
A Review of Coal and Biomass Hydrogasification: Process Layouts, Hydrogasifiers, and Catalysts




Abstract
1. Introduction
- Lower operative temperatures and higher thermal efficiency (with the minimized contribution of exothermic reactions);
- Unnecessary expensive oxygen plants typical of gasification units and very low production of harmful chemicals such as PCDDs (polychlorinated diben-zo-p-dioxins) [17];
- Higher fraction of finally produced methane and lower carbon monoxide yield;
- Chance of eliminating the residual CO2 through its combination with H2 and further methane production;
- Higher process compactness (gasification reaction and exothermic pathways occur in the same reactor instead of the two separate reactors of indirect gasifiers).
2. Process Configurations
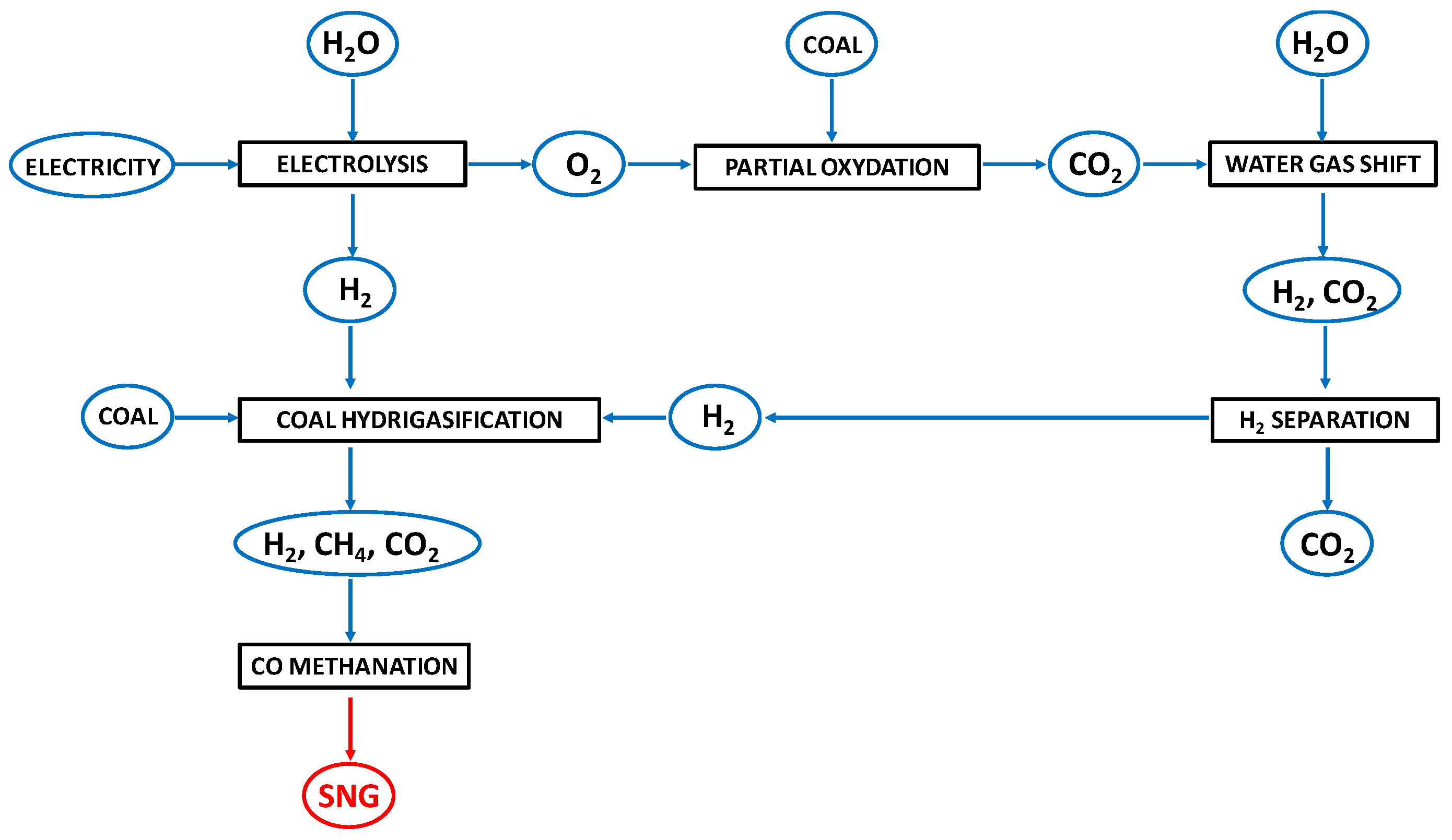
2.1. Process Layouts for the Hydrogasification of Coal-Derived Materials
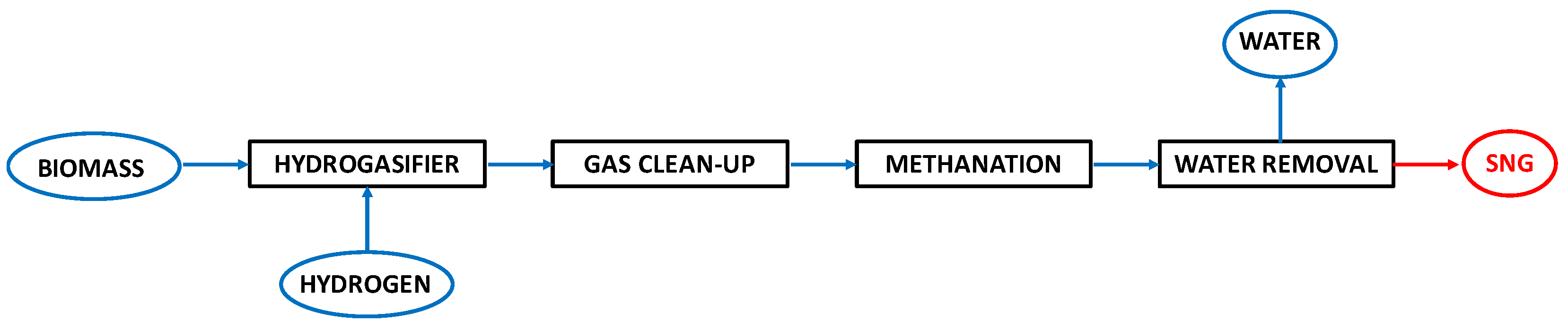
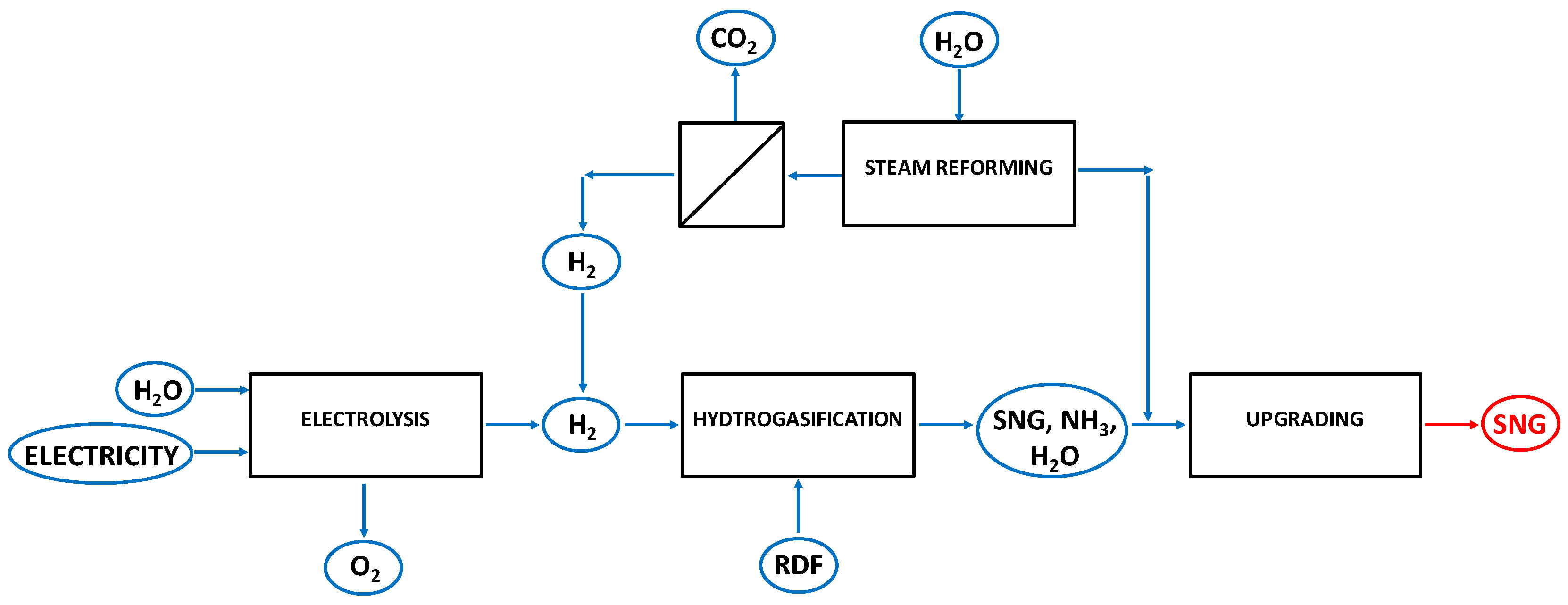
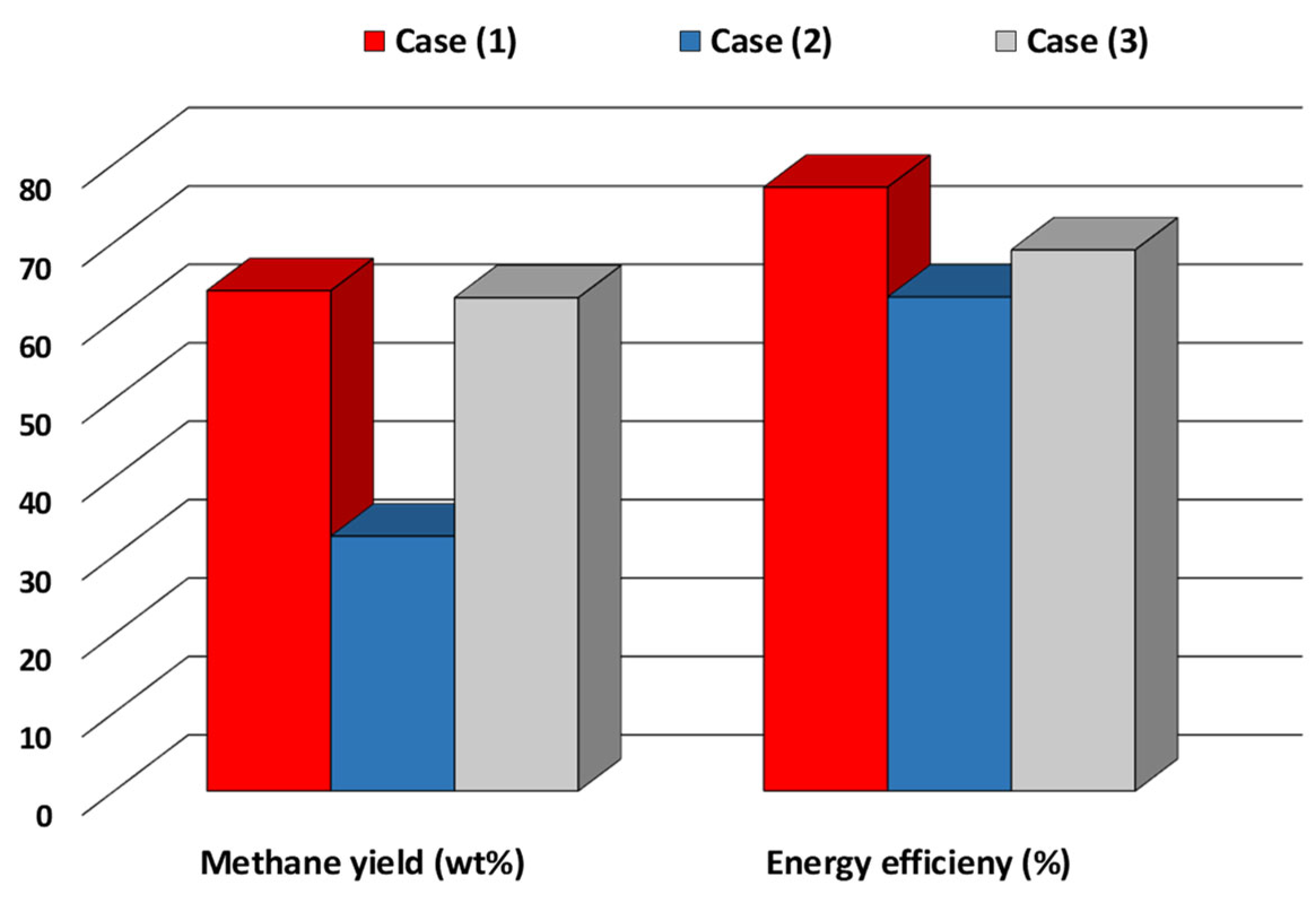
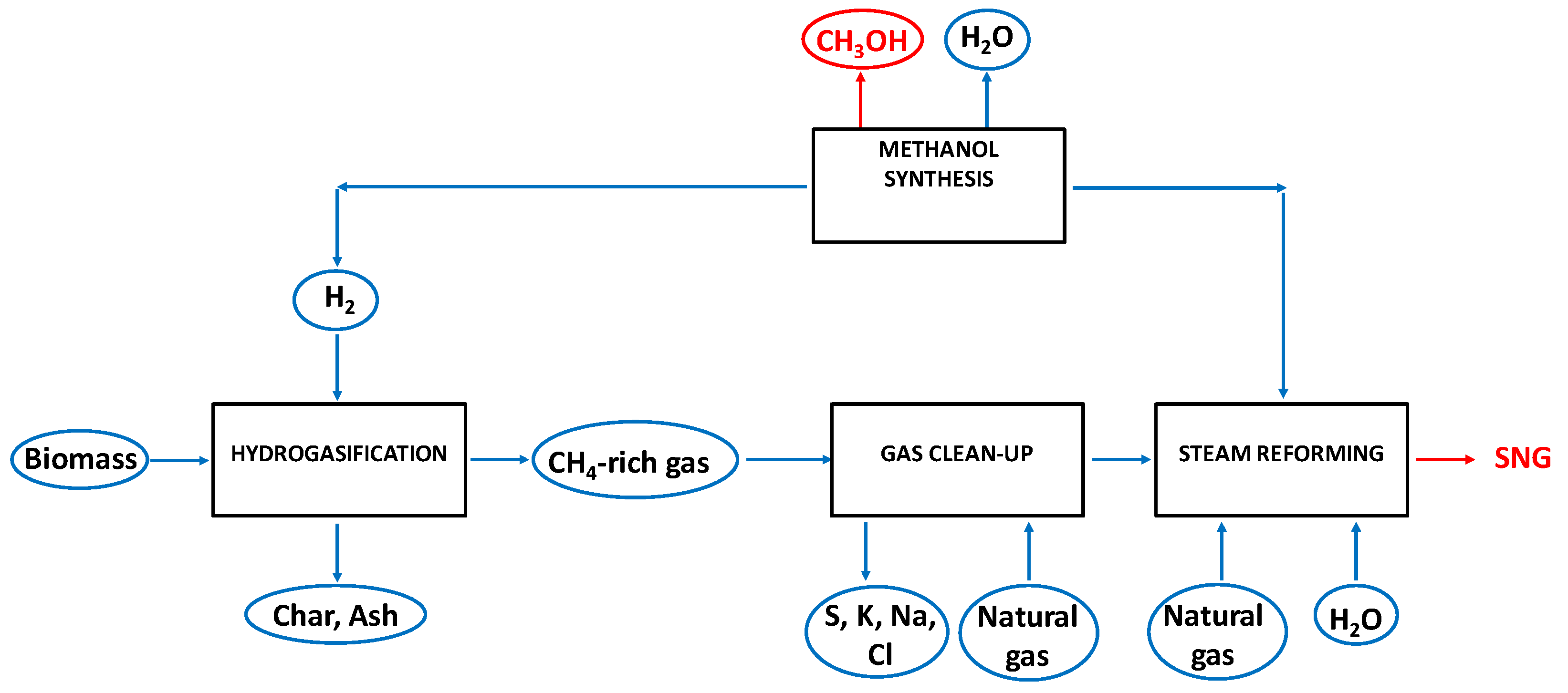
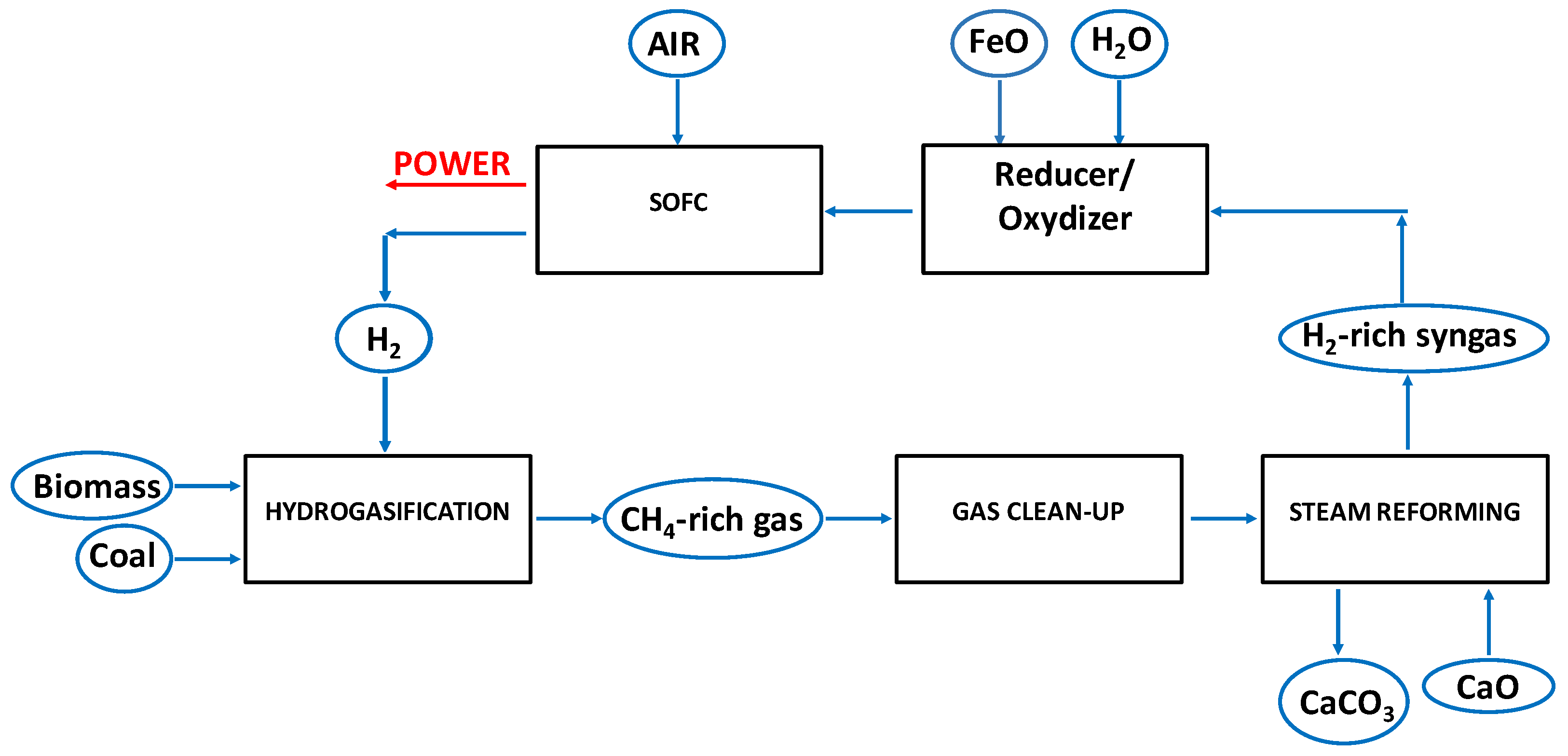
2.2. Process Layouts for the Hydrogasification of Biomass-Derived Materials
3. Catalysts for Coal and Biomass Hydrogasification
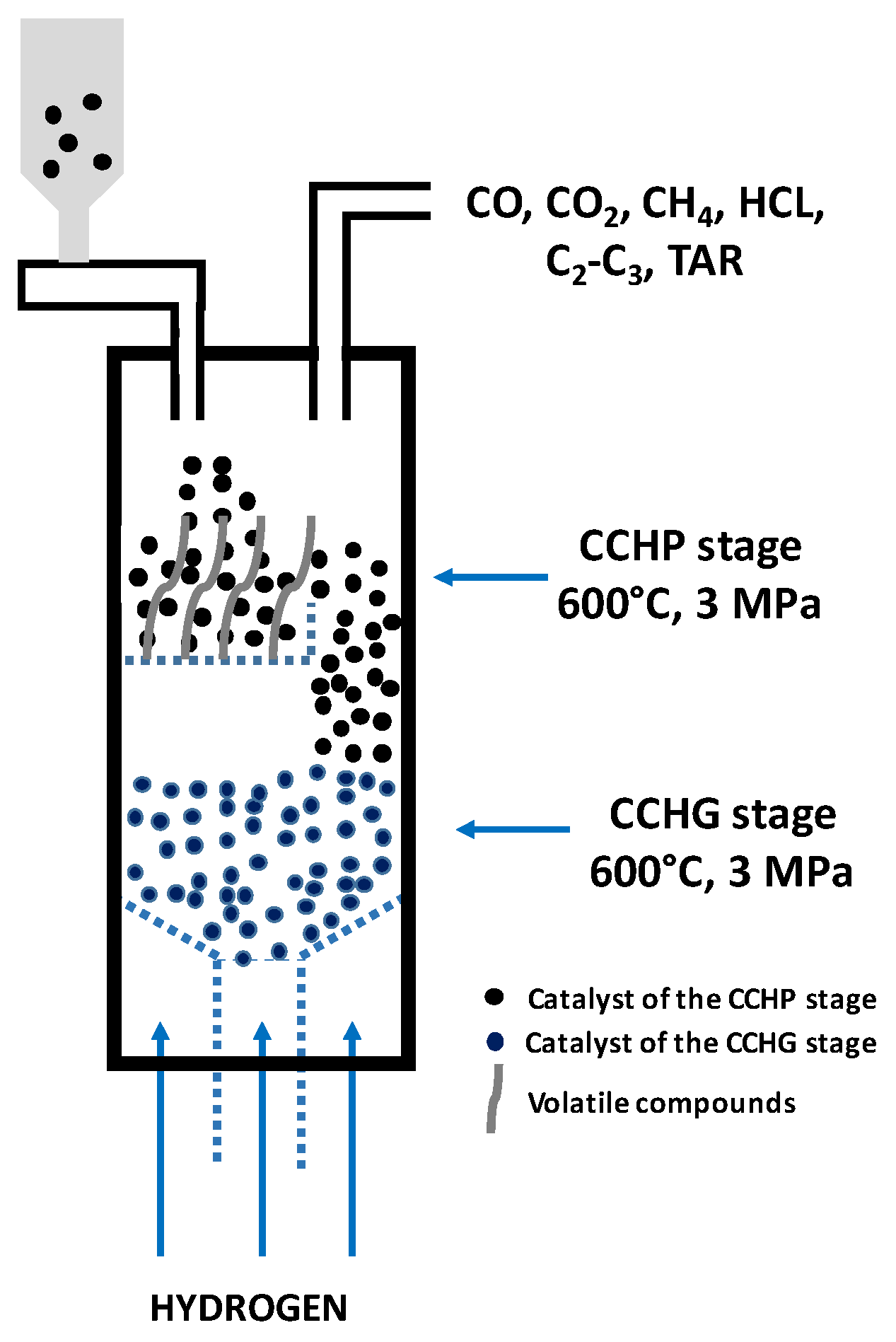
3.1. Catalytic Hydrogasification of Coal
3.1.1. Iron-Group Metals-Based Catalysts
3.1.2. Copper Catalysts and Composite Catalysts
3.1.3. Alkaline- and Alkaline-Earth-Based Catalysts
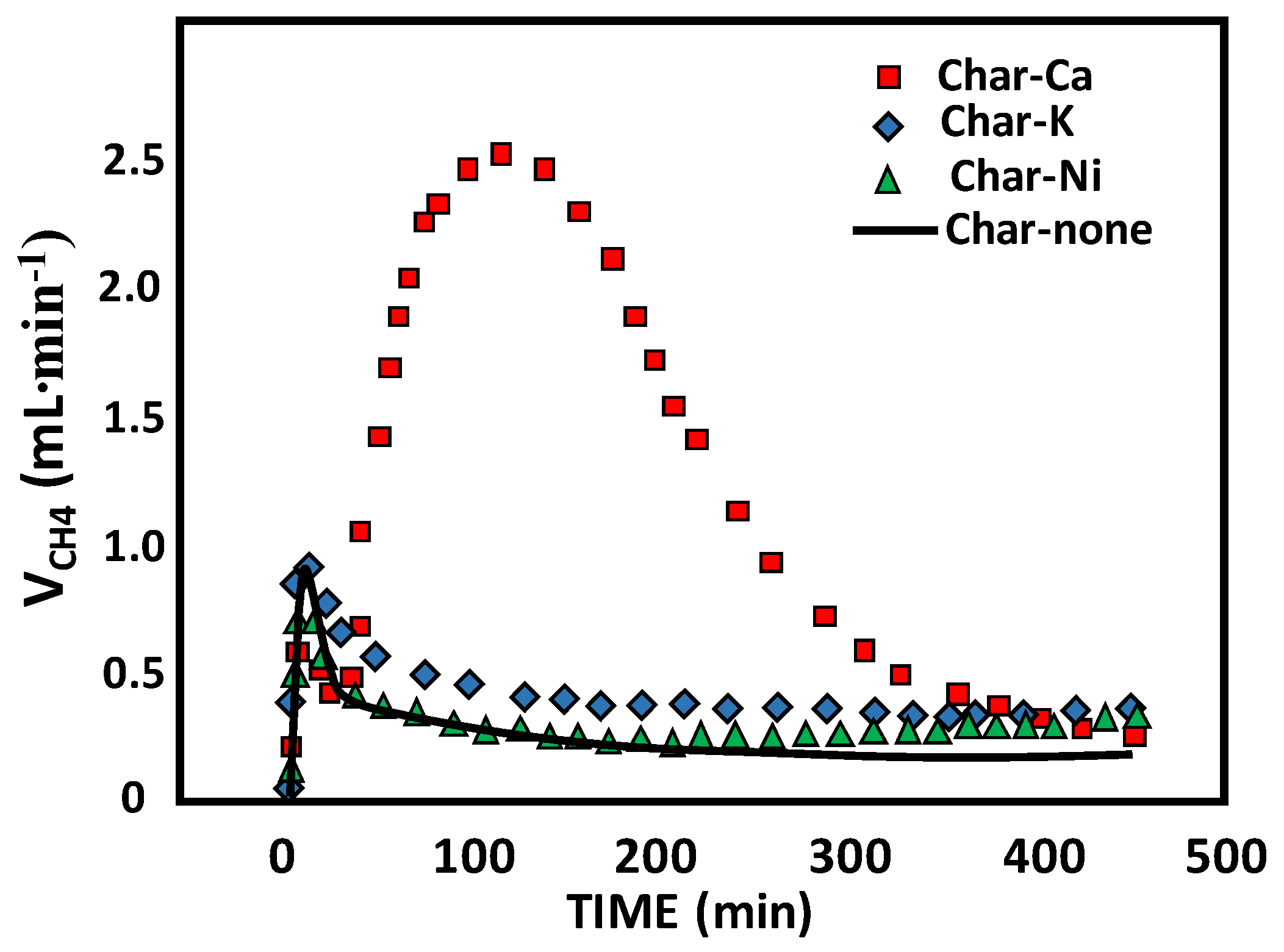
3.1.4. Char-Based Catalysts
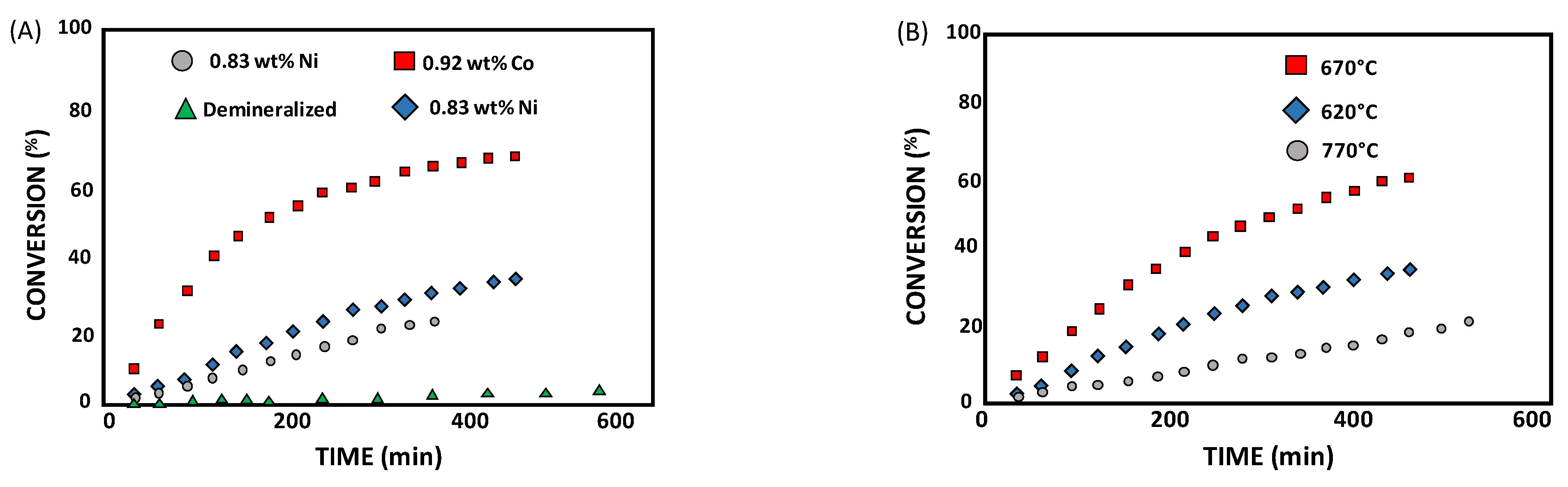
3.2. Catalytic Hydrogasification of Biomass
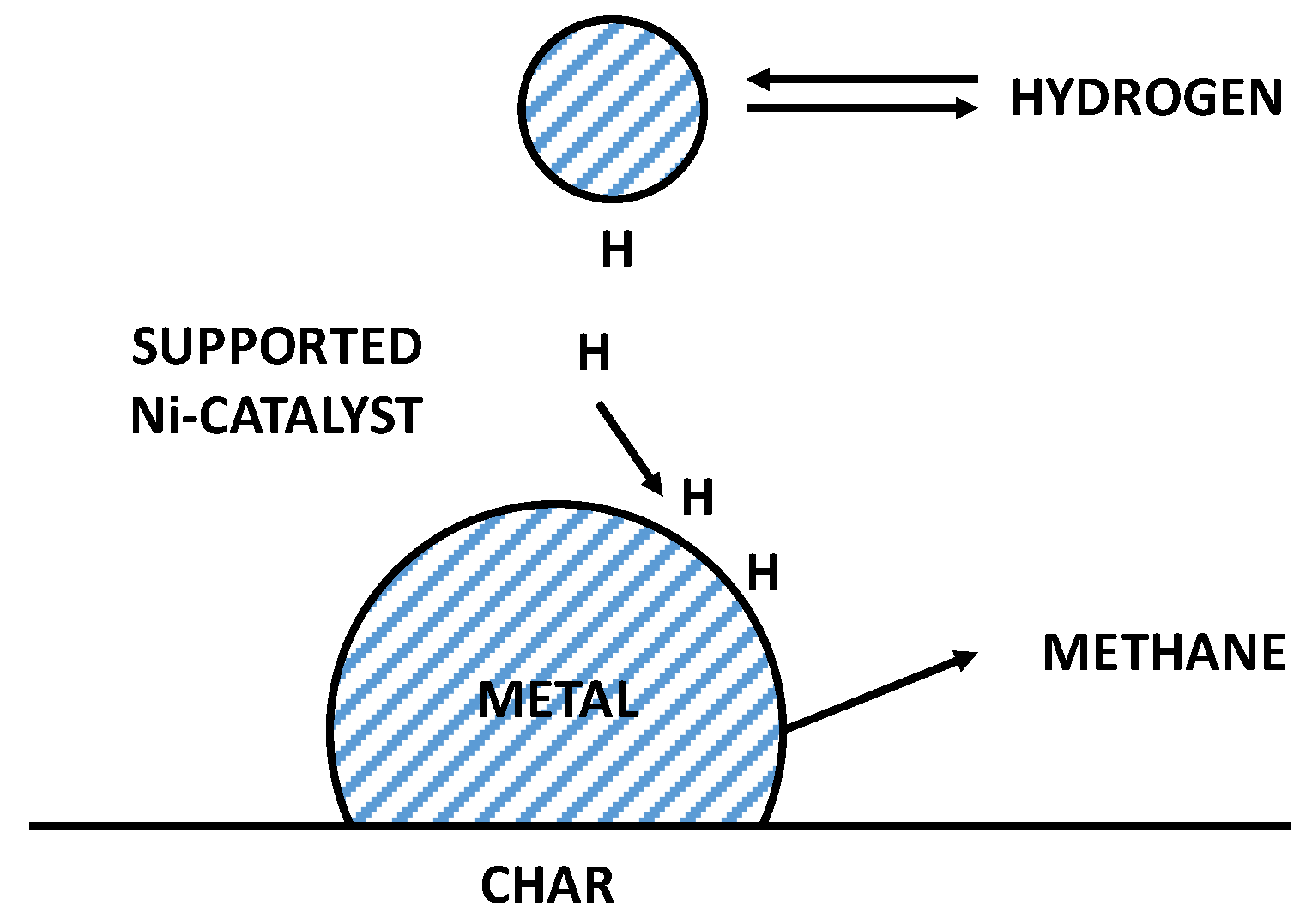
3.3. Reaction Mechanism
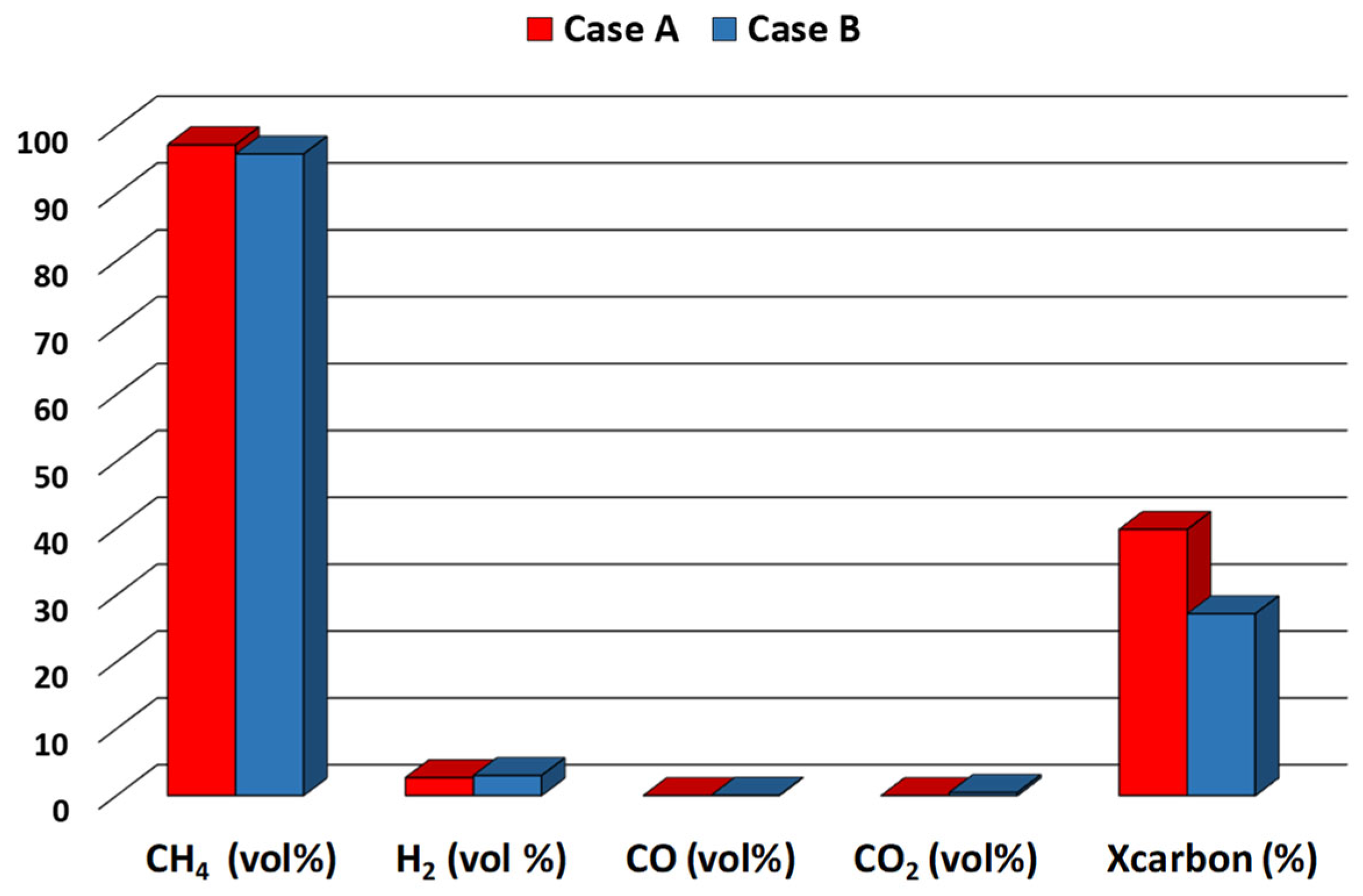
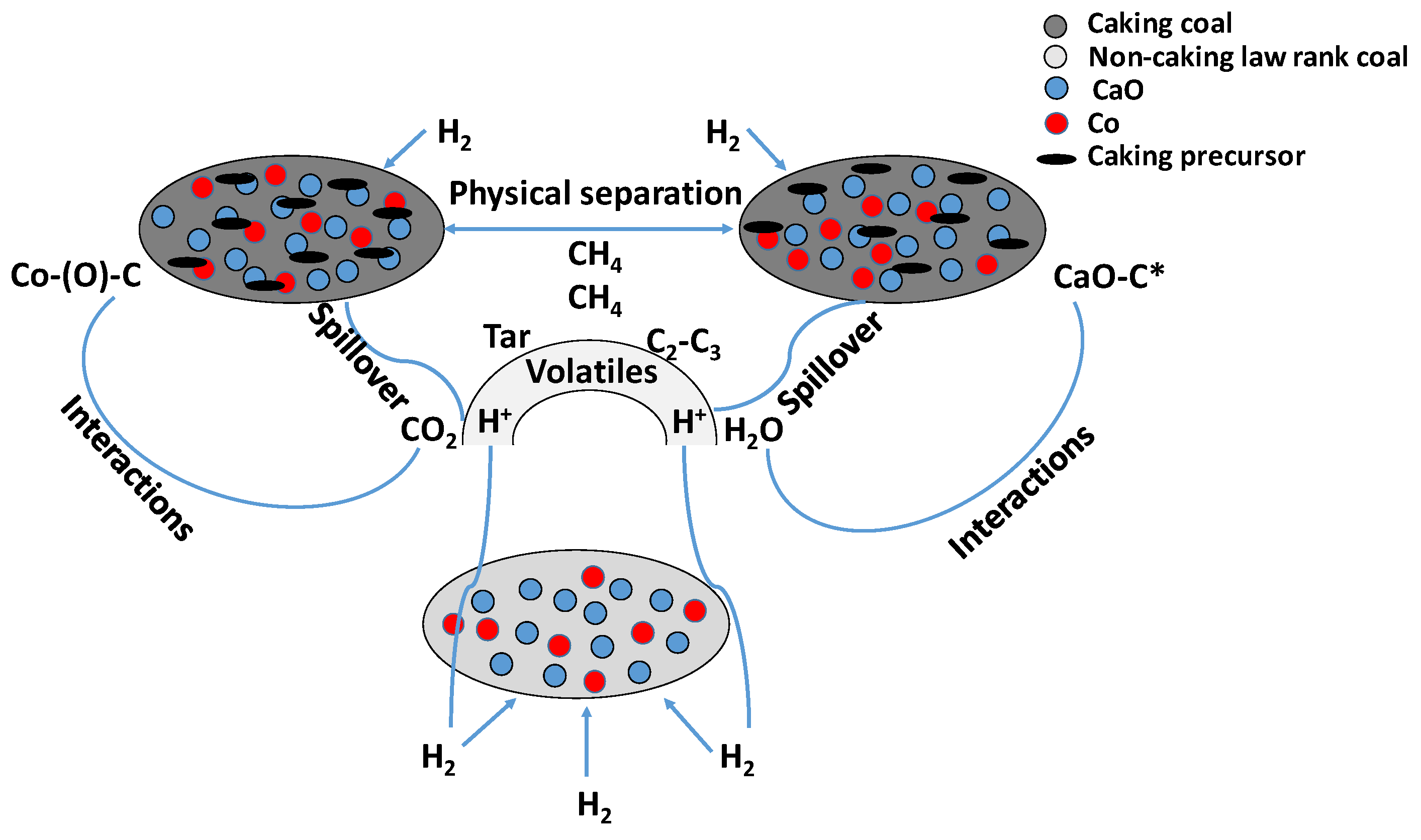
- For medium-low-rank coals with non-caking behavior and low sulfur content, the probable mechanism could concern the supplying of active hydrogen by the Co-Ca catalyst and the C-C bonds’ catalytic fracturing, which is the determining step (Figure 16);
- For caking coal, the caking agglomeration properties were damaged in situ by means of the blending of the coal, which improved coal reactivity due to the reduced contact between the cohesive coal particles (physical separation) as well as the activated hydrogen spillover, thus promoting the activity of the Co-Ca catalyst in the presence of volatile catalyst-coal interactions (Figure 16).
- For high-rank coal with non-caking behavior and low/high sulfur content, the suitable amount of Ca compounds (CaO/CaCO3) influenced the hydrogenation of graphite carbon and limited the deactivation effect of sulfur on the Co-Ca catalyst; indeed, the H2S strongly absorbed onto the Co surface through its fixing on Co particles, which promote the catalyst activity (Equation (8)).
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAEM | Alkali and alkaline rare metals |
| BTX | Benzene, toluene, and xylene |
| C* | Activated carbon |
| Ca | Amorphous carbon |
| Cg | Graphite carbon |
| CCHG | Coal catalytic hydrogasification |
| CCHP | Coal catalytic hydropyrolysis |
| CEC | Cation exchange capacity |
| CnHm | Light hydrocarbons |
| HCL | High-value-added liquid hydrocarbons |
| LAH | Light aromatic hydrocarbons |
| MCFC | Molten Carbonate Fuel Cells |
| MSCF | One thousand standard cubic feet |
| Numberfluidization | Fluidization number |
| PCDDS | Polychlorinated diben-zo-p-dioxins |
| PCX | Phenol, cresol, and xylenol |
| RDF | Refuse-derived fuel |
| SEM | Scanning Electron Microscopy |
| SNG | Substitute Natural Gas |
| SOFC | Solid Oxide Fuel Cells |
| Timereaction | Reaction time |
| Timeresidence | Residence time |
| U | Gas velocity |
| UCG | Underground Coal Hydrogasification |
| Umf | Minimum fluidization velocity |
| VCH4 | Methane formation rate |
| VER | Variable Energy Resources |
| WGS | Water Gas Shift |
| Xchar | Char conversion |
| Xcarbon | Carbon conversion |
| XRD | X-ray diffraction |
| YCH4 | Methane yield |
| ZEC | Zero Emission Carbon |
References
- Alshorifi, F.T.; Ali, S.L.; Salama, R.S. Promotional Synergistic Effect of Cs–Au NPs on the Performance of Cs–Au/MgFe2O4 Catalysts in Catalysis 3,4-Dihydropyrimidin-2(1H)-Ones and Degradation of RhB Dye. J. Inorg. Organomet. Polym. Mater. 2022, 32, 3765–3776. [Google Scholar] [CrossRef]
- Alshorifi, F.T.; Alswat, A.A.; Salama, R.S. Gold-selenide quantum dots supported onto cesium ferrite nanocomposites for the efficient degradation of rhodamine B. Heliyon 2022, 8, e09652. [Google Scholar] [CrossRef]
- El-Hakam, S.A.; Alshorifi, F.T.; Salama, R.S.; Gamal, S.; El-Yazeed, W.S.A.; Ibrahim, A.A.; Ahmed, A.I. Application of nanostructured mesoporous silica/ bismuth vanadate composite catalysts for the degradation of methylene blue and brilliant green. J. Mater. Res. Technol. 2022, 18, 1963–1976. [Google Scholar] [CrossRef]
- Keefer, B.G.; Babicki, M.L.; Sellars, B.G.; Ng, E. Method and System for Biomass Hydrogasification. U.S. Patent 9,394,171 B2, 19 July 2016. [Google Scholar]
- García-Mateos, F.J.; Moulefera, I.; Rosas, J.M.; Benyoucef, A.; Rodríguez-Mirasol, J.; Cordero, T. Alcohol Dehydrogenation on Kraft Lignin-Derived Chars with Surface Basicity. Catalysts 2017, 7, 308. [Google Scholar] [CrossRef]
- Yin, H.; Yip, A.C.K. A Review on the Production and Purification of Biomass-Derived Hydrogen Using Emerging Membrane Technologies. Catalysts 2017, 7, 297. [Google Scholar] [CrossRef]
- Yong, Z.J.; Bashir, M.J.K.; Ng, C.A.; Sethupathi, S.; Lim, J.W.; Show, P.L. Sustainable Waste-to-Energy Development in Malaysia: Appraisal of Environmental, Financial, and Public Issues Related with Energy Recovery from Municipal Solid Waste. Processes 2019, 7, 676. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, Q.; Liu, Z. Superior catalytic effect of calcium oxide on the hydrogasification of char for CH4. Fuel 2016, 180, 737–742. [Google Scholar] [CrossRef]
- Liu, Z.; Norbeck, J.M.; Raju, A.S.K.; Kim, S.; Park, C.S. Synthetic natural gas production by sorption enhanced steam hydrogasification based processes for improving CH4 yield and mitigating CO2 emissions. Energy Convers. Manag. 2016, 126, 256–265. [Google Scholar] [CrossRef]
- Pankiewicz-Sperka, M.; Kapusta, K.; Basa, W.; Stolecka, K. Characteristics of Water Contaminants from Underground Coal Gasification (UCG) Process—Effect of Coal Properties and Gasification Pressure. Energies 2021, 14, 6533. [Google Scholar] [CrossRef]
- Kapusta, K.; Wiatowski, M.; Thomas, H.R.; Zagorščak, R.; Sadasivam, S.; Masum, S.; Kempka, T.; Otto, C.; Basa, W.; Szyja, M.; et al. Experimental simulations of methane-oriented underground coal gasification using hydrogen—The effect of coal rank and gasification pressure on the hydrogasification process. Int. J. Hydrogen Energy 2023, 48, 921–932. [Google Scholar] [CrossRef]
- Burchart, D.; Gazda-Grzywacz, M.; Grzywacz, P.; Burmistrz, P.; Zarębska, K. Life Cycle Assessment of Hydrogen Production from Coal Gasification as an Alternative Transport Fuel. Energies 2023, 16, 383. [Google Scholar] [CrossRef]
- Zhang, A.; Kaiho, M.; Yasuda, H.; Zabat, M.; Nakano, K.; Yamada, O. Fundamental studies on hydrogasification of Taiheiyo coal. Energy 2005, 30, 2243–2250. [Google Scholar] [CrossRef]
- Yan, L.; Yue, G.; He, B. Exergy analysis of a coal/biomass co-hydrogasification based chemical looping power generation system. Energy 2015, 93, 1778–1787. [Google Scholar] [CrossRef]
- Wang, X.; Yao, K.; Huang, X.; Chen, X.; Yu, G.; Liu, H.; Wang, F.; Fan, M. Effect of CaO and biomass ash on catalytic hydrogasification behavior of coal char. Fuel 2019, 249, 103–111. [Google Scholar] [CrossRef]
- Mozaffarian, M.; Zwart, R.W.R. Production of Substitute Natural Gas by Biomass Hydrogasification. In Progress in Thermochemical Biomass Conversion; Blackwell Science Ltd.: Oxford, UK, 2001; pp. 405–419. [Google Scholar] [CrossRef]
- Safavi, A.; Richter, C.; Unnthorsson, R. Dioxin Formation in Biomass Gasification: A Review. Energies 2022, 15, 700. [Google Scholar] [CrossRef]
- Steinberg, M. Hydro-Gasification of Coal for Hydrogen Production with Reduced CO2 Emission. In Proceedings of the WHEC16: 16. World Hydrogen Energy Conference, Lyon, France, 13–16 June 2006. [Google Scholar]
- Zhu, Y.; Wang, Q.; Li, K.; Cen, J.; Fang, M.; Ying, C. Study on pressurized isothermal pyrolysis characteristics of low-rank coal in a pressurized micro-fluidized bed reaction analyzer. Energy 2022, 240, 122475. [Google Scholar] [CrossRef]
- Frigo, S.; Spazzafumo, G. Cogeneration of power and substitute of natural gas using biomass and electrolytic hydrogen. Int. J. Hydrogen Energy 2018, 43, 11696–11705. [Google Scholar] [CrossRef]
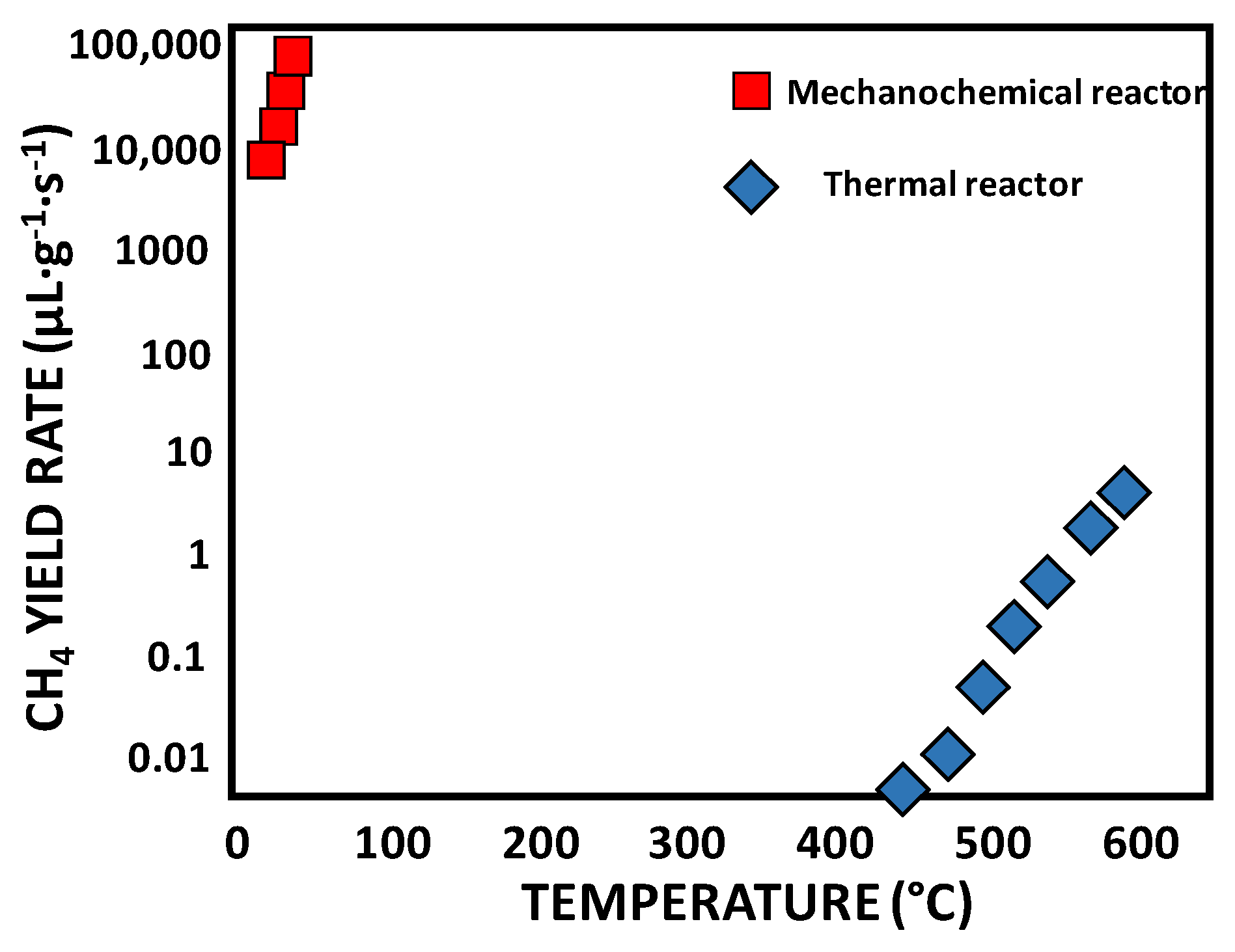
- Han, G.-F.; Zhang, P.; Scholzen, P.; Noh, H.-J.; Yang, M.; Kweon, D.H.; Jeon, J.-P.; Kim, Y.H.; Kim, S.-W.; Han, S.-P.; et al. Extreme Enhancement of Carbon Hydrogasification via Mechanochemistry. Angew. Chem. Int. Ed. 2022, 61, e202117851. [Google Scholar] [CrossRef]
- Gil, S.; Smoliński, A. Experimental Study of Hydrogasification of Lignite and Subbituminous Coal Chars. Sci. World J. 2015, 2015, 867030. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, Y.; Wang, W.X.; Zheng, C.G. An Experimental Investigation on Hydrogasification of Coal Chars in a Fixed Bed Reactor under High Pressure. In Advanced Materials Research; Trans Tech Publications: Zurich, Switzerland, 2012; pp. 863–870. [Google Scholar]
- Tomeczek, J.; Gil, S. The kinetics of coal chars hydrogasification. Fuel Process. Technol. 2010, 91, 1564–1568. [Google Scholar] [CrossRef]
- Miller, D.J. Enhanced Coal Hydrogasification via Oxidative Pretreatment; Michigan State University: East Lansing, MI, USA, 1992; 79p. [Google Scholar] [CrossRef]
- Woźniak, G.; Longwic, R.; Szydło, K.; Kryłowicz, A.; Kryłowicz, J.; Juszczak, R. The EFFICIENCY of the process of Coal Gasification in the Presence of Hydrogen. In E3S Web of Conferences; EDP Sciences: Les Ulis, France, 2018; p. 00030. [Google Scholar]
- Liu, X.; Xiong, B.; Huang, X.; Ding, H.; Zheng, Y.; Liu, Z.; Zheng, C. Effect of catalysts on char structural evolution during hydrogasification under high pressure. Fuel 2017, 188, 474–482. [Google Scholar] [CrossRef]
- González, J.F.; Ramiro, A.; Sabio, E.; Encinar, J.M.; González, C.M. Hydrogasification of Almond Shell Chars. Influence of Operating Variables and Kinetic Study. Ind. Eng. Chem. Res. 2002, 41, 3557–3565. [Google Scholar] [CrossRef]
- Toomajian, M.E.; Lussier, M.G.; Miller, D.J. Effect of oxidation and other treatments on hydrogasification rate of coal char. Fuel 1992, 71, 1055–1061. [Google Scholar] [CrossRef]
- Malekshahian, M.; Hill, J.M. Potassium catalyzed CO2 gasification of petroleum coke at elevated pressures. Fuel Process. Technol. 2013, 113, 34–40. [Google Scholar] [CrossRef]
- Zhang, Y.; Wan, L.; Guan, J.; Xiong, Q.; Zhang, S.; Jin, X. A Review on Biomass Gasification: Effect of Main Parameters on Char Generation and Reaction. Energy Fuels 2020, 34, 13438–13455. [Google Scholar] [CrossRef]
- Mishra, S.; Upadhyay, R.K. Review on biomass gasification: Gasifiers, gasifying mediums, and operational parameters. Mater. Sci. Energy Technol. 2021, 4, 329–340. [Google Scholar] [CrossRef]
- Tezer, Ö.; Karabağ, N.; Öngen, A.; Çolpan, C.Ö.; Ayol, A. Biomass gasification for sustainable energy production: A review. Int. J. Hydrogen Energy 2022, 47, 15419–15433. [Google Scholar] [CrossRef]
- Akbarian, A.; Andooz, A.; Kowsari, E.; Ramakrishna, S.; Asgari, S.; Cheshmeh, Z.A. Challenges and opportunities of lignocellulosic biomass gasification in the path of circular bioeconomy. Bioresour. Technol. 2022, 362, 127774. [Google Scholar] [CrossRef]
- Shahabuddin, M.; Alam, T. Gasification of solid fuels (coal, biomass and MSW): Overview, challenges and mitigation strategies. Energies 2022, 15, 4444. [Google Scholar] [CrossRef]
- Tosti, S.; Spazzafumo, G.; Capobianco, D.; Buceti, G.; Pozio, A.; Bartucca, S. EU scenarios of renewable coal hydro-gasification for SNG production. Sustain. Energy Technol. Assess. 2016, 16, 43–52. [Google Scholar] [CrossRef]
- Bargiacchi, E.; Candelaresi, D.; Valente, A.; Spazzafumo, G.; Frigo, S. Life Cycle Assessment of Substitute Natural Gas production from biomass and electrolytic hydrogen. Int. J. Hydrogen Energy 2021, 46, 35974–35984. [Google Scholar] [CrossRef]
- Veringa, H. Feasibility of Sng Production by Biomass Hydrogasification. In Proceedings of the 12th European Conference and Technology Exhibition on Biomass for Energy, Industry and Climate Protection, Amsterdam, The Netherlands, 17–21 June 2002. [Google Scholar]
- Zhang, J.; Wang, X.; Wang, F.; Wang, J. Investigation of hydrogasification of low-rank coals to produce methane and light aromatics in a fixed-bed reactor. Fuel Process. Technol. 2014, 127, 124–132. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, J.; Yan, X.; Pan, D.; Ren, H.; Qu, X. Catalytic coal hydrogasification by cobalt-calcium catalyst in a pressurized fluidized bed: Role of hydropyrolysis and catalysis process. J. Anal. Appl. Pyrolysis 2018, 135, 251–259. [Google Scholar] [CrossRef]
- Tromp, P.J.J.; Kapteijn, F.; Moulijn, J.A. Characterization of coal pyrolysis by means of differential scanning calorimetry. 2. Quantitative heat effects in a H2 and in a CO2 atmosphere. Fuel Process. Technol. 1989, 23, 63–74. [Google Scholar] [CrossRef]
- Higman, C.; Tam, S. Advances in Coal Gasification, Hydrogenation, and Gas Treating for the Production of Chemicals and Fuels. Chem. Rev. 2014, 114, 1673–1708. [Google Scholar] [CrossRef]
- Scharf, H.-J.; Schrader, L.; Teggers, H. Results from the operation of a semi-technical test plant for brown coal hydrogasification. Nucl. Eng. Des. 1984, 78, 223–231. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.G.; Kim, J.H.; Choi, Y.C. Hydrogasification characteristics of bituminous coals in an entrained-flow hydrogasifier. Fuel 2006, 85, 803–806. [Google Scholar] [CrossRef]
- Minutillo, M.; Perna, A. Renewable energy storage system via coal hydrogasification with co-production of electricity and synthetic natural gas. Int. J. Hydrogen Energy 2014, 39, 5793–5803. [Google Scholar] [CrossRef]
- Perdikaris, N.; Panopoulos, K.D.; Fryda, L.; Kakaras, E. Design and optimization of carbon-free power generation based on coal hydrogasification integrated with SOFC. Fuel 2009, 88, 1365–1375. [Google Scholar] [CrossRef]
- Hobbs, R. Development of a Hydrogasification Process for Co-Production of Substitute Natural Gas (SNG) and Electric Power from Western Coals-Phase I; Arizona Public Service Company: Phoenix, AZ, USA, 2007. [Google Scholar] [CrossRef]
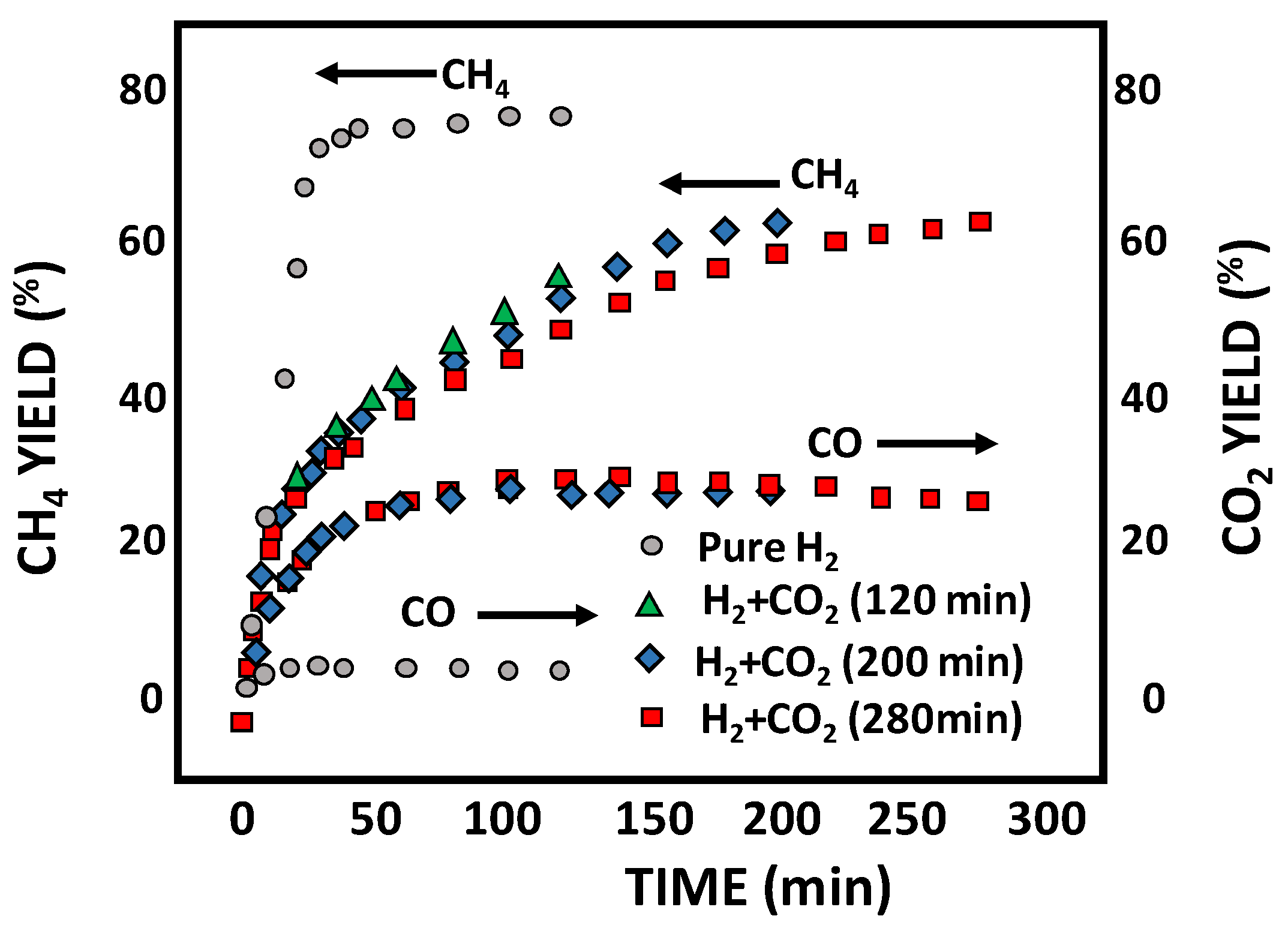
- Feng, J.; Yan, S.; Zhang, R.; Gu, S.; Qu, X.; Bi, J. Characteristics of Co–Ca catalyzed coal hydrogasification in a mixture of H2 and CO2 atmosphere. Fuel 2022, 324, 124486. [Google Scholar] [CrossRef]
- Nourozieh, H.; Kariznovi, M.; Chen, Z.; Abedi, J. Simulation Study of Underground Coal Gasification in Alberta Reservoirs: Geological Structure and Process Modeling. Energy Fuels 2010, 24, 3540–3550. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamada, O.; Kaiho, M.; Nakagome, H. Effect of polyethylene addition to coal on hydrogasification enhancement. J. Mater. Cycles Waste Manag. 2014, 16, 151–155. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamada, O.; Zhang, A.; Nakano, K.; Kaiho, M. Hydrogasification of coal and polyethylene mixture. Fuel 2004, 83, 2251–2254. [Google Scholar] [CrossRef]
- HYGAS: 1964 to 1972. In Pipeline Gas from Coal—Hydrogenation (IGT Hydrogasification Process); Institute of Gas Technology: Chicago, IL, USA, 1975; Volume 4, 588p. [CrossRef]
- Michael, E. An Analysis of Coal Hydrogasification Processes; Department of Energy: Washington, DC, USA, 1978; pp. 12–100. [Google Scholar]
- Friedman, J. Development of a Single-Stage, Entrained-Flow, Short-Residence-Time Hydrogasifier. Rep. US Dep. Energy FE 1979, 2518, 24. [Google Scholar]
- Falk, A.; Schuman, M.; Kahn, D. Advancement of Flash Hydrogasification: Task VIII. Performance Testing; Rockwell International Corp: Canoga Park, CA, USA, 1986. [Google Scholar]
- Stroud, H.; Noguchi, F. 55th Autumn Meeting of Institute of Gas Engineers; Institute of Gas Engineering Commission: Derby, UK, 1989; pp. 1403–1406. [Google Scholar]
- Yuan, S.; Bi, J.; Qu, X.; Lu, Q.; Cao, Q.; Li, W.; Yao, G.; Wang, Q.; Wang, J. Coal hydrogasification: Entrained flow bed design-operation and experimental study of hydrogasification characteristics. Int. J. Hydrogen Energy 2018, 43, 3664–3675. [Google Scholar] [CrossRef]
- Xia, Z.; Yan, S.; Chen, C.; Qu, X.; Bi, J. Three-dimensional simulation of coal catalytic hydrogasification in a pressurized bubbling fluidized bed. Energy Convers. Manag. 2021, 250, 114874. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, X.; Qian, Y.; Wang, J. Formation behaviors of gas and liquid products during the two-stage hydrogasification of low-rank coal. Fuel 2015, 150, 217–225. [Google Scholar] [CrossRef]
- Yan, S.; Qu, X.; Xia, Z.; Chen, C.; Bi, J. Effect of experimental variables on coal catalytic hydrogasification in a pressurized fluidized bed. Fuel 2022, 307, 121761. [Google Scholar] [CrossRef]
- Hüttinger, K.J.; Michenfelder, A. Hydrogasification of Brown Coal-Chemism, Kinetics and Technical Aspects. In Carbon and Coal Gasification; Springer: Dordrecht, The Netherlands, 1986; pp. 485–507. [Google Scholar]
- Yan, L.; He, B.; Pei, X.; Wang, C.; Li, X.; Duan, Z. Kinetic Models for Coal Hydrogasification and Analyses of Hydrogasification Characteristics in Entrained-Flow Gasifiers. Energy Fuels 2013, 27, 6388–6396. [Google Scholar] [CrossRef]
- Tosti, S.; Sousa, M.A.; Buceti, G.; Madeira, L.M.; Pozio, A. Process analysis of refuse derived fuel hydrogasification for producing SNG. Int. J. Hydrogen Energy 2019, 44, 21470–21480. [Google Scholar] [CrossRef]
- Steinberg, M.; Grohse, E.W.; Tung, Y. A Feasibility Study for the Coprocessing of Fossil Fuels with Biomass by the Hydrocarb Process; Environmental Protection Agency: Washington, DC, USA, 1991; 92p. [Google Scholar]
- Buceti, G.; Tosti, S.; Pozio, A. Idrogassificazione di rifiuti—Metano da energie rinnovabili e combustibile derivato dai rifiuti: Un’alternativa ai processi di incenerimento e termovalorizzazione. La Chimica e L’industria—Rivista Della Società Chimica Italiana 2017, 4, 18–26. [Google Scholar] [CrossRef]
- Barbuzza, E.; Buceti, G.; Pozio, A.; Santarelli, M.; Tosti, S. Gasification of wood biomass with renewable hydrogen for the production of synthetic natural gas. Fuel 2019, 242, 520–531. [Google Scholar] [CrossRef]
- Frigo, S.; Spazzafumo, G. Comparison of different system layouts to generate a substitute of natural gas from biomass and electrolytic hydrogen. Int. J. Hydrogen Energy 2020, 45, 26166–26178. [Google Scholar] [CrossRef]
- Yan, L.; He, B.; Pei, X.; Wang, C.; Duan, Z.; Song, J.; Li, X. Design and comparisons of three biomass based hydrogen generation systems with chemical looping process. Int. J. Hydrogen Energy 2014, 39, 17540–17553. [Google Scholar] [CrossRef]
- Steinberg, M. Process for Conversion of Coal to Substitute Natural Gas (SNG); HCE LLC: Hawthorne, CA, USA, 2005. [Google Scholar]
- Gu, J.; Yang, S.; Kokossis, A. Modeling and Analysis of Coal-Based Lurgi Gasification for LNG and Methanol Coproduction Process. Processes 2019, 7, 688. [Google Scholar] [CrossRef]
- Steinberg, M. Co-Processing Coal and Natural Gas by the Hynol Process for Enhanced Methanol Production and Reduced CO2 Emissions; Department of Energy: Washington, DC, USA, 1997; 27p. [Google Scholar] [CrossRef]
- Ogunjemilusi, D.; Omotayo, F.; Igbinosun, E. Hynol Process for the Gasification of Microbial in Biomass for the Production of Methanol Fuel (CH 4 OH). Int. J. Multidiscip. Sci. Adv. Technol. 2021, 2, 9–19. [Google Scholar] [CrossRef]
- Sellars, B.G.; Babicki, M.L.; Keefer, B.G.; Ng, E. Method of hydrogasification of Biomass to Methane with Low Depositable Tars. U.S. Patent, 8,383,871, 26 February 2013. [Google Scholar]
- Scott, D.S. Hydrogasification of Biomass to Produce High Yields of Methane. U.S. Patent, 4,822,935, 18 April 1989. [Google Scholar]
- You, S.-H.; Jang, H.-T.; Cha, W.-S. Hydrogasification of Mixture Composed of Plastics and Biomass. In Proceedings of the KAIS Fall Conference; The Korea Academia-Industrial Cooperation Society: Cheonan, Republic of Korea, 2011; pp. 283–285. [Google Scholar]
- Porada, S. A comparison of basket willow and coal hydrogasification and pyrolysis. Fuel Process. Technol. 2009, 90, 717–721. [Google Scholar] [CrossRef]
- Oruc, O.; Dincer, I. Evaluation of hydrogen production with iron-based chemical looping fed by different biomass. Int. J. Hydrogen Energy 2020, 45, 34557–34565. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, N.; Wang, J. Co-hydrogasification of lignocellulosic biomass and swelling coal. IOP Conf. Ser. Earth Environ. Sci. 2016, 40, 012042. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, X.; Chen, X.; Yu, G. Effect of Biomass Char Additives on the Hydrogasification Behavior of a Bituminous Coal. BioResources 2016, 11, 9002–9016. [Google Scholar] [CrossRef]
- Islam, M.W. A review of dolomite catalyst for biomass gasification tar removal. Fuel 2020, 267, 117095. [Google Scholar] [CrossRef]
- Valderrama Rios, M.L.; González, A.M.; Lora, E.E.S.; Almazán del Olmo, O.A. Reduction of tar generated during biomass gasification: A review. Biomass Bioenergy 2018, 108, 345–370. [Google Scholar] [CrossRef]
- Skodras, G.; Panagiotidou, S.; Kokorotsikos, P.; Serafidou, M. Potassium catalyzed hydrogasification of low-rank coal for synthetic natural gas production. Open Chem. 2016, 14, 92–109. [Google Scholar] [CrossRef]
- Qu, X.; Wang, Q.-F.; Yan, S.; Feng, J.; Zhang, J.-S.; Zhang, R.; Bi, J.-C. The behavior of the different catalysts for model carbon hydrogasification. J. Fuel Chem. Technol. 2022, 50, 1–10. [Google Scholar] [CrossRef]
- Maneewan, K.; Krerkkaiwan, S.; Sunphorka, S.; Vitidsant, T.; Kuchonthara, P. Catalytic Effect of Biomass Pyrolyzed Char on the Atmospheric Pressure Hydrogasification of Giant Leucaena (Leucaena leucocephala) Wood. Ind. Eng. Chem. Res. 2014, 53, 11913–11919. [Google Scholar] [CrossRef]
- Tomita, A.; Tamai, Y. Hydrogenation of carbons catalyzed by transition metals. J. Catal. 1972, 27, 293–300. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, H.; Bi, J.; Qu, X.; Yan, S.; Zhang, J.; Zhang, J. The evolution of Fe and Fe-Ca catalysts during char catalytic hydrogasification. Fuel 2019, 257, 116040. [Google Scholar] [CrossRef]
- Matsumoto, S. Catalyzed hydrogasification of Yallourn char in the presence of supported hydrogenation nickel catalyst. Energy Fuels 1991, 5, 60–63. [Google Scholar] [CrossRef]
- Murakami, K.; Arai, M.; Shirai, M. Hydrogasification of Loy Yang brown coal by ion-exchanged nickel species. Energy Fuels 2000, 14, 1240–1244. [Google Scholar] [CrossRef]
- Haga, T.; Ozaki, J.-I.; Suzuki, K.; Nishiyama, Y. Role of MgO and CaO promoters in Ni-catalyzed hydrogenation reactions of CO and carbon. J. Catal. 1992, 134, 107–117. [Google Scholar] [CrossRef]
- Haga, T.; Nishiyama, Y. Promotion of iron-group catalysts by a calcium salt in hydrogasification of carbons at elevated pressures. Ind. Eng. Chem. Res. 1987, 26, 1202–1206. [Google Scholar] [CrossRef]
- Yan, S.; Bi, J.; Qu, X. The behavior of catalysts in hydrogasification of sub-bituminous coal in pressured fluidized bed. Appl. Energy 2017, 206, 401–412. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, N.; Qu, X.; Bi, J.; Cao, Q.; Wang, J. Promoted catalysis of calcium on the hydrogasification reactivity of iron-loaded subbituminous coal. Fuel 2017, 200, 153–161. [Google Scholar] [CrossRef]
- Qu, X.; Yan, S.; Yan, X.; Zhang, J.; Zheng, Q.; An, Y. Role of cobalt and calcium on coal catalytic hydrogasification in a pressurized fluidized bed. Fuel 2019, 239, 347–356. [Google Scholar] [CrossRef]
- Feng, J.; Yan, S.; Zhang, R.; Gu, S.; Qu, X.; Bi, J. Recycling and reuse performance of cobalt catalyst for coal hydrogasification. Fuel 2023, 335, 126939. [Google Scholar] [CrossRef]
- Yuan, S.; Qu, X.; Zhang, R.; Bi, J. Effect of calcium additive on product yields in hydrogasification of nickel-loaded Chinese sub-bituminous coal. Fuel 2015, 147, 133–140. [Google Scholar] [CrossRef]
- Bridier, B.; Perez-Ramirez, J. Cooperative effects in ternary Cu−Ni−Fe catalysts lead to enhanced alkene selectivity in alkyne hydrogenation. J. Am. Chem. Soc. 2010, 132, 4321–4327. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, F.; Bi, J.; Qu, X.; Yan, S.; Zhang, J.; Zhang, J. Study of cu–Ni–ca composite catalysts in catalytic hydrogasification of char. Energy Fuels 2019, 33, 9661–9670. [Google Scholar] [CrossRef]
- Liu, H.; Huang, Z.; Kang, H.; Li, X.; Xia, C.; Chen, J.; Liu, H. Efficient bimetallic NiCu-SiO2 catalysts for selective hydrogenolysis of xylitol to ethylene glycol and propylene glycol. Appl. Catal. B Environ. 2018, 220, 251–263. [Google Scholar] [CrossRef]
- Freitas, I.C.; Manfro, R.L.; Souza, M.M. Hydrogenolysis of glycerol to propylene glycol in continuous system without hydrogen addition over Cu-Ni catalysts. Appl. Catal. B Environ. 2018, 220, 31–41. [Google Scholar] [CrossRef]
- Gandarias, I.; Requies, J.; Arias, P.; Armbruster, U.; Martin, A. Liquid-phase glycerol hydrogenolysis by formic acid over Ni–Cu/Al2O3 catalysts. J. Catal. 2012, 290, 79–89. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, R.; Chen, J. Deoxygenation of methyl laurate as a model compound on Ni-Zn alloy and intermetallic compound catalysts: Geometric and electronic effects of oxophilic Zn. Appl. Catal. B Environ. 2018, 224, 88–100. [Google Scholar] [CrossRef]
- Han, Q.; Rehman, M.U.; Wang, J.; Rykov, A.; Gutiérrez, O.Y.; Zhao, Y.; Wang, S.; Ma, X.; Lercher, J.A. The synergistic effect between Ni sites and Ni-Fe alloy sites on hydrodeoxygenation of lignin-derived phenols. Appl. Catal. B Environ. 2019, 253, 348–358. [Google Scholar] [CrossRef]
- Li, C.; Xu, G.; Zhai, Y.; Liu, X.; Ma, Y.; Zhang, Y. Hydrogenation of biomass-derived ethyl levulinate into γ-valerolactone by activated carbon supported bimetallic Ni and Fe catalysts. Fuel 2017, 203, 23–31. [Google Scholar] [CrossRef]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Bi, J.; Zhang, R.; Qu, X.; Zhang, J.; Zhang, J. Investigating the Cu-based catalysts for char catalytic hydrogasification and its recovery. Fuel 2021, 294, 120567. [Google Scholar] [CrossRef]
- Zoheidi, H.; Miller, D.J. Role of oxygen surface groups in catalysis of hydrogasification of carbon black by potassium carbonate. Carbon 1987, 25, 809–819. [Google Scholar] [CrossRef]
- Gardner, N.; Samuels, E.; Wilks, K. Catalyzed Hydrogasification of Coal Chars. In Coal Gasification; American Chemical Society: New York, NY, USA, 1974; Volume 131, pp. 217–236. [Google Scholar] [CrossRef]
- Kokorotsikos, P.; Stavropoulos, G.; Sakellaropoulos, G. Effect of catalyst impregnation conditions on Greek lignite hydrogasification. Fuel 1986, 65, 1462–1465. [Google Scholar] [CrossRef]
- Sheth, A.C.; Sastry, C.; Yeboah, Y.D.; Xu, Y.; Agarwal, P. Catalytic gasification of coal using eutectic salts: Reaction kinetics for hydrogasification using binary and ternary eutectic catalysts. Fuel 2004, 83, 557–572. [Google Scholar] [CrossRef]
- Karcz, A.; Porada, S. The influence of coal rank on formation of gaseous hydrocarbons in hydrogasification of coal. Fuel 1996, 75, 641–645. [Google Scholar] [CrossRef]
- Chin, G.; Guifen, L.; Qushi, D. New approach for gasification of coal char. Fuel 1987, 66, 859–863. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Yang, Z.; Yan, Y.; Mao, Y.; Bi, J. Effect of bauxite additives on ash sintering characteristics during the K2CO3-catalyzed steam gasification of lignite. RSC Adv. 2015, 5, 6720–6727. [Google Scholar] [CrossRef]
- Hong, B.; Wang, X.; Zhou, Z.; Yu, G. A Comparison of the Gas-Product-Release Characteristics from Coal Pyrolysis and Hydrogasification. Energy Technol. 2013, 1, 449–456. [Google Scholar] [CrossRef]
- Cypres, R.; Furfari, S. Fixed-bed pyrolysis of coal under hydrogen pressure at low heating rates. Fuel 1981, 60, 768–778. [Google Scholar] [CrossRef]
- Arendt, P.; van Heek, K.-H. Comparative investigations of coal pyrolysis under inert gas and H2 at low and high heating rates and pressures up to 10 MPa. Fuel 1981, 60, 779–787. [Google Scholar] [CrossRef]
- He, X.; Jin, L.; Wang, D.; Zhao, Y.; Zhu, S.; Hu, H. Integrated Process of Coal Pyrolysis with CO2 Reforming of Methane by Dielectric Barrier Discharge Plasma. Energy Fuels 2011, 25, 4036–4042. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, Z.; Liu, Q. Synergetic Catalysis of Calcium Oxide and Iron in Hydrogasification of Char. Energy Fuels 2017, 31, 198–204. [Google Scholar] [CrossRef]
- Skodras, G.; Kokorotsikos, P.; Serafidou, M. Cation exchange capability and reactivity of low-rank coal and chars. Cent. Eur. J. Chem. 2014, 12, 33–43. [Google Scholar] [CrossRef]
- Hüttinger, K.J.; Krauss, W. Catalytic activity of coal minerals in hydrogasification of coal. Fuel 1981, 60, 93–102. [Google Scholar] [CrossRef]
- Yan, L.; He, B.; Ma, L.; Pei, X.; Wang, C.; Li, M.; Song, J. Numerical study with ChemKin for hydrogasification mechanism of pulverized coal and Hg speciation transformation inside a hydrogasifier. Strojarstvo Časopis Za Teoriju i Praksu u Strojarstvu 2013, 55, 73–85. [Google Scholar]
- Balagurumurthy, B.; Bhaskar, T. Hydropyrolysis of lignocellulosic biomass: State of the art review. Biomass Convers. Biorefinery 2014, 4, 67–75. [Google Scholar] [CrossRef]
- Krerkkaiwan, S.; Fushimi, C.; Yamamoto, H.; Tsutsumi, A.; Kuchonthara, P. Influences of heating rate during coal char preparation and AAEMs on volatile–char interaction with different sources of biomass volatile. Fuel Process. Technol. 2014, 119, 10–18. [Google Scholar] [CrossRef]
- Krerkkaiwan, S.; Tsutsumi, A.; Kuchonthara, P. Biomass derived tar decomposition over coal char bed. ScienceAsia 2013, 39, 511. [Google Scholar] [CrossRef][Green Version]
- Zhou, X.; Yang, X.; Li, J.; Zhao, J.; Song, S.; Hao, Z.; Li, C.; Zhang, J.; Yu, Z.; Fang, Y. High-pressure rapid hydrogasification of pinewood for methane production using calcium looping concept. Energy Convers. Manag. 2020, 203, 112247. [Google Scholar] [CrossRef]
- Suzuki, T.; Iwasaki, J.; Tanka, K.; Okazaki, N.; Funaki, M.; Yamada, T. Influence of calcium on the catalytic behavior of nickel in low temperature hydrogasification of wood char. Fuel 1998, 77, 763–767. [Google Scholar] [CrossRef]
- Keep, C.W.; Terry, S.; Wells, M. Studies of the nickel-catalyzed hydrogenation of graphite. J. Catal. 1980, 66, 451–462. [Google Scholar] [CrossRef]
- Tamai, Y.; Watanabe, H.; Tomita, A. Catalytic gasification of carbon with steam, carbon dioxide and hydrogen. Carbon 1977, 15, 103–106. [Google Scholar] [CrossRef]
- Basu, P. Chapter 3—Biomass Characteristics. In Biomass Gasification, Pyrolysis and Torrefaction, 2nd ed.; Basu, P., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 47–86. [Google Scholar] [CrossRef]
- van Heek, K.H.; Mühlen, H.J. Chemical Kinetics of Carbon and Char Gasification. In Fundamental Issues in Control of Carbon Gasification Reactivity; Lahaye, J., Ehrburger, P., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 1–34. [Google Scholar] [CrossRef]
- Zolin, A.; Jensen, A.; Jensen, P.A.; Frandsen, F.; Dam-Johansen, K. The Influence of Inorganic Materials on the Thermal Deactivation of Fuel Chars. Energy Fuels 2001, 15, 1110–1122. [Google Scholar] [CrossRef]
- Yan, S.; Qu, X.; Xia, Z.; Chen, C.; Bi, J. Catalytic hydrogasification characteristic of coal with diverse properties in a pressurized fluidized bed. Fuel 2021, 296, 120652. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Gong, H.; Luo, Z.; Zhan, D.; Shen, Z.; Thong, J.T. Cobalt-mediated crystallographic etching of graphite from defects. Small 2012, 8, 2515–2523. [Google Scholar] [CrossRef] [PubMed]
- Renda, S.; Ricca, A.; Palma, V. Study of the effect of noble metal promotion in Ni-based catalyst for the Sabatier reaction. Int. J. Hydrogen Energy 2021, 46, 12117–12127. [Google Scholar] [CrossRef]
- Renda, S.; Ricca, A.; Palma, V. Precursor salts influence in Ruthenium catalysts for CO2 hydrogenation to methane. Appl. Energy 2020, 279, 115767. [Google Scholar] [CrossRef]
- Ruocco, C.; Palma, V.; Ricca, A. Experimental and kinetic study of oxidative steam reforming of ethanol over fresh and spent bimetallic catalysts. Chem. Eng. J. 2019, 377, 119778. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Steam-Oxygen Gasification | Catalytic Steam Gasification | Hydrogasification |
|---|---|---|---|
| Coal feedstock | Mined-crushed-transported | Mined-crushed-transported | Mined-crushed-transported |
| Oxygen plant | Yes | No | No |
| Gasifier type | Steam-oxygen | Steam | Hydrogen |
| Steam methane reforming | No | Yes | Yes |
| Thermal efficiency | 63.2 | 71.5 | 73.2 |
| CO2 emissions (lbsCO2∙MSCF a H2−1) | 102 | 90 | 87 |
| % Increase in efficiency from steam-oxygen | 0 | 13 | 16 |
| Capital investment (dollars∙MSCF a H2−1∙day−1) | 1729 | 1729 | 1383 |
| Production costs (dollars∙MSCF a H2−1) | 1.87 | 1.94 | 1.53 |
| % Cost reduction from steam-oxygen | 0 | 4% increase | 18 |
| Temperature (°C) | H2 | H2 + 10 vol % CO2 |
|---|---|---|
| 700 | 3.8% | 9% |
| 800 | 4.7% | 13% |
| 900 | 10% | 20% |
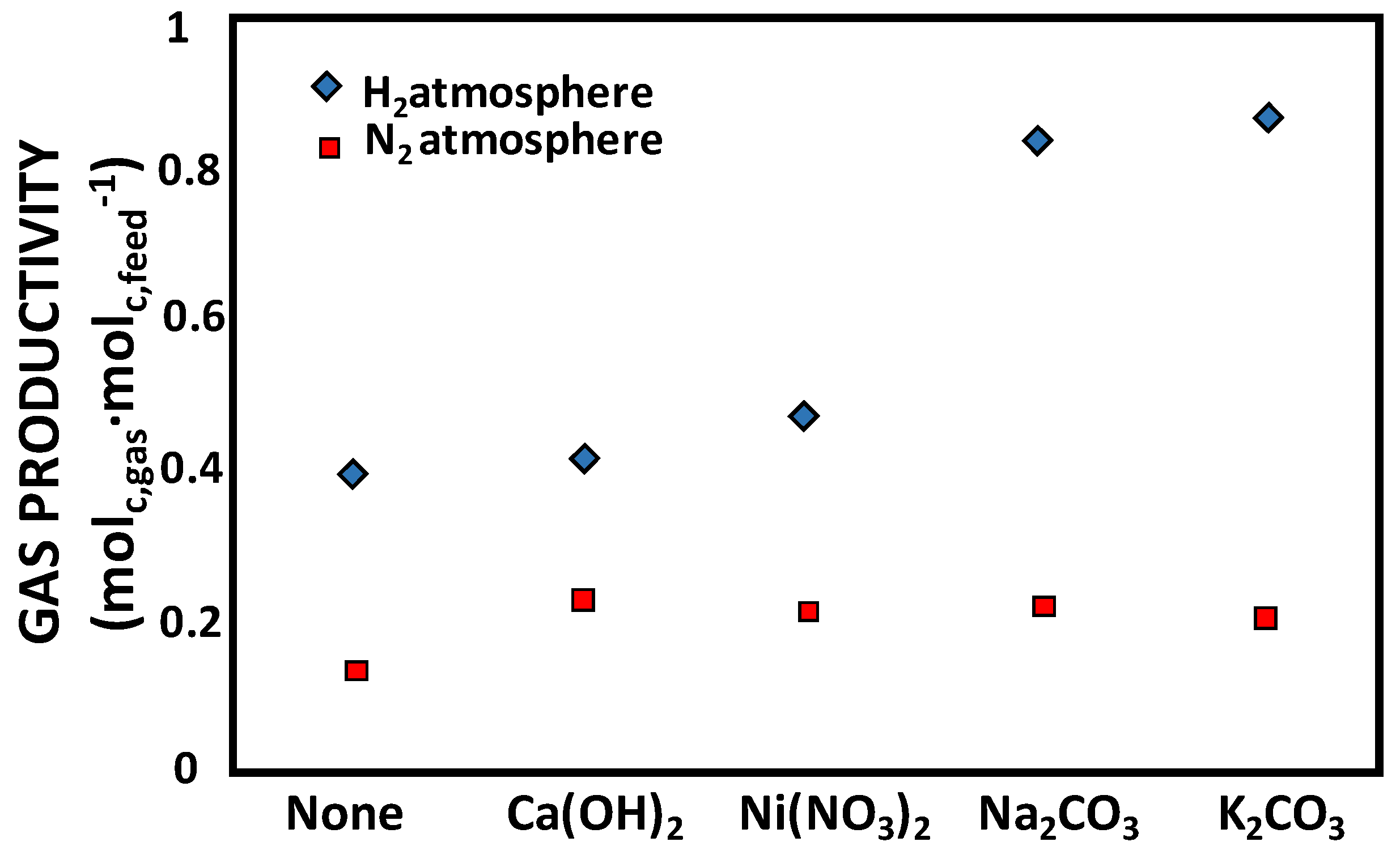
| Sample | Gas Yield under N2 (%) | Gas Yield under H2 (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| CO2 | CO | CH4 | CnHm | CO2 | CO | CH4 | CnHm | |
| None | 2.99 | 3.04 | 5.47 | 1.88 | 0.91 | 1.73 | 29.33 | 7.25 |
| Ca(OH)2 | 5.35 | 9.77 | 5.75 | 2.49 | 2.85 | 2.71 | 29.00 | 7.00 |
| Ni(NO3)2 | 8.66 | 6.17 | 4.17 | 1.39 | 4.13 | 2.39 | 33.02 | 7.81 |
| Na2CO3 | 5.89 | 8.56 | 5.83 | 2.10 | 3.70 | 2.13 | 67.10 | 10.86 |
| KOH | 6.24 | 6.95 | 5.61 | 2.10 | 3.14 | 1.89 | 73.95 | 7.91 |
| Carbon Matrix | Catalyst | Reactor Type | Reaction Conditions | Xchar and YCH4 | CH4 Production | Ref. |
|---|---|---|---|---|---|---|
| 50 g, Low-ash bituminous coal | Co-Ca/Co-K/Co-Mg Better catalyst 5 wt% Co–1 wt% Ca | Pressurized fluidized bed | 12 L∙min−1 of H2600–850 °C 3.0 MPa U∙Umf−1: 1.9–15 Timeresidence: 1–8 s Timereaction: 30 min | XChar: 27–91.3 wt% Y CH4: 9.6–78.8 wt% | 72 mL∙(min∙gcoal)−1 | [60] |
| 100 mg, pitch-based activated carbon | 2 wt%Fe/Co/Ni/K/Ca/Mg Order of catalyst activity: Co ≈ Ni > Fe >> Ca ≈ Mg > K. Better catalyst: 1 wt% Ca–2 wt% Co | Pressurized thermo-gravimetric analyzer | 200 mL·min−1 of H2 850 °C 3 MPa Timereaction: 90 min | Xchar: 98% | - | [91] |
| 0.5 g, sub-bituminous coal char | 5 wt% Cu–1 wt% Ca catalyst | Pressurized fixed-bed reactor | 50 mL·min−1 of H2 800 °C 2 MPa Timereaction: 450 min | Y CH4: 61% | 6.65 mL∙(min⋅gchar)−1 | [105] |
| 1 g, sub-bituminous coal char | 5 wt% Fe/1 wt% Ca/ 5 wt% Fe–1 wt% Ca | Pressurized fixed-bed reactor | 50 mL·min of H2750 °C 2 MPa Timereaction: 450 min | -No catalyst: Y CH4: 7.76% -5 wt% Fe: Y CH4: 11.58% -1 wt% Ca Y CH4: 12.20% -5 wt% Fe–1 wt% Ca Y CH4: 53.40% | For 5 wt% Fe–1 wt% Ca: 3.2 mL·(min·gchar)−1 | [86] |
| 1 g, sub-bituminous coal char | 2.0% Cu−1.0 wt% Ca, 1.5% Cu−0.5% Ni−1.0% Ca, 1.0% Cu−1.0% Ni−1.0% Ca, 0.5% Cu−1.5% Ni−1.0% Ca, 2.0% Ni−1.0% Ca Better catalyst: 2.5% Cu–2.5% Ni–1.0% Ca | Pressurized fixed-bed reactor | 50 mL·min−1 of H2 800 °C 2 MPa Timereaction: 450 min | YCH4: 88.3% | 12 mL·(min·gchar)−1 | [97] |
| 50 g, sub-bituminous coal char | 5 wt% Co–1 wt% Ca | Pressurized fluidized bed reactor | 12.0 L·min−1 of H2 850 °C 3 MPa Timereaction: 100 min | X char: 90.0 wt% Y CH4: 77.3 wt% | 70 mL·(min·gcoal)−1 | [93] |
| 50 g, sub-bituminous coal | 5 wt% Co–1 wt% Ca | Pressurized fluidized bed reactor | 11.7 NL ·min−1 of H2 800 °C 1 MPa Timeresidence: 30 min Timereaction: 100 min | X char: 91.3 wt% Y CH4: 77.3 wt% | 67 mL· (min·gcoal)−1 | [42] |
| 2 g, coal char mixed with catalyst | 10 wt% CaO/10 wt% CaCO3/ 10 wt% Na2CO3/ 10 wt% K2CO3 | High-pressure fixed-bed tube reactor | 1.5 NL ·min−1 850 °C 5 MPa Timereaction: 40 min | -No catalyst: X char: 18.13% -10 wt% CaO: X char: 32.25% -10 wt% Na2CO3: X char: 71.49% -10 wt% K2CO3: X char80% | - | [29] |
| 0.5 g, bituminous coal char | CaO-0.258 FeS2 | Pressurized fixed-bed reactor | 50 mL·min−1 800 °C 3.0 MPa Timereaction: 200 min | Y CH4: 77.9 wt% in 7.5 h. | 1.8 mL∙(min·gchar)−1 | [117] |
| 100 g, sub-bituminous coal | Co-Ca/Ni-Ca/Fe-Ca Order of catalytic activity: 5% Co–1% Ca > 5% Ni–1% Ca > 5% Fe–1% Ca | Pressurize fluidized bed | 11.7 L(STP) ·min−1 850 °C 3 MPa U∙Umf−1: 2 | 5% Co-1% Ca X Char: 91.3 wt%, Y CH4: 78.8 wt% | 5% Co-1% Ca: 69 mL·(min·g coal)1 1.2 Nm3CH4 · kgcoal−1 | [91] |
| Low-rank coal (lignite) | 20 wt% K2CO3 | Tubular fixed-bed reactor | 850 °C 1 MPa Timereaction:100 min | Xchar: 40% | 2 mL·(g· min)−1 | [82] |
| Sub-bituminous coal | 5 wt% Ni–1 wt% Ca | Fixed-bed reactor | 4.4 L·min−1 of H2 750 °C 1 MPa Timereaction:60 min | Xchar: 65–75 wt% YCH4: 25 wt% | - | [95] |
| 10 g, lignite | 10 wt% K2CO3/10 wt% Ca(OH)2/10 wt% Ni (NO3)2 Sequence of activity: K2CO3 > Ni (NO3)2 > Ca (OH)2 | Pressurized fixed-bed reactor | 1 L·min−1 of H2 800 °C 4.0 MPa Timereaction:60 min | -Raw coal: Y CH4: 29.3% -Ca(OH)2: Y CH4: 29% -Ni(NO3)2: Y CH4: 33.0% -Na2CO3: Y CH467.1% -KOH: YCH4: 78% K > Na > Ni > Ca | KOH catalyst: 0.348 L·g coal | [113] |
| 0.5 g, Giant Leucaena wood | Slow (SC) and fast (FC) pyrolyzed char from Giant Leucaena wood Better catalyst for the tar reduction: char obtained by slow pyrolysis at 750 °C | Two-stage fixed-bed reactor | 8.33% v/v H2 in Ar 700 °C 0.01 MPa | Xchar: 20% Gas yield: 45% with the SC at 750 °C while 42% with the other | - | [84] |
| 0.5 g, brown coal char | Fe/Ni/Co 45 wt% Ni/diatomite | Quartz tube reactor | 100 mL·min−1 of H2 850 °C 0.01 MPa Timereaction:400 min | Ni/diatomite Xchar: 70% | - | [87] |
| 2.5–4 g, combination of rice husk with swelling coal | Potassium of coal | Two-stage fixed-bed reactor | 100 and 500 mL·min−1 of H2 1–5 MPa 500–700 °C | Xchar: 50% Y gas:<20% | - | [78] |
| 50 g, bituminous coal | 3 wt% Co–2 wt% Ca | Batch-pressurized fluidized bed reactor | 12.0 L∙min−1 of H2 and CO2 (10 vol%) 850 °C 3 MPa Timereaction: 280 min Numberfluidization: 3 | Y gas: 83.3–90.7% Y CH4: 54.8–77.4% | 34.6–57.1 mL·(g·min)−1 | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraceno, E.; Ruocco, C.; Palma, V. A Review of Coal and Biomass Hydrogasification: Process Layouts, Hydrogasifiers, and Catalysts. Catalysts 2023, 13, 417. https://doi.org/10.3390/catal13020417
Saraceno E, Ruocco C, Palma V. A Review of Coal and Biomass Hydrogasification: Process Layouts, Hydrogasifiers, and Catalysts. Catalysts. 2023; 13(2):417. https://doi.org/10.3390/catal13020417
Chicago/Turabian StyleSaraceno, Emilia, Concetta Ruocco, and Vincenzo Palma. 2023. "A Review of Coal and Biomass Hydrogasification: Process Layouts, Hydrogasifiers, and Catalysts" Catalysts 13, no. 2: 417. https://doi.org/10.3390/catal13020417
APA StyleSaraceno, E., Ruocco, C., & Palma, V. (2023). A Review of Coal and Biomass Hydrogasification: Process Layouts, Hydrogasifiers, and Catalysts. Catalysts, 13(2), 417. https://doi.org/10.3390/catal13020417