

Natural Chiral Ligand Strategy: Metal-Catalyzed Reactions with Ligands Prepared from Amino Acids and Peptides



Abstract
1. Introduction
2. Scope of Review
3. Applications of Amino Acid and Peptide Ligands in Metal Catalysis
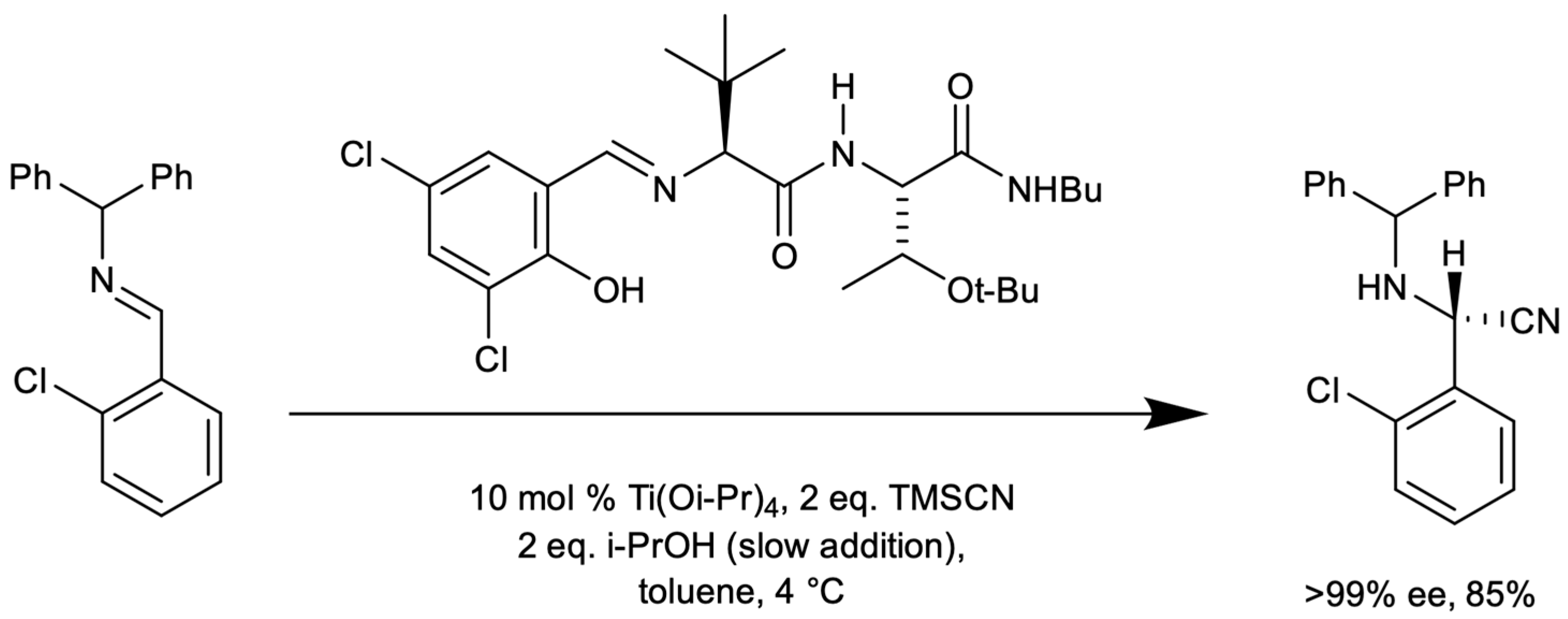
3.1. Ti-Catalyzed Cyanide Addition to Imines
3.2. Cu-Catalyzed Asymmetric Conjugate Addition of Alkylzinc to Disubstituted Cyclic Enones
3.3. Zr- and Hf-Catalyzed Enantioselective Additions of R2Zn to Imines
3.4. Ru-Catalyzed Enantioselective Reduction of Acetophenone
3.5. Pd-Catalyzed Sonogashira and Suzuki Cross Coupling Reactions
3.6. Cu-Catalyzed Enantioselective Cross-Coupling C−O Bond Formation
3.7. Mono-Nitrogen-Protected Amino Acids (MPAAs) as Bidentate Ligands for Pd-Catalyzed C−H Functionalization
3.8. Vanadium-Based Catalysts for Asymmetric Epoxidation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gung, B.W.; Holmes, M.; Jones, C.; Ma, R.; Barnes, C. Structure-enantioselectivity correlation in NHC-Au(I) catalysis for 1,6-enynecyclization. Tetrahedron Lett. 2016, 57, 3912–3915. [Google Scholar] [CrossRef]
- Deng, H.; Isler, M.; Snapper, M.; Hoveyda, A. Aluminum-Catalyzed Asymmetric Addition of TMSCN to Aromatic and Aliphatic Ketones Promoted by an Easily Accessible and Recyclable Peptide Ligand. Angew. Chem. Int. Ed. 2002, 41, 1009–1012. [Google Scholar] [CrossRef]
- Krueger, C.; Kuntz, K.; Dzierba, C.; Wirschun, W.; Gleason, J.; Snapper, M.; Hoveyda, A. Ti-Catalyzed Enantioselective Addition of Cyanide to Imines. A Practical Synthesis of Optically Pure α-Amino Acids. J. Am. Chem. Soc. 1999, 121, 4284–4285. [Google Scholar] [CrossRef]
- Josephson, N.; Kuntz, K.; Snapper, M.; Hoveyda, A. Mechanism of enantioselective Ti-catalyzed Strecker reaction: Peptide-based metal complexes as bifunctional catalysts. J. Am. Chem. Soc. 2001, 123, 11594–11599. [Google Scholar] [CrossRef]
- Luchaco-Cullis, C.; Mizutani, H.; Murphy, K.; Hoveyda, A. Modular Pyridinyl Peptide Ligands in Asymmetric Catalysis: Enantioselective Synthesis of Quaternary Carbon Atoms Through Copper-Catalyzed Allylic Substitutions. Angew. Chem. Int. Ed. 2001, 40, 1456–1460. [Google Scholar] [CrossRef]
- Hoveyda, A.; Hird, A.; Kacprzynski, M. Small Peptides as Ligands for Catalytic Asymmetric Alkylations of Olefins. Rational Design of Catalysts or of Searches that Lead to Them. Chem. Commun. 2004, 16, 1779–1785. [Google Scholar] [CrossRef]
- Degrado, S.; Mizutani, H.; Hoveyda, A. Modular Peptide-Based Phosphine Ligands in Asymmetric Catalysis: Efficient and Enantioselective Cu-Catalyzed Conjugate Additions to Five-, Six-, and Seven-Membered Cyclic Enones. J. Am. Chem. Soc. 2001, 123, 755–756. [Google Scholar] [CrossRef]
- Hird, A.; Hoveyda, A. Catalytic Enantioselective Alkylations of Tetrasubstituted Olefins. Synthesis of All-Carbon Quaternary Stereogenic Centers through Cu-Catalyzed Asymmetric Conjugate Additions of Alkylzinc Reagents to Enones. J. Am. Chem. Soc. 2005, 127, 14988–14989. [Google Scholar] [CrossRef]
- Hird, A.; Hoveyda, A. Cu-Catalyzed Enantioselective Conjugate Additions of Alkyl Zinc Reagents to Unsaturated N-Acyl Oxazolidinones Promoted by a Chiral Triamide Phosphane. Angew. Chem. Int. Ed. 2003, 42, 1276–1279. [Google Scholar] [CrossRef]
- Akullian, L.; Porter, J.; Traverse, J.; Snapper, M.; Hoveyda, A. Asymmetric Synthesis of Acyclic Amines Through Zr- and Hf-Catalyzed Enantioselective Alkylzinc Reagents to Imines. Adv. Synth. Catal. 2005, 347, 417–425. [Google Scholar] [CrossRef]
- Cole, B.; Shimizu, K.; Krueger, C.; Harrity, J.; Snapper, M.; Hoveyda, A. Discovery of Chiral Catalysts through Ligand Diversity: Ti-Catalyzed Enantioselective Addition of TMSCN to meso Epoxides. Angew. Chem. Int. Ed. 1996, 35, 1668–1671. [Google Scholar] [CrossRef]
- Fu, P.; Snapper, M.; Hoveyda, A. Catalytic Asymmetric Alkylations of Ketoimines. Enantioselective Synthesis of N-Substituted Quaternary Carbon Stereogenic Centers by Zr- Catalyzed Additions of Dialkylzinc Reagents to Aryl-, Alkyl-, and Trifluoroalkyl-Substituted Ketoimines. J. Am. Chem. Soc. 2008, 130, 5530–5541. [Google Scholar] [CrossRef]
- Murphy, K.; Hoveyda, A. Catalytic Enantioselective Synthesis of Quaternary All-Carbon Stereogenic Centers. Preparation of α,α‘-Disubstituted β,γ-Unsaturated Esters through Cu-Catalyzed Asymmetric Allylic Alkylations. Org. Lett. 2005, 7, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Wirschun, W.; Kuntz, K.; Snapper, M.; Hoveyda, A. Ti-Catalyzed Regio- and Enantioselective Synthesis of Unsaturated α-Amino Nitriles, Amides, and Acids. Catalyst Identification through Screening of Parallel Libraries. J. Am. Chem. Soc. 2000, 122, 2657–2658. [Google Scholar] [CrossRef]
- Chinn, A.; Kim, B.; Kwon, Y.; Miller, S. Enantioselective Intermolecular C–O Bond Formation in the Desymmetrization of Diarylmethines Employing a Guadinylated Peptide-Based Catalyst. J. Am. Chem. Soc. 2017, 139, 18107–18114. [Google Scholar] [CrossRef]
- Bøgevig, A.; Pastor, I.; Adolfson, H. Highly Enantioselective Ruthenium-Catalyzed Reduction of Ketones Employing Readily Available Peptide Ligands. Chem. Eur. J. 2004, 10, 294–302. [Google Scholar] [CrossRef]
- Worm-Leonhard, K.; Meldal, M. Green Catalysts: Solid-Phase Peptide Carbene Ligands in Aqueous Transition-Metal Catalysis. Eur. J. Org. Chem. 2008, 2008, 5244–5253. [Google Scholar] [CrossRef]
- de Almeida, A.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Ali, R.; Meng, J.; Khan, M.; Jiang, X. Machine learning advancements in organic synthesis: A focused exploration of artificial intelligence applications in chemistry. J. AI Chem. 2024, 2, 100049. [Google Scholar] [CrossRef]
- Shi, B.; Maugel, N.; Zhang, Y.; Yu, J. Pd(II)-Catalyzed Enantioselective Activation of C(sp2)H and C(sp3)H Bonds Using Mono Protected Amino Acids as Chiral Ligands. Angew. Chem. Int. Ed. 2008, 47, 4882. [Google Scholar] [CrossRef]
- Shao, Q.; Wu, K.; Zhuang, Z.; Qian, S.; Yu, J. From Pd(OAc)2 to Chiral Catalysts: The Discovery and Development of Bifunctional Mono-N-Protected Amino Acid Ligands for Diverse C–H Functionalization Reactions. Acc. Chem. Res. 2020, 53, 833. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.; Wang, D.; Yu, J. Ligand-Accelerated C−H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 460. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Zhang, T.; Lin, Z.; Shi, B. Transition Metal-Catalyzed Enantioselective C−H Functionalization via Chiral Transient Directing Group Strategies. Angew. Chem. Int. Ed. 2020, 59, 9594. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Panja, S.; Grover, J.; Hazra, P.; Pandit, S.; Bairagi, Y.; Zhang, X.; Maiti, D. Dual Ligand Enabled Nondirected C–H Chalcogenation of Arenes and Heteroarenes. J. Am. Chem. Soc. 2022, 144, 12032. [Google Scholar] [CrossRef]
- Wang, H.; Li, H.; Chen, X.; Zhou, C.; Li, S.; Yang, Y.; Li, G. Asymmetric Remote meta-C–H Activation Controlled by a Chiral Ligand. ACS Catal. 2022, 12, 13435. [Google Scholar] [CrossRef]
- Losada, P.; Goiocechea, L.; Mascareñas, J.; Gulías, M. Axially Chiral 2-Hydroxybiaryls by Palladium-Catalyzed Enantioselective C–H Activation. ACS Catal. 2023, 13, 13994. [Google Scholar] [CrossRef]
- Metrano, A.J.; Shugrue, C.R.; Kim, B.; Chinn, A.J.; Stone, E.A.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev. 2020, 120, 11479. [Google Scholar] [CrossRef]
- Hoshino, Y.; Yamamoto, H. Novel α-Amino Acid-Based Hydroxamic Acid Ligands for Vanadium-Catalyzed Asymmetric Epoxidation of Allylic Alcohols. J. Am. Chem. Soc. 2000, 122, 10452–10453. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide [Reference] | Metal | N-Terminus Function | Reaction Type |
|---|---|---|---|
| (S)-Val-(S)-Glu-Gly [2] | Al | imine | Addition of TMSCN to ketone |
| (S)-Val-(S)-Phe-Gly [5] | Cu | Pyridinyl-imine | allylic substitution |
| (S)-Val-(S)-Phe-Gly [5] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Phe-Gly [5] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | Addition of RZnR to ketones |
| (S)-Val [5] | Cu | Pyridinyl-imine | Allylic substitution |
| (S)-Val [5] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val [5] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to acyclic enones |
| (S)-Val [4] | Ti | 2-hydroxy-5-methyoxy-phenyl-imine | Strecker cyanohydrin formation |
| (S)-Val [10] | Zr | 2-hydroxyphenyl-imine | Imine alkylation |
| (S)-Val [16] | Ru | N-Boc | Reduction of acetophenone |
| (S)-Val [16] | Ru | N-Boc | Reduction of acetophenone |
| (R)-Val [16] | Ru | N-Boc | Reduction of acetophenone |
| (R)-Val [16] | Ru | N-Boc | Reduction of acetophenone |
| (S)-Thr-(R)-Val [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to heterocycle |
| (S)-Thr-(R)-Val [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Ala-(S)-Val [6] | Cu | 2-hydroxynaphthalene-1-imine | Allylic alkylations |
| (S)-Val-(S)-Ala [6] | Cu | 2-(Hydroxy)phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Ala [6] | Cu | 2-(Amino)phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Ala [6] | Cu | 2-(Methylamino)phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Leu-(S)-Ala [6] | Cu | 2-(Methylamino)phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Leu-(S)-Trp [6] | Cu | 2-(Methylamino)phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Thr [4] | Ti | 2-hydroxy-3,5-dichloro-phenyl-imine | Strecker cyanohydrin formation |
| (S)-Val-(R)-Thr [4] | Ti | 2-hydroxy-5-methyoxy-phenyl-imine | Strecker cyanohydrin formation |
| (S)-Val-(S)-Thr [4] | Ti | 2-hydroxy-3-fluoro-phenyl-imine | CN addition to epoxides |
| (S)-Val-(S)-Thr [4] | Ti | 2-hydroxy-3-fluorophenyl-imine | Addition of TMSCM to epoxides |
| (S)-Val-Gly [4] | Ti | 2-hydroxy-5-methyoxy-phenyl-imine | Strecker cyanohydrin formation |
| (S)-Val-Gly [10] | Zr | 2-hydroxyphenyl-imine | Imine alkylation |
| (S)-Phe-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of (R)2Zn to cyclic enones |
| (S)-Ala-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of (R)2Zn to cyclic enones |
| (S)-Thr-(R)-Thr [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of (R)2Zn to cyclic enones |
| (S)-Asp-(S)-Phe [13] | Cu | 2-hydroxy-5-methylphenyl-imine | Allylic alkylations |
| (S)-Asp-(S)-Phe [13] | Cu | 2-hydroxy-5-tertbutylphenyl-imine | Allylic alkylations |
| (S)-Thr-(S)-Trp [13] | Cu | 2-hydroxynaphthalene-imine | Allylic alkylations of olefins |
| (S)-Val-(S)-Thr-Gly [13] | Cu | 2-hydroxyphenyl-imine | Addition of CN to arylimines |
| (S)-Val-(S)-Thr-Gly [14] | Ti | 2-hydroxyphenyl-imine | Addition of CN to arylimines |
| (S)-Val-(S)-Thr-Gly [14] | Ti | 2-hydroxy-5-methoxyphenyl-imine | Addition of CN to alkenylimines |
| (S)-Val-(S)-Thr-Gly [14] | Ti | 2-hydroxy-3,5-dichlorophenyl-imine | Cyanide addition to imines |
| (S)-Val-(S)-Phe [5] | Cu | Pyridinyl-imine | Allylic substitution |
| (S)-Val-(S)-Phe [5] | Cu | Isopropoxy-Pyridinyl-imine | Allylic substitution of alkenes |
| (S)-Val-(S)-Phe [5] | Cu | Isopropoxy-Pyridinyl-imine | Allylic substitution of alkenes |
| (S)-Val-(S)-Phe [5] | Cu | Isopropoxy-Pyridinyl-imine | Allylic substitution of alkenes |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enone |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to nitroalkenes |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to acyclic enamine |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to acyclic enone |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | Addition of RZnR to ketones |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | Addition of RZnR to ketones |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Phe [9] | Cu | 2-(Diphenyl)phosphino-phenyl-imine | ACA of RZnR to cyclic enones |
| (S)-Val-(S)-Phe [10] | Hf | 2-hydroxy-5-methoxy-amine | Imine alkylation |
| (S)-Val-(S)-Phe [11] | Ti | 2-hydroxy-3,5-ditertbutylphenyl-imine | Addition of TMSCM to epoxides |
| (S)-Val-(S)-Phe [11] | Ti | 2-hydroxynaphthalenephenyl-imine | Addition of TMSCM to epoxides |
| (S)-Val-(S)-Phe [10] | Zr | 2-hydroxy-5-methoxyphenyl-amine | Alkylation of ketoimine esters |
| (S)-Val-(S)-Phe [10] | Zr | 2-hydroxyphenyl-imine | Imine alkylation |
| (S)-Val-(S)-Phe [10] | Zr | 2-hydroxy-3,5-ditertbutylphenyl-amine | Imine alkylation |
| (S)-Val-(S)-Phe [10] | Zr | 2-hydroxyphenyl-amine | Imine alkylation |
| (S)-Val-(S)-Phe [10] | Zr | 2-hydroxy-5-methoxyphenyl-amine | Alkylation of ketoimine esters |
| (S)-Val-(S)-Phe [13] | Cu | 2-hydroxyphenyl-imine | Allylic alkylations of olefins |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-5-methoxyphenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-dichlorophenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-dibromophenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3-nitro-5-bromophenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-5-nitrophenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-ditertbutylphenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxynaphthalenephenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-ditertbutylphenyl-amine | Alkylation of ketoimines |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-5-methoxyphenyl-amine | Alkylation of ketoimine esters |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-ditertbutylphenyl-amine | Alkylation of ketoimine esters |
| (S)-Val-(S)-Phe [12] | Zr | 2-hydroxy-3,5-ditertbutylphenyl-amine | Synthesis of N-heterocycles |
| (S)-Ala [16] | Ru | N-Boc | Reduction of acetophenone |
| (S)-Leu [16] | Ru | N-Boc | Reduction of acetophenone |
| (S)-Ile [16] | Ru | N-Boc | Reduction of acetophenone |
| (S)-Phe [16] | Ru | N-Boc | Reduction of acetophenone |
| Phe-Val [17] | Pd | NHC-pyridine | Sonogashira cross-coupling |
| Phe-Val [17] | Pd | NHC-pyridine | Suzuki cross-coupling |
| Phe-Val [17] | Pd | NHC-pyridine | Suzuki cross-coupling |
| Asp-(D)-Pro [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Cle [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-(D)-Ala [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Gly [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Acpc [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Ala [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib-(D)-Ala-Ala [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib-(D)-Ala [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib-Ala [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib-(D)-Leu [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
| Asp-(D)-Pro-Aib-(D)-Phe [15] | Cu | TETRAMETHYLGUANIDINE | C-O bond formation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gung, B.W.; Kubesch, C.; Bernstein, G. Natural Chiral Ligand Strategy: Metal-Catalyzed Reactions with Ligands Prepared from Amino Acids and Peptides. Catalysts 2024, 14, 813. https://doi.org/10.3390/catal14110813
Gung BW, Kubesch C, Bernstein G. Natural Chiral Ligand Strategy: Metal-Catalyzed Reactions with Ligands Prepared from Amino Acids and Peptides. Catalysts. 2024; 14(11):813. https://doi.org/10.3390/catal14110813
Chicago/Turabian StyleGung, Benjamin W., Cole Kubesch, and Gavriella Bernstein. 2024. "Natural Chiral Ligand Strategy: Metal-Catalyzed Reactions with Ligands Prepared from Amino Acids and Peptides" Catalysts 14, no. 11: 813. https://doi.org/10.3390/catal14110813
APA StyleGung, B. W., Kubesch, C., & Bernstein, G. (2024). Natural Chiral Ligand Strategy: Metal-Catalyzed Reactions with Ligands Prepared from Amino Acids and Peptides. Catalysts, 14(11), 813. https://doi.org/10.3390/catal14110813