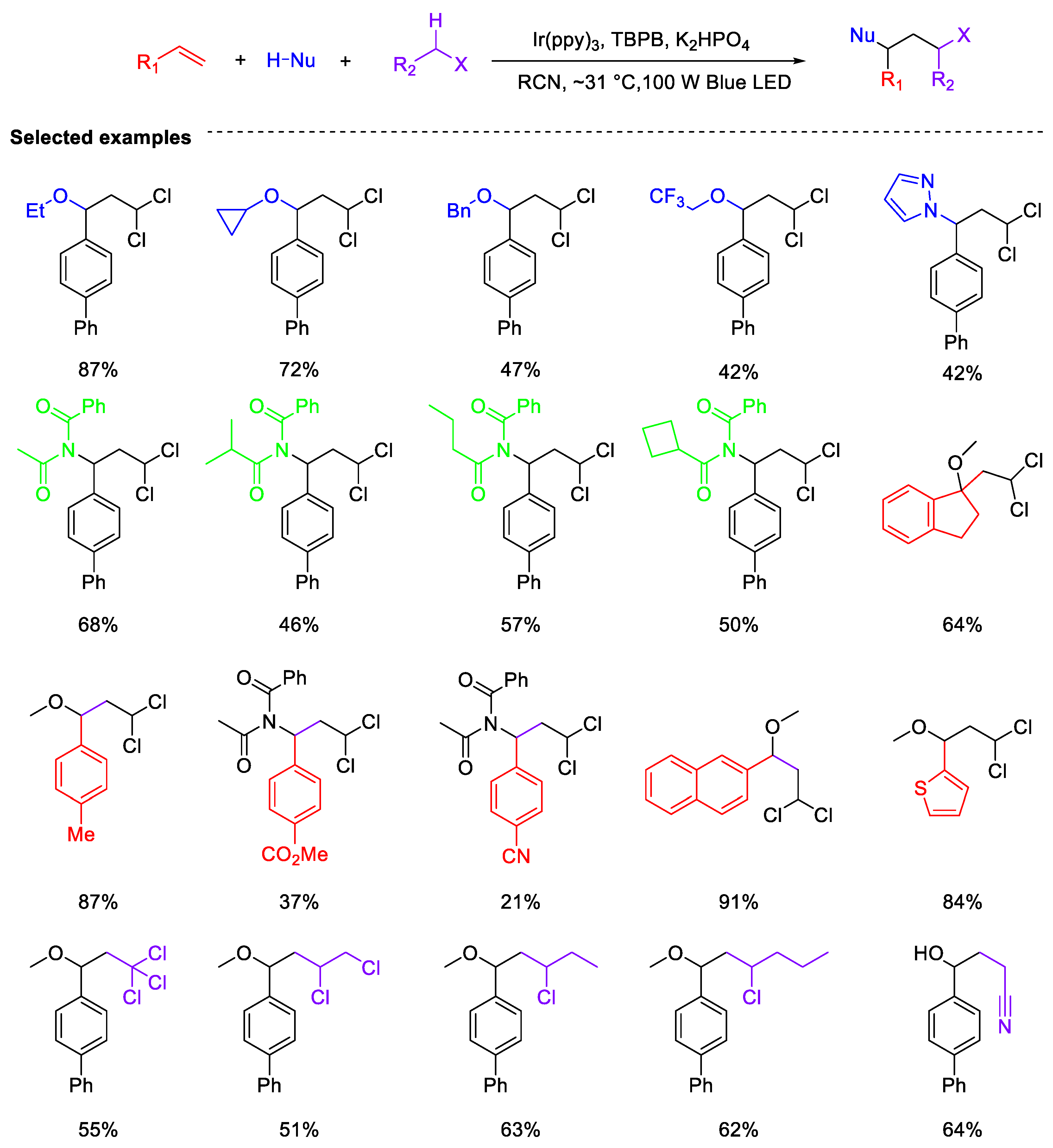
- Hao, T.; Shen, J.; Wei, Y.; Shi, M. Alkene Difunctionalization Enabled by Photocatalytic Hydrogen Atom Transfer from Haloalkane α-C(sp3)–H Bonds. Chem. Catal. 2023, 3, 100807. [Google Scholar] [CrossRef]
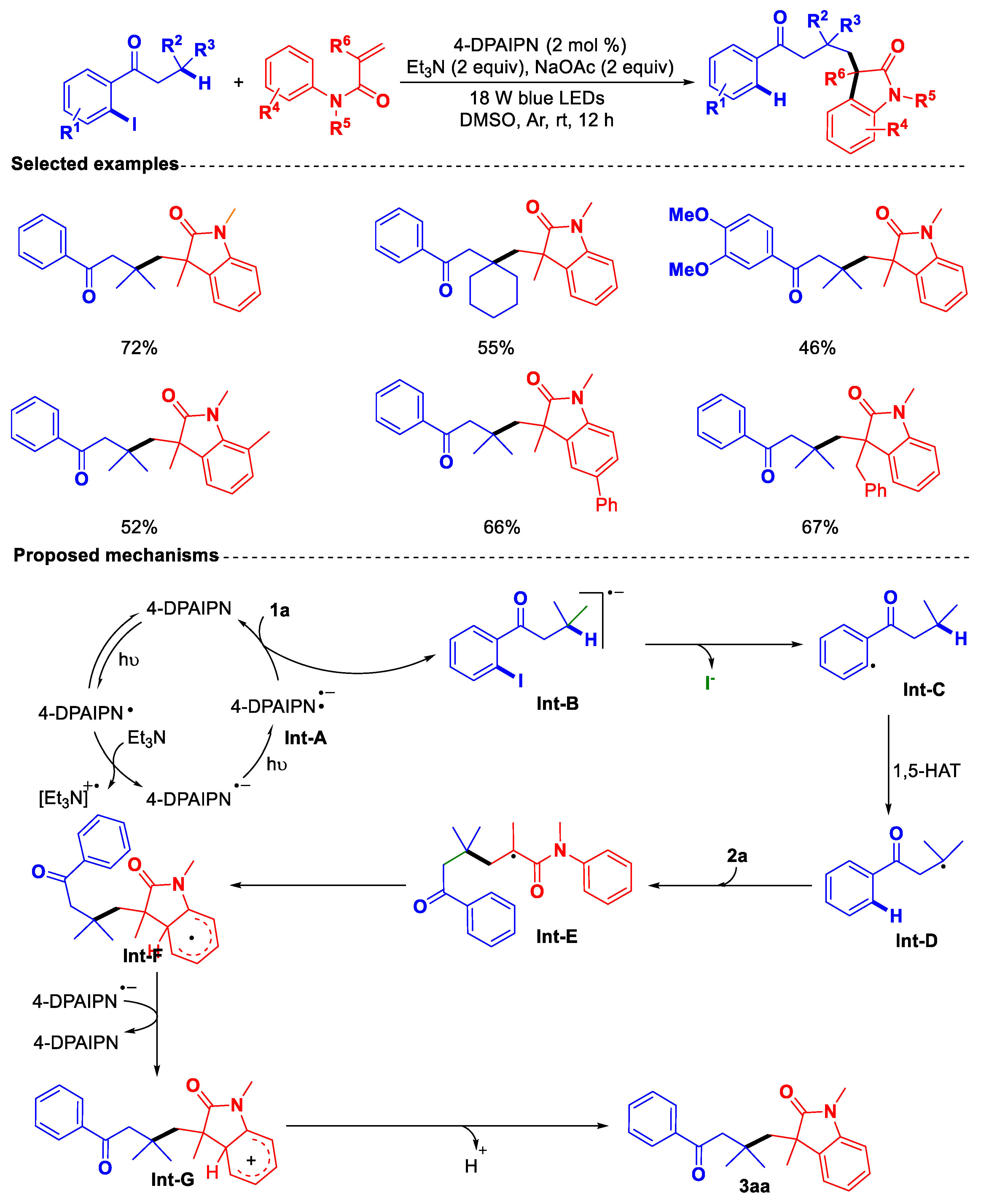
- Lu, Z.-R.; Zeng, L.; Wu, S.; Hu, M.; Li, J.-H. Photoredox Site-Selective C(sp3)−H Alkylation of 1-(o-Iodoaryl) alkan-1-ones with Activated Alkenes Enabled by Hydrogen Atom Transfer. Org. Lett. 2025, 27, 2970–2974. [Google Scholar] [CrossRef]
- Tong, S.; Limouni, A.; Wang, Q.; Wang, M.-X.; Zhu, J. Catalytic Enantioselective Double Carbopalladation/C−H Functionalization with Statistical Amplification of Product Enantiopurity: A Convertible Linker Approach. Angew. Chem. Int. Ed. 2017, 129, 14380–14384. [Google Scholar] [CrossRef]
- Ping, Y.; Wang, K.; Pan, Q.; Ding, Z.; Zhou, Z.; Guo, Y.; Kong, W. Ni-Catalyzed Regio-and Enantioselective Domino Reductive Cyclization: One-Pot Synthesis of 2, 3-Fused Cyclopentannulated Indolines. ACS Catal. 2019, 9, 7335–7342. [Google Scholar] [CrossRef]
- Li, Y.; Ding, Z.; Lei, A.; Kong, W. Ni-Catalyzed Enantioselective Reductive Aryl-Alkenylation of Alkenes: Application to the Synthesis of (+)-Physovenine and (+)-Physostigmine. Org. Chem. Front. 2019, 6, 3305–3309. [Google Scholar] [CrossRef]
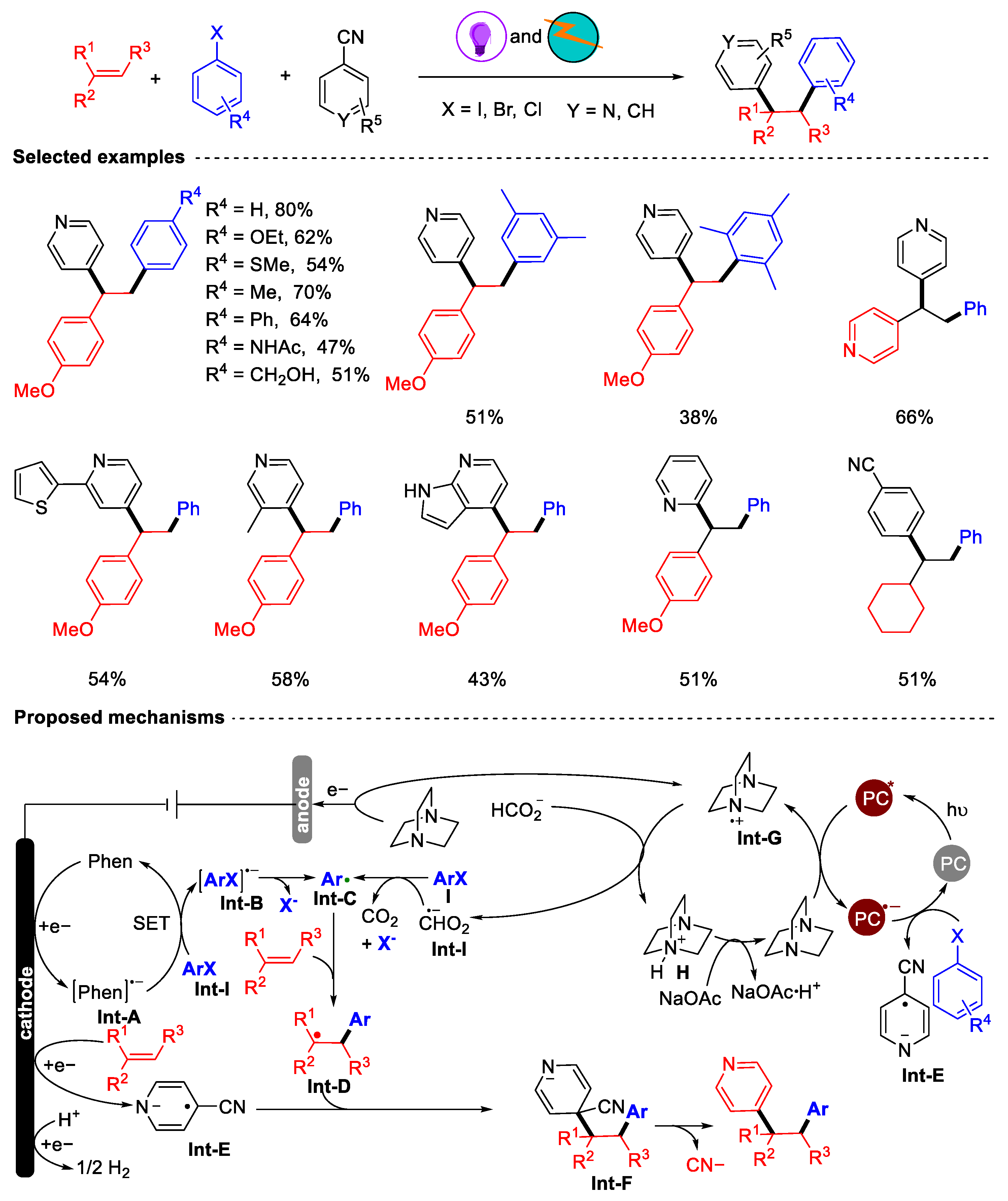
- Zeng, L.; Qin, J.-H.; Lv, G.-F.; Hu, M.; Sun, Q.; Ouyang, X.-H.; He, D.-L.; Li, J.-H. Electrophotocatalytic Reductive 1, 2-Diarylation of Alkenes with Aryl Halides and Cyanoaromatics. Chin. J. Chem. 2023, 41, 1921–1930. [Google Scholar] [CrossRef]
- Ouyang, X.-H.; Li, Y.; Song, R.-J.; Hu, M.; Luo, S.; Li, J.-H. Intermolecular Dialkylation of Alkenes with Two Distinct C (sp3)—H Bonds Enabled by Synergistic Photoredox Catalysis and Iron Catalysis. Sci. Adv. 2019, 5, eaav9839. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-H.; Lv, G.-F.; Qin, J.-H.; Xu, X.-H.; Li, J.-H. Visible-Light-Induced Photoredox 1,2-Dialkylation of Styrenes with α-Carbonyl Alkyl Bromides and Pyridin-1-Ium Salts. J. Org. Chem. 2024, 89, 281–290. [Google Scholar] [CrossRef]
- Tuhin, P.; Peter, B.; Felix, S.; Frank, G. Photosensitized Intermolecular Carboimination of Alkenes through the Persistent Radical Effect. Angew. Chem. Int. Ed. 2020, 59, 3172–3177. [Google Scholar]
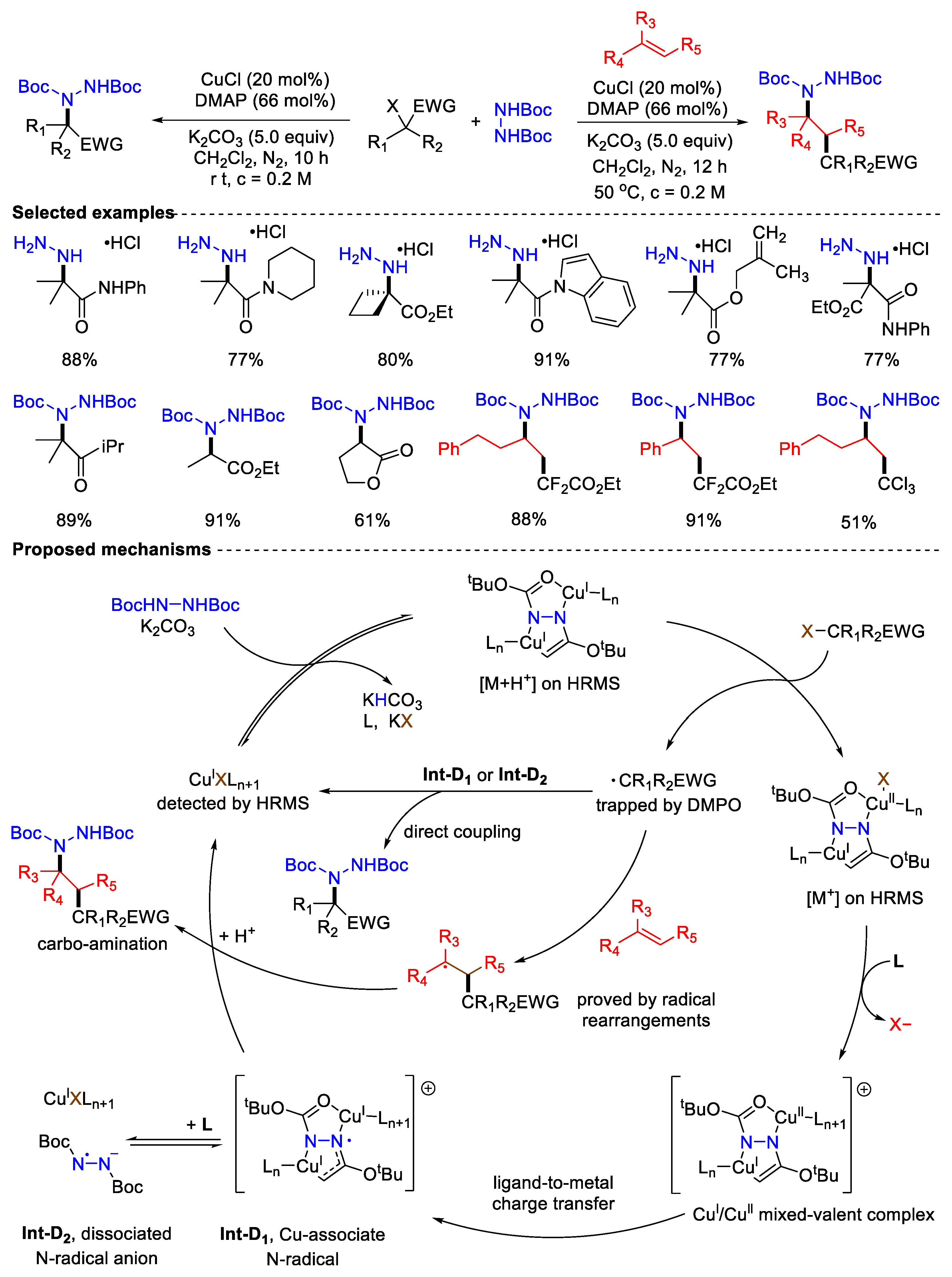
- Lu, D.; Li, Y.; Wang, P.; Wang, Z.; Yang, D.; Gong, Y. Cu-Catalyzed C(sp3)–N Coupling and Alkene Carboamination Enabled by Ligand-Promoted Selective Hydrazine Transfer to Alkyl Radicals. ACS Catal. 2022, 12, 3269–3278. [Google Scholar] [CrossRef]
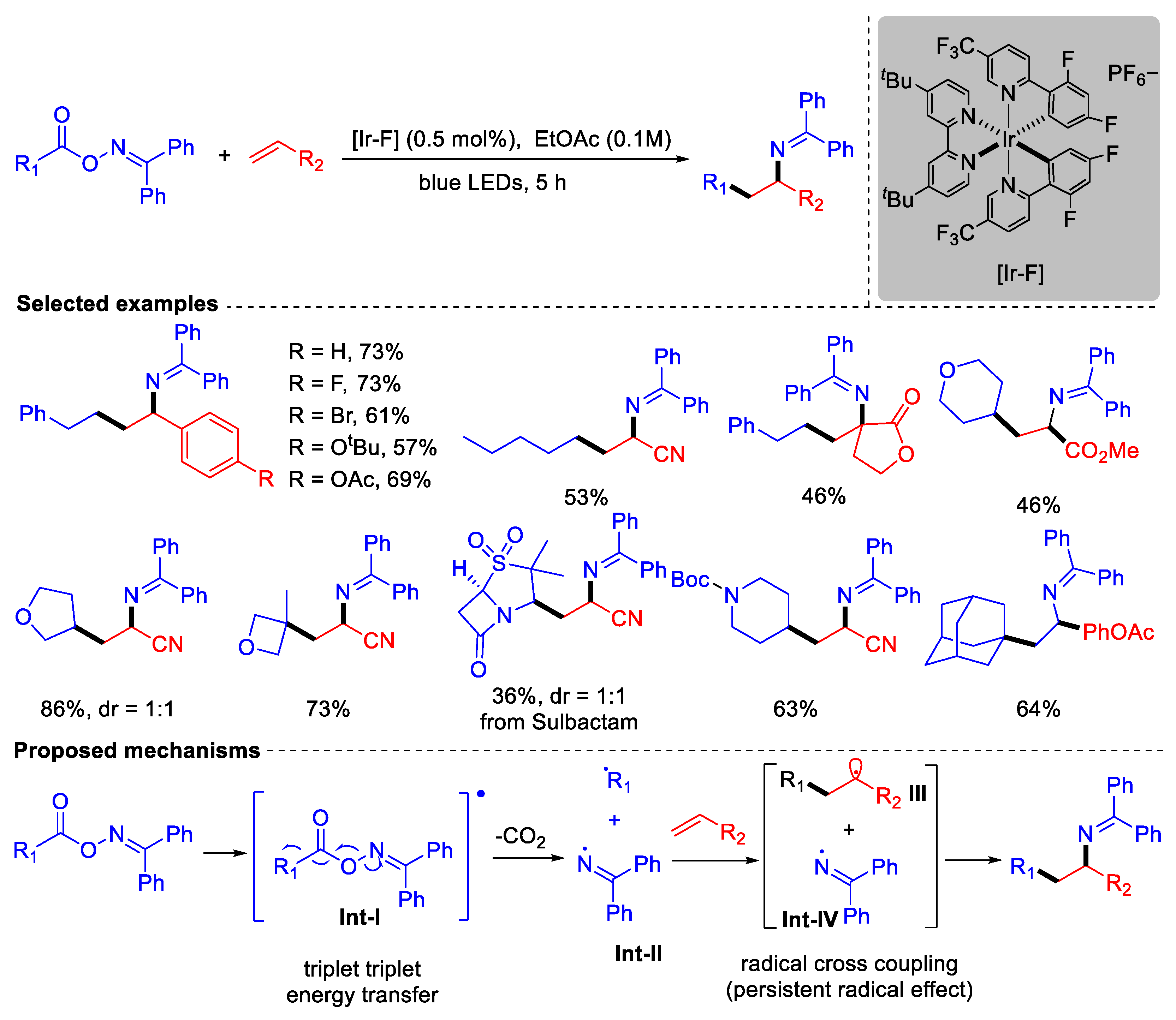
- Huang, H.-M.; Bellotti, P.; Pflüger, P.M.; Schwarz, J.L.; Heidrich, B.; Glorius, F. Three-Component, Interrupted Radical Heck/Allylic Substitution Cascade Involving Unactivated Alkyl Bromides. J. Am. Chem. Soc. 2020, 142, 10173–10183. [Google Scholar] [CrossRef]
- Fujita, T.; Fuchibe, K.; Ichikawa, J. Transition-Metal-Mediated and-Catalyzed C− F Bond Activation by Fluorine Elimination. Angew. Chem. Int. Ed. 2019, 58, 390–402. [Google Scholar] [CrossRef]
- Ai, H.-J.; Ma, X.; Song, Q.; Wu, X.-F. C-F Bond Activation under Transition-Metal-Free Conditions. Sci. China Chem. 2021, 64, 1630–1659. [Google Scholar] [CrossRef]
- Ma, X.; Song, Q. Recent Progress on Selective Deconstructive Modes of Halodifluoromethyl and Trifluoromethyl-Containing Reagents. Chem. Soc. Rev. 2020, 49, 9197–9219. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-Z.; Xue, Q.; Sun, Q.; Li, Y.; Hu, M.; Xu, C.-H.; Li, J.-H. Radical-Mediated [3 + 2 + 1] Annulation of α-Polyfluoromethyl Alkenes with Arylisocyanates Enabled by C(sp3)–F Activation. Org. Chem. Front. 2024, 11, 1305–1313. [Google Scholar] [CrossRef]









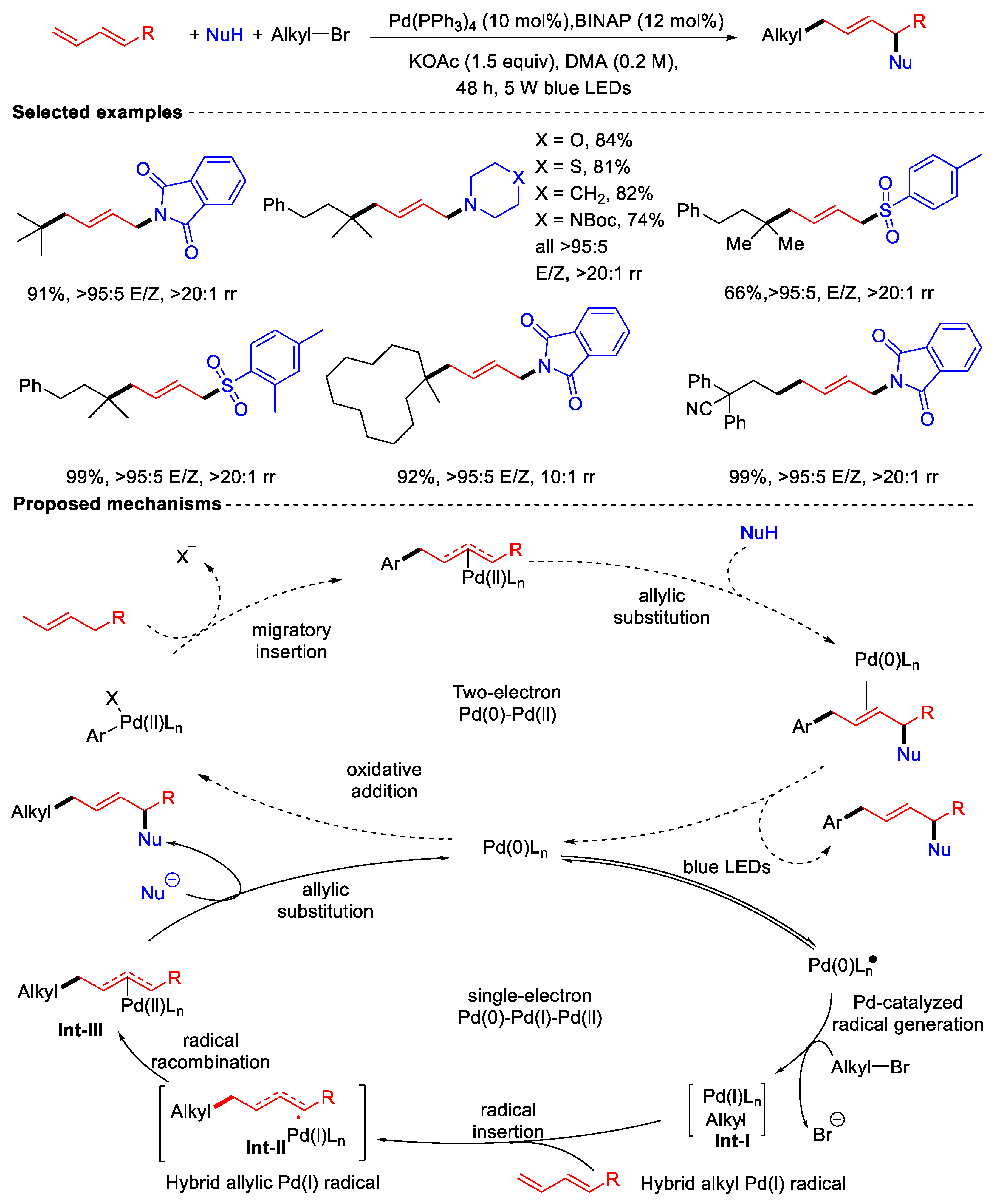
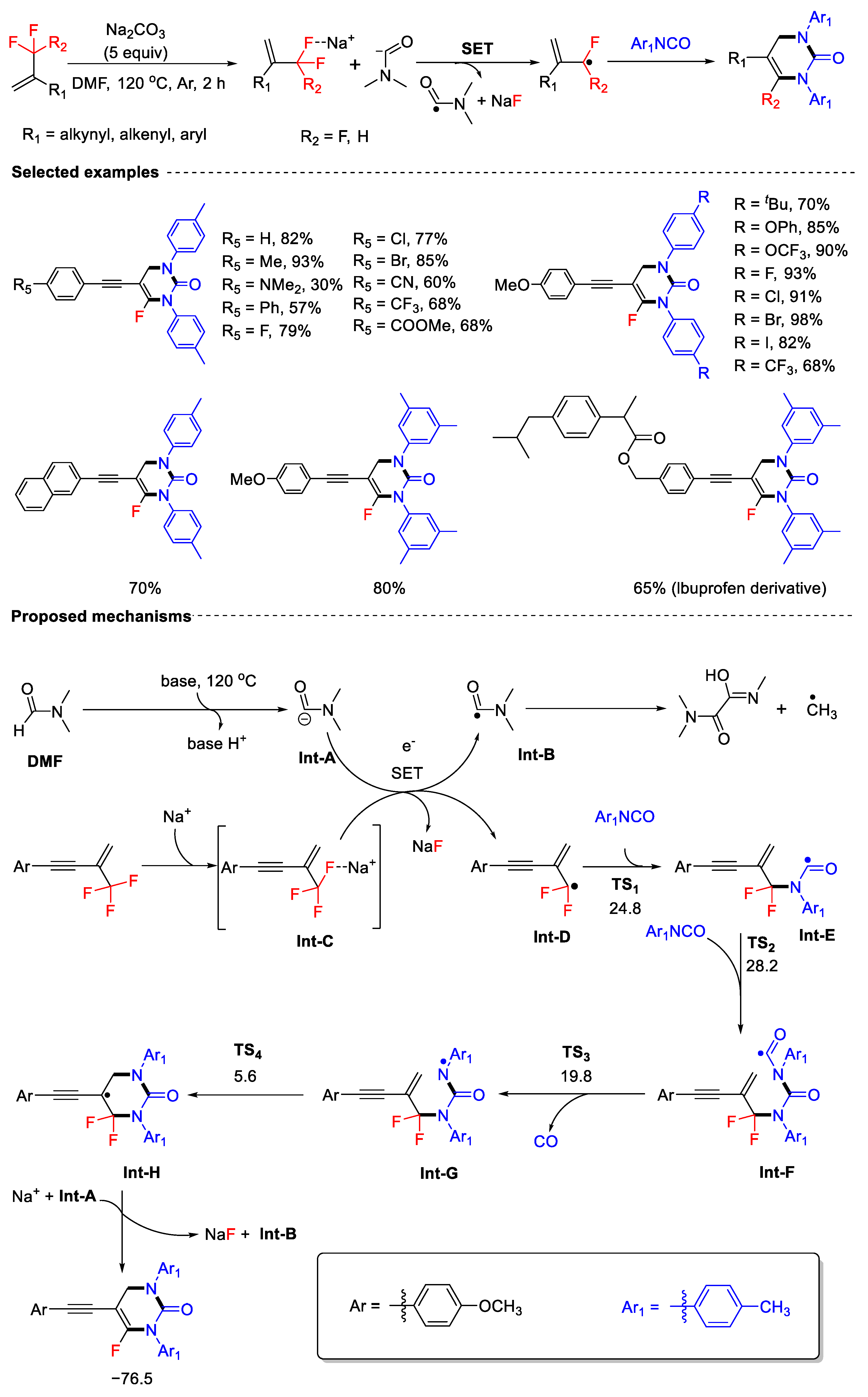
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, H.; Hu, M.; Li, J. Recent Advances in Carbon-Centered Radical-Initiated Olefin Transformation Chemistry. Catalysts 2025, 15, 461. https://doi.org/10.3390/catal15050461
Ren H, Hu M, Li J. Recent Advances in Carbon-Centered Radical-Initiated Olefin Transformation Chemistry. Catalysts. 2025; 15(5):461. https://doi.org/10.3390/catal15050461
Chicago/Turabian StyleRen, Huazhan, Ming Hu, and Jinheng Li. 2025. "Recent Advances in Carbon-Centered Radical-Initiated Olefin Transformation Chemistry" Catalysts 15, no. 5: 461. https://doi.org/10.3390/catal15050461
APA StyleRen, H., Hu, M., & Li, J. (2025). Recent Advances in Carbon-Centered Radical-Initiated Olefin Transformation Chemistry. Catalysts, 15(5), 461. https://doi.org/10.3390/catal15050461