
Hydride Generation on the Cu-Doped CeO2(111) Surface and Its Role in CO2 Hydrogenation Reactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calculation Methods
3. Results and Discussion
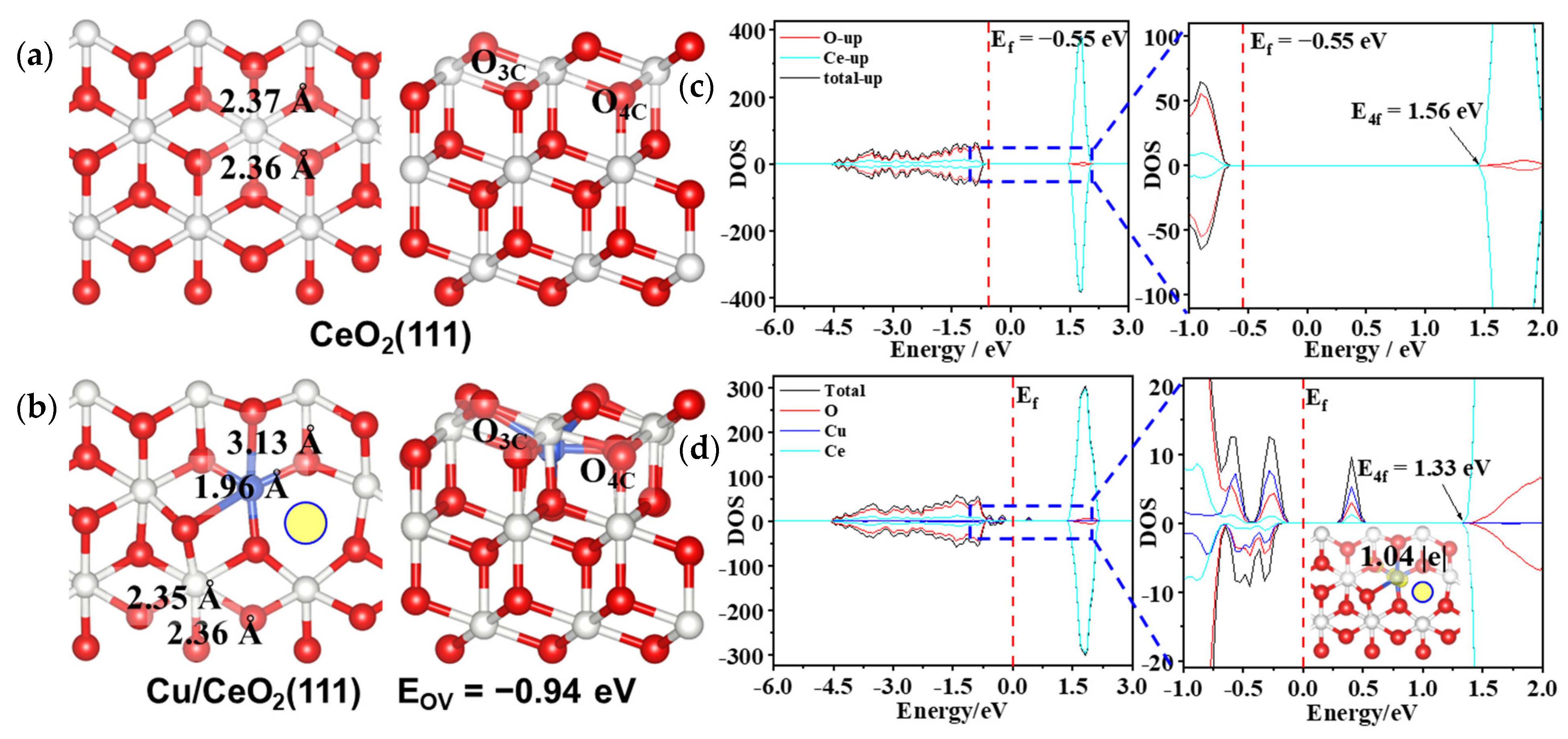
3.1. Structural and Electronic Properties
3.2. H Adsorption
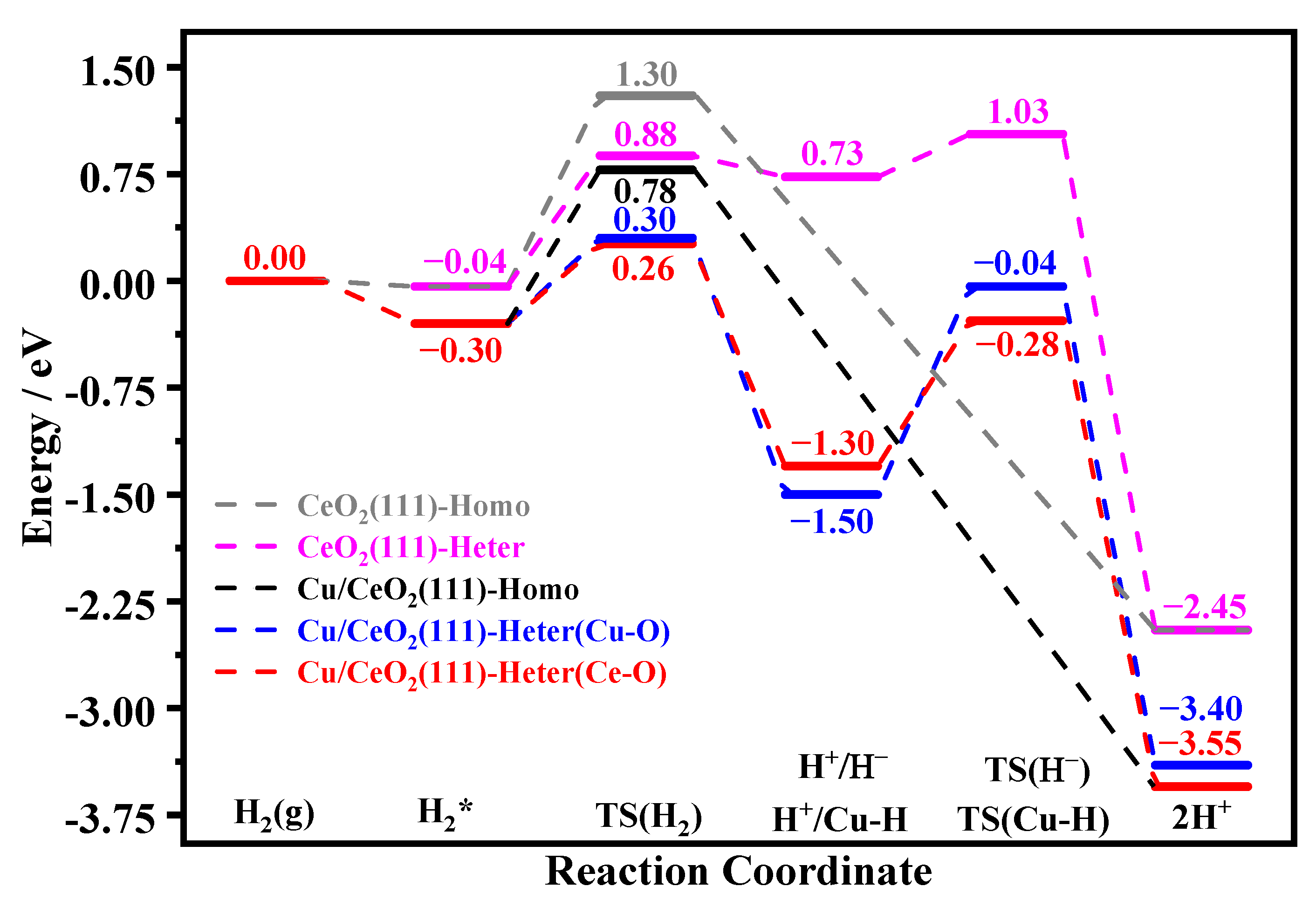
3.3. H2 Dissociation and H- Formation
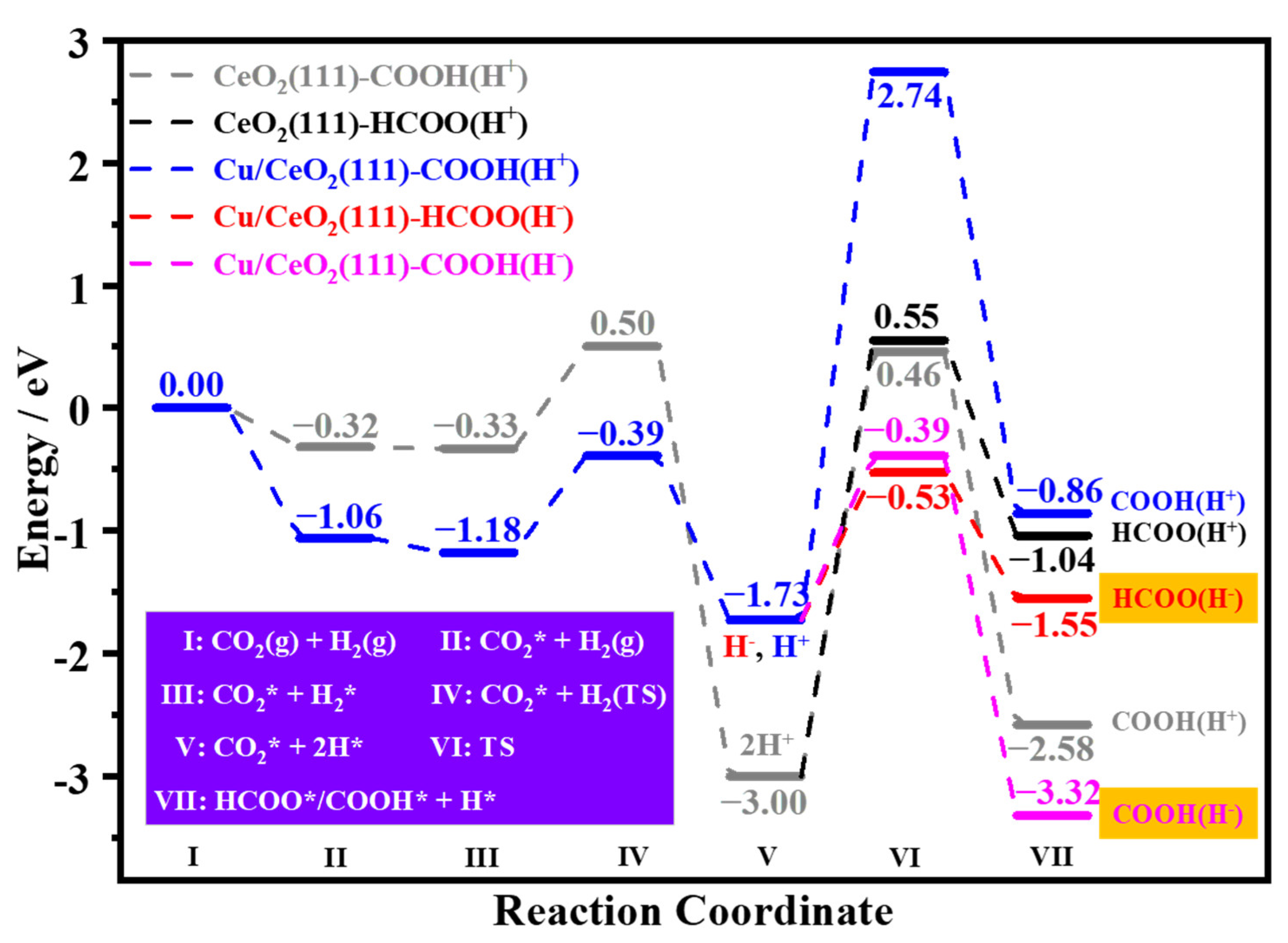
3.4. Selective Hydrogenation of CO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gianvito, V.; Patrick, D.; Julia, V.; Miguel, B.; Sebastián, C.; Mónica, C.; Adrian, B.; Javier, P.-R. Promoted Ceria Catalysts for Alkyne Semi-hydrogenation. J. Catal. 2015, 324, 69–78. [Google Scholar]
- Esrafilzadeh, D.; Zavabeti, A.; Jalili, R.; Atkin, P.; Choi, J.; Carey, B.J.; Brkljača, R.; O’Mullane, A.P.; Dickey, M.D.; Officer, D.L.; et al. Room Temperature CO2 Reduction to Solid Carbon Species on Liquid Metals Featuring Atomically Thin Ceria Interfaces. Nat. Commun. 2019, 10, 865. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.; Palomino, R.M.; Senanayake, S.D. Ceria-based Model Catalysts: Fundamental Studies on the Importance of the Metal—Ceria Interface in CO Oxidation, the Water—Gas Shift, CO2 Hydrogenation, and Methane and Alcohol Reforming. Chem. Soc. Rev. 2017, 46, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in Hydrogenation Catalysis: High Selectivity in the Conversion of Alkynes to Olefins. Angew. Chem. Int. Ed. 2012, 51, 8620–8623. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.; Zhou, S.; Kunwar, D.; Riva, A.D.L.; Peterson, E.; Payne, R.; Gao, L.; Lin, S.; Guo, H.; Datye, A. Design of Effective Catalysts for Selective Alkyne Hydrogenation by Doping of Ceria with a Single-atom Promotor. J. Am. Chem. Soc. 2018, 140, 12964–12973. [Google Scholar] [CrossRef]
- James, K.; Jisue, M.; Wu, Z.L. A Review of the Interactions between Ceria and H2 and the Applications to Selective Hydrogenation of Alkynes. Chin. J. Catal. 2020, 41, 901–914. [Google Scholar]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based Heterogeneous Catalysts toward Catalytic Conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Chang, K.; Zhang, H.C.; Cheng, M.-J.; Lu, Q. Application of Ceria in CO2 Conversion Catalysis. ACS Catal. 2020, 10, 613–631. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, S.S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.B.; Liu, X.H. Insights into the Influence of CeO2 Crystal Facet on CO2 Hydrogenation to Methanol over Pd/CeO2 Catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Cheng, Z.; Lo, C.S. Mechanistic and Microkinetic Analysis of CO2 Hydrogenation on Ceria. Phys. Chem. Chem. Phys. 2016, 18, 7987–7996. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Haider, M.A.; Agarwal, M.; Sinha, N.; Basu, S. Role of Reduced CeO2 Surface for CO2 Reduction to CO and Methanol. J. Phys. Chem. C 2016, 120, 16626–16635. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, X.-L.; Xiao, H.; Lei, M.; Li, J. Mechanistic Investigations on Thermal Hydrogenation of CO2 to Methanol by Nanostructured CeO2: The Crystal-Plane Effect on Catalytic Reactivity. J. Phys. Chem. C 2019, 123, 11763–11771. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Yang, Y.; Mims, C.; Peden, C.H.F.; Li, J.; Mei, D.H. Insight into Methanol Synthesis from CO2 Hydrogenation on Cu(111): Complex Reaction Network and the Effects of H2O. J. Catal. 2011, 281, 199–211. [Google Scholar] [CrossRef]
- Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C.F.; Helveg, S.; Chorkendorff, I.; Sehested, J. Quantifying the Promotion of Cu Catalysts by ZnO for Methanol Synthesis. Science 2016, 352, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Kattel, S.; Ramırez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active Sites for CO2 Hydrogenation to Methanol on Cu/ZnO Catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef]
- Liu, S.P.; Zhao, M.; Gao, W.; Jiang, Q.; Jacob, T. Theoretical Studies on the CO2 Reduction to CH3OH on Cu(211). Electrocatalysis 2017, 8, 647–656. [Google Scholar] [CrossRef]
- Xu, D.; Ding, M.Y.; Hong, X.L.; Liu, G.L. Mechanistic Aspects of the Role of K Promotion on Cu−Fe-based Catalysts for Higher Alcohol Synthesis from CO2 Hydrogenation. ACS Catal. 2020, 10, 14516–14526. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.Q.; Ding, M.Y.; Hong, X.L.; Liu, G.L.; Tsang, S.C.E. Advances in Higher Alcohol Synthesis from CO2 Hydrogenation. Chem 2020, 7, 849–881. [Google Scholar] [CrossRef]
- Liu, H.X.; Li, S.Q.; Wang, W.W.; Yu, W.-Z.; Zhang, W.-J.; Ma, C.; Jia, C.-J. Partially Sintered Copper–Ceria as Excellent Catalyst for the High-Temperature Reverse Water Gas Shift Reaction. Nat. Commun. 2022, 13, 867. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.B.; Zhang, L.J.; Cai, J.; Peng, Z.; Cheng, P.H.; Li, X.B.; Zhang, H.; Yang, F.; Liu, Z. Formation and Activity Enhancement of Surface Hydrides by the Metal–Oxide Interface. Adv. Mater. Interfaces 2021, 8, 2002169. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, M.H.; Li, M.; Wu, X.J.; Xia, C.R. Synergistic Effects towards H2 Oxidation on the Cu–CeO2 Electrode: A Combination Study with DFT Calculations and Experiments. J. Mater. Chem. A 2016, 4, 5745–5754. [Google Scholar] [CrossRef]
- Wan, Q.; Wei, F.F.; Wang, Y.Q.; Wang, F.T.; Zhou, L.S.; Lin, S.; Xie, D.Q.; Guo, H. Single Atom Detachment from Cu Clusters, and Diffusion and Trapping on CeO2: Implications in Ostwald Ripening and Atomic Redispersion. Nanoscale 2018, 10, 17893–17901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Chen, Z.; Han, P.; Du, Y.H.; Gu, Z.X.; Xu, X.; Zheng, Z.F. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Yang, T.; Mao, X.N.; Zhang, Y.; Wu, X.P.; Wang, L.; Chu, M.Y.; Pao, C.-W.; Yang, S.Z.; Xu, Y.; Huang, X.Q. Coordination Tailoring of Cu Single Sites on C3N4 Realizes Selective CO2 Hydrogenation at Low Temperature. Nat. Commun. 2021, 12, 6022. [Google Scholar] [CrossRef] [PubMed]
- Schweke, D.; Shelly, L.; David, R.B.; Danon, A.; Kostirya, N.; Hayun, S. Comprehensive Study of the Ceria H2 System: Effect of the Reaction Conditions on the Reduction Extent and Intermediates. J. Phys. Chem. C 2020, 124, 6180–6187. [Google Scholar] [CrossRef]
- Fernandez-Torre, D.; Carrasco, J.; Ganduglia-Pirovano, M.V.; Perez, R. Hydrogen Activation, Diffusion, and Clustering on CeO2: A DFT + U Study. J. Chem. Phys. 2014, 141, 014703. [Google Scholar] [CrossRef] [PubMed]
- García-Melchor, M.; López, N. Homolytic Products from Heterolytic Paths in H2 Dissociation on Metal Oxides: The Example of CeO2. J. Phys. Chem. C 2014, 118, 10921–10926. [Google Scholar] [CrossRef]
- Duchoň, T.; Hackl, J.; Mueller, D.N.; Kullgren, J.; Du, D.; Senanayake, S.D.; Mouls, C.; Gottlob, D.; Khan, M.M.I.; Cramm, S.; et al. Establishing Structure-sensitivity of Ceria Reducibility: Real-time Observations of Surface-hydrogen Interactions. J. Mater. Chem. A 2020, 8, 5501–5507. [Google Scholar] [CrossRef]
- Vilé, G.; Colussi, S.; Krumeich, F.; Trovarelli, A.; Pérez-Ramírez, J. Opposite Face Sensitivity of CeO2 in Hydrogenation and Oxidation Catalysis. Angew. Chem. Int. Ed. 2014, 53, 12069–12072. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-P.; Gong, X.-Q.; Lu, G. Role of Oxygen Vacancies in the Surface Evolution of H at CeO2: A Charge Modification Effect. Phys. Chem. Chem. Phys. 2015, 17, 3544–3549. [Google Scholar] [CrossRef]
- Li, Z.; Werner, K.; Qian, K.; You, R.; Płucienik, A.; Jia, A.; Wu, L.; Zhang, L.; Pan, H.; Kuhlenbeck, H.; et al. Oxidation of Reduced Ceria by Incorporation of Hydrogen. Angew. Chem. Int. Ed. 2019, 131, 14828–14835. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, Y.; Tao, F.; Daemen, L.; Foo, G.S.; Nguyen, L.; Zhang, X.; Beste, A.; Ramirez-Cuesta, A.J. Direct Neutron Spectroscopy Observation of Cerium Hydride Species on a Cerium Oxide Catalyst. J. Am. Chem. Soc. 2017, 139, 9721–9727. [Google Scholar] [CrossRef] [PubMed]
- Werner, K.; Weng, X.; Calaza, F.; Sterrer, M.; Kropp, T.; Paier, J.; Sauer, J.; Wilde, M.; Fukutani, K.; Shaikhutdinov, S.; et al. Toward an Understanding of Selective Alkyne Hydrogenation on Ceria: On the Impact of O Vacancies on H2 Interaction with CeO2. J. Am. Chem. Soc. 2017, 139, 17608–17616. [Google Scholar] [CrossRef] [PubMed]
- Coperet, C.; Estes, D.P.; Larmier, K.; Searles, K. Isolated Surface Hydrides: Formation, Structure, and Reactivity. Chem. Rev. 2016, 116, 8463–8505. [Google Scholar] [CrossRef]
- Moon, J.; Cheng, Y.; Daemen, L.L.; Li, M.; Polo-Garzon, F.; Ramirez-Cuesta, A.J.; Wu, Z. Discriminating the Role of Surface Hydride and Hydroxyl for Acetylene Semi-Hydrogenation over Ceria through in Situ Neutron and Infrared Spectroscopy. ACS Catal. 2020, 10, 5278–5287. [Google Scholar] [CrossRef]
- Wang, Q.R.; Guo, J.P.; Chen, P. The Power of Hydrides. Joule 2020, 4, 705–709. [Google Scholar] [CrossRef]
- Li, Z.R.; Kristin, W.; Chen, L.; Jia, A.P.; Qian, K.; Zhong, J.Q.; You, R.; Wu, L.H.; Zhang, L.Y.; Pan, H.B.; et al. Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk. Chem. Eur. J. 2021, 27, 5268–5276. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Chu, D.-R.; Zhou, H.; Wu, X.-P.; Gong, X.-Q. Role of Low-Coordinated Ce in Hydride Formation and Selective Hydrogenation Reactions on CeO2 Surfaces. ACS Catal. 2022, 12, 624–632. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total–Energy Calculations Using a Pane–Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented–wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, J.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Teter, M.P.; Payne, M.C.; Allan, D.C. Solution of Schrodinger’s Equation for Large Systems. Phys. Rev. B 1989, 40, 12255–12263. [Google Scholar] [CrossRef] [PubMed]
- Kümmerle, E.; Heger, G. The Structures of C–Ce2O3+δ, Ce7O12, and Ce11O20. J. Solid State Chem. 1999, 147, 485–500. [Google Scholar] [CrossRef]
- Jerratsch, J.-F.; Shao, X.; Nilius, N.; Freund, H.-J.; Popa, C.; Ganduglia-Pirovano, M.V.; Burow, A.M.; Sauer, J. Electron Localization in Defective Ceria Films: A Study with Scanning-Tunneling Microscopy and Density-Functional Theory. Phys. Rev. Lett. 2011, 106, 246801. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Parker, S.C.; Watson, G.W. The Electronic Structure of Oxygen Vacancy Defects at the Low Index Surfaces of Ceria. Surf. Sci. 2005, 595, 223–232. [Google Scholar] [CrossRef]
- Nolan, M.; Grigoleit, S.; Sayle, D.C.; Parker, S.C.; Watson, G.W. Density Functional Theory Studies of the Structure and Electronic Structure of Pure and Defective Low Index Surfaces of Ceria. Surf. Sci. 2005, 576, 217–229. [Google Scholar] [CrossRef]
- Vicario, G.; Balducci, G.; Fabris, S.; Gironcoli, S.D.; Baroni, S. Interaction of Hydrogen with Cerium Oxide Surfaces: A Quantum Mechanical Computational Study. J. Phys. Chem. B 2006, 110, 19380–19385. [Google Scholar] [CrossRef]
- Michaelides, A.; Liu, Z.P.; Zhang, C.J.; Alavi, A.; King, D.A.; Hu, P. Identification of General Linear Relationships between Activation Energies and Enthalpy Changes for Dissociation Reactions at Surfaces. J. Am. Chem. Soc. 2003, 125, 3704–3705. [Google Scholar] [CrossRef]
- Alavi, A.; Hu, P.; Deutsch, T.; Silvestrelli, P.L.; Hutter, J. CO Oxidation on Pt(111): An Ab Initio Density Functional Theory Study. Phys. Rev. Lett. 1998, 80, 3650–3653. [Google Scholar] [CrossRef]
- Liu, Z.P.; Hu, P. General Rules for Predicting Where a Catalytic Reaction Should Occur on Metal Surfaces: A Density Functional Theory Study of C–H and C–O Bond Breaking/making on Flat, Stepped, and Kinked Metal Surfaces. J. Am. Chem. Soc. 2003, 125, 1958–1967. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Li, Y.M. Introduction to Surface Chemistry and Catalysis; Wiley-VCH: Berkeley, CA, USA, 2010. [Google Scholar]
- Yang, Z.X.; He, B.L.; Lu, Z.S.; Hermansson, K. Physisorbed, Chemisorbed, and Oxidized CO on Highly Active Cu−CeO2. J. Phys. Chem. C 2010, 114, 4486–4494. [Google Scholar] [CrossRef]
- Zhou, S.L.; Wan, Q.; Lin, S. Cu/O Frustrated Lewis Pairs on Cu Doped CeO2 for Acetylene Hydrogenation: A First-Principles Study. Catalysts 2022, 12, 74. [Google Scholar] [CrossRef]
- Guo, C.; Wei, S.; Zhou, S.; Zhang, T.; Wang, Z.; Ng, S.-P.; Lu, X.; Wu, C.-M.L.; Guo, W. Initial Reduction of CO2 on Pd-, Ru-, and Cu-Doped CeO2 Surfaces: Effects of Surface Modification on Catalytic Activity and Selectivity. ACS Appl. Mater. Interfaces 2017, 9, 26107–26117. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martínez-Arias, A.; Fernández-García, A. Unusual Physical and Chemical Properties of Cu in Ce1-xCuxO2 Oxides. J. Phys. Chem. B 2005, 109, 19595–19603. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Wang, D.; Gong, X.-Q. Strategies to Improve the Activity while Maintaining the Selectivity of Oxidative Coupling of Methane at La2O3: A Density Functional Theory Study. ACS Catal. 2020, 10, 586–594. [Google Scholar] [CrossRef]
- Wexler, R.B.; Gautam, G.S.; Stechel, E.B.; Carter, E.A. Factors Governing Oxygen Vacancy Formation in Oxide Perovskites. J. Am. Chem. Soc. 2021, 143, 13212–13227. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.-Q.; Liu, H.-H.; Wu, X.-P.; Hu, P.; Gong, X.-Q. Hydride Generation on the Cu-Doped CeO2(111) Surface and Its Role in CO2 Hydrogenation Reactions. Catalysts 2022, 12, 963. https://doi.org/10.3390/catal12090963
Wang Z-Q, Liu H-H, Wu X-P, Hu P, Gong X-Q. Hydride Generation on the Cu-Doped CeO2(111) Surface and Its Role in CO2 Hydrogenation Reactions. Catalysts. 2022; 12(9):963. https://doi.org/10.3390/catal12090963
Chicago/Turabian StyleWang, Zhi-Qiang, Hui-Hui Liu, Xin-Ping Wu, Peijun Hu, and Xue-Qing Gong. 2022. "Hydride Generation on the Cu-Doped CeO2(111) Surface and Its Role in CO2 Hydrogenation Reactions" Catalysts 12, no. 9: 963. https://doi.org/10.3390/catal12090963
APA StyleWang, Z.-Q., Liu, H.-H., Wu, X.-P., Hu, P., & Gong, X.-Q. (2022). Hydride Generation on the Cu-Doped CeO2(111) Surface and Its Role in CO2 Hydrogenation Reactions. Catalysts, 12(9), 963. https://doi.org/10.3390/catal12090963