Abstract
The development of an efficient catalyst especially with a high productivity for decarboxylation of L-lysine to cadaverine, is of both industrial and economic significance. Here, we reported the synthesis of RuO2 well-confined in the supercage of FAU zeolite (RuO2@FAU) through in situ hydrothermal strategies. A set of characterizations, such as XRD, Raman, TEM, XPS, NH3-TPD and N2 physical adsorption, confirmed the successful encapsulation of RuO2 clusters (~1.5 nm) inside the FAU zeolite. RuO2@FAU had the higher cadaverine productivity of 120.9 g/L/h/mmol cat., which was almost six times that of traditionally supported ruthenium oxide catalysts (21.2 g/L/h/mmol cat.). RuO2@FAU catalysts with different ammonia exchange degrees, as well as different Si/Al ratios were further evaluated. After optimization, the highest cadaverine productivity of 480.3 g/L/h/mmol cat. was obtained. Deep analysis of the electronic properties of RuO2@FAU indicated that the surface defect structures, such as oxygen vacancies, played a vital role in the adsorption or activation of L-lysine which finally led to a boosted performance. Furthermore, the mechanism of decarboxylation of L-lysine to cadaverine was proposed.
1. Introduction
Cadaverine is a prime important monomer for the development of new and high-performance materials including polyamide (such as nylon 5X) and polyurethane, which can be widely used as engineering plastics and fibrous polymers [1]. Synthesis of cadaverine from bio-based L-lysine is a sustainable and economic process (Scheme 1), which is of great potential to substitute the key monomer, hexamethylenediamine, in petrol-based nylon 66. The biological decarboxylation of L-lysine with decarboxylase as a catalyst has been traditionally applied to produce cadaverine [2,3,4]. Considering the expensive separation process and the toxicity of cadaverine for the cells [5,6,7], the industrial-scale process is still pursuing an economic, reusable, and stable catalyst for the decarboxylation of L-lysine.

Scheme 1.
Synthesis of cadaverine by decarboxylation of L-lysine.
Recently, the chemo-catalytic heterogeneous process has attracted more and more interests in decarboxylation of amino acids from both academics and industry. De Vos et al. reported decarboxylation of pyroglutamic acid to 2-pyrrolidone (70% yield) and proline to pyrrolidine (95% selectivity) with supported palladium (Pd) catalysts [8,9]. However, supported Pd catalysts performed poorly in the decarboxylation of L-lysine with a yield of cadaverine of less than 10%. Later, Verduyckt et al. reported the decarboxylation of L-lysine with commercial Ru/C as the catalyst and obtained cadaverine with a yield of 32% [10]. Though the yield was not ideal, it certainly provided a significantly meaningful approach to producing cadaverine. Xie et al. studied the decarboxylation of L-lysine on Ru/C in detail and after reaction conditions were optimal, the yield of cadaverine was significantly improved to 40% [11]. It indicated that a Ru-based catalyst was a candidate for the decarboxylation of L-lysine to cadaverine. In our previous works [12,13], Ru-based catalysts with different kinds of supports were prepared by the traditional impregnation method and further evaluated in the decarboxylation of L-lysine. Among those tested supports, zeolite supported ruthenium oxide catalyst performed much better with a greatly enhanced selectivity of cadaverine (54%). Structure characterization showed that RuO2 was the main phase of ruthenium oxide nanoparticles. The under-coordinated Ru-O bonds in ruthenium oxide catalysts were regarded as active surface sites leading to the dissociative adsorption of H2, which greatly contributed to the hydrogenation–decarboxylation process of L-lysine. Unfortunately, a serious agglomeration of ruthenium oxide nanoparticles was observed for those supported catalysts, which might be largely to blame for the unremarkable performance in the decarboxylation of L-lysine. Still, more effort needs to be devoted to developing an ultrafine ruthenium oxide catalyst for further industrial application.
The encapsulation of metal clusters in zeolite micropores or cavities is an attractive approach to preparing ultrafine metal nanoparticles. The confinement of zeolite micropores provided a physical restriction for small metal clusters, which finally improved stability against the leaching of active metal species during catalysis [14,15]. Additionally, an improved catalytic performance would be obtained via the synergistic effects between the metal sites and functional sites of zeolites. RuO2 clusters have been successfully encapsulated in LTA (8 MR) for methanol oxidation [16], in MFI (10 MR) for phenol hydrodeoxygenation [17] and in FAU (12 MR) for aerobic alcohol oxidation [18]. Yang et al. [19] found that metal–acid intimacy played an essential role in enhancing the production of cyclohexane. Particularly, few works have been reported on the application of encapsulated RuO2 clusters in the decarboxylation of L-lysine. Herein, well-dispersed RuO2 clusters (~1.5 nm) encapsulated in supercages of FAU zeolites have been developed by an in situ hydrothermal synthesis approach in this study. The successful encapsulation of ruthenium oxide clusters in obtained RuO2@FAU catalyst has been proven by high-resolution transmission electron microscopy (HRTEM). The homemade RuO2@FAU catalyst was evaluated for the decarboxylation of L-lysine and after conditions were optimized, a significantly enhanced productivity of cadaverine (480.3 g/L/h/mmol cat.) was obtained.
2. Results and Discussion
2.1. Synthesis and Characterization for RuO2@FAU Catalysts
Supported metal oxide catalysts generally met a problem of serious agglomeration of nano-sized metal particles which seriously inhibited its evaluation performance. To solve this problem, we prepared ruthenium oxide encapsulated in zeolite channels by the in situ synthesis method. RuCl3 was added to the synthesis system during the aging period. After vigorous stirring, the mixture was transferred to the oven (100 °C) for 12 h for crystallization. Finally, RuO2@FAU samples were obtained after centrifugation.
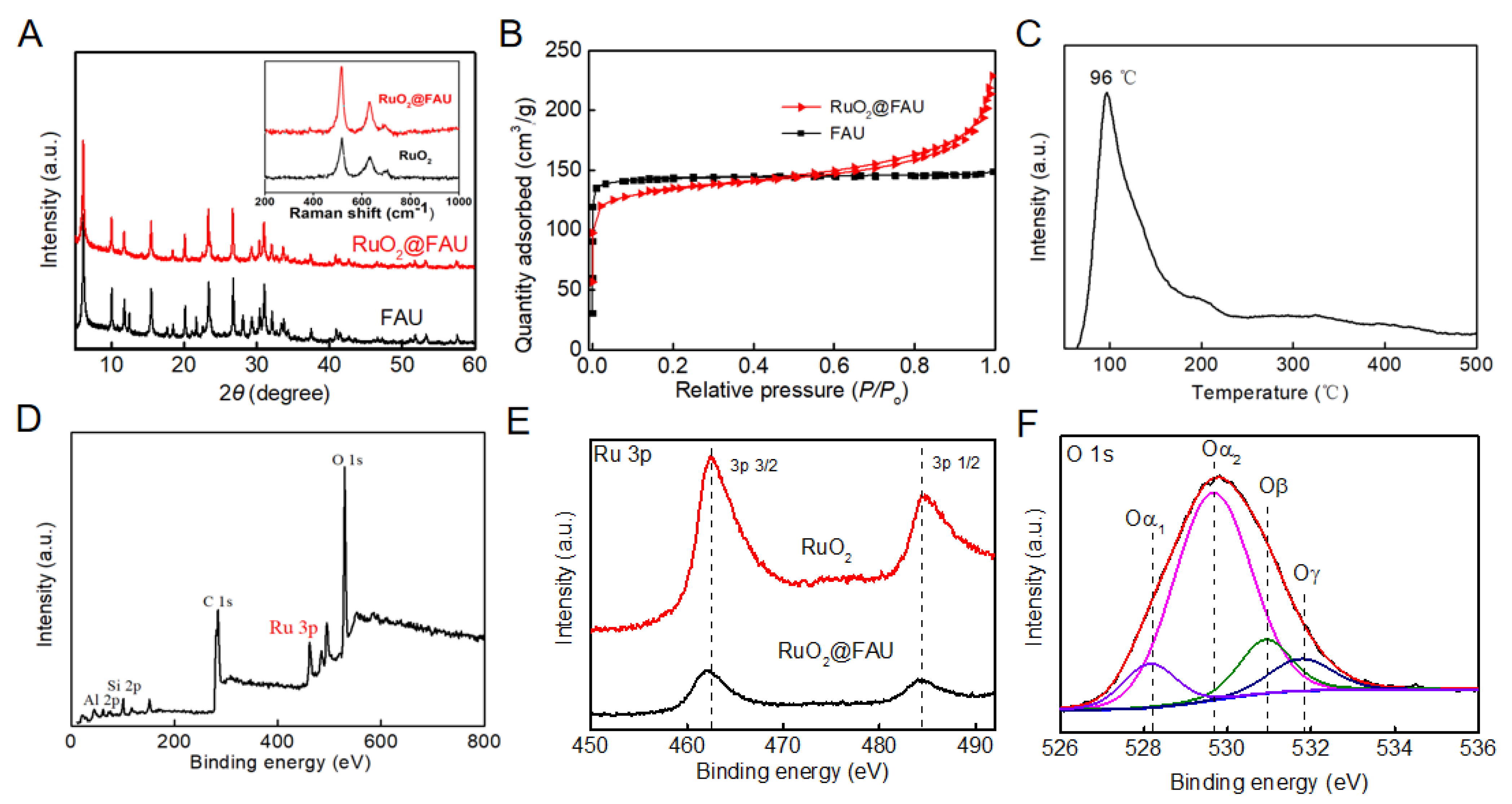
X-ray powder diffraction (XRD) results indicated that RuO2@FAU had the same structure as the unmodified FAU zeolite (Figure 1A). Both diffraction patterns match well with typical faujasite zeolites [20]. Diffraction patterns of RuO2 were not observed, indicating that no crystal phase was formed when it came to the zeolite-confined RuO2 compound. The Raman spectrum of RuO2@FAU was given as an inserted graph in Figure 1A and characteristic peaks centered at 518 cm−1, 634 cm−1 and 706 cm−1 were all assigned to RuO2 for both amorphous and crystalline samples [21,22]. Raman characteristic lines indicated the existence of RuO2 in the catalyst. Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES) analysis showed that the Si/Al ratio of RuO2@FAU was 1.26 and the loading amount of the ruthenium element was 1.48 wt.%. Figure 1B showed the nitrogen adsorption and desorption isotherms of FAU and RuO2@FAU. Compared with FAU, RuO2@FAU exhibited an obvious decrease in the micropore volume from 0.217 to 0.177 cm3∙g−1 and surface area from 506 to 424 m2∙g−1 (Table 1). It indicated that ruthenium particles were successfully encapsulated into FAU cages [19]. The hydrogen temperature-programmed reduction result of RuO2@FAU was given in Figure 1C. The reduction peak at 96 °C was assigned to RuO2 clusters in RuO2@FAU, which was lower than its bulk compounds (102 °C) [23]. It had been reported that the reduction temperature of highly dispersed metal oxide was much lower than that of its bulk counterparts [24]. The shift toward a lower reduction temperature indicated a high dispersion of RuO2 nanoparticles within FAU zeolites.
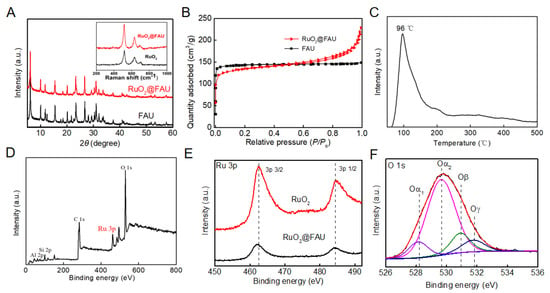
Figure 1.
Phase characterizations. (A) XRD patterns of FAU and RuO2@FAU catalysts, and the inserted graph was Raman spectrum of RuO2 and RuO2@FAU; (B) nitrogen adsorption-desorption isotherms of FAU and RuO2@FAU catalyst; (C) hydrogen temperature-programmed reduction curves of RuO2@FAU; (D) XPS survey spectrum of RuO2@FAU; (E) high-resolution spectra of XPS of RuO2 and RuO2@FAU for Ru 3p and (F) high-resolution spectra of RuO2@FAU for O 1s.

Table 1.
The textural and acidic properties of different RuO2@FAU catalysts.
X-ray photoelectron spectroscopy (XPS) was applied to study the surface chemical environment of ruthenium oxide confined in zeolite channels. Figure 1D was the survey spectrum of RuO2@FAU, and a Ru 3p peak was observed. To obtain the exact valence of the ruthenium element in RuO2@FAU, the high-resolution XPS spectrum of Ru 3p was further given in Figure 1E. Peaks at 462.6 and 484.4 eV were assigned to Ru 3p3/2 and Ru 3p1/2 for Ru4+. Compared with the RuO2 standard, Ru 3p in RuO2@FAU lightly transfers about 0.3 eV. It indicated that the zeolite affected the electronic structures of RuO2 [25,26,27,28]. Figure 1F depicted the high-resolution O 1s spectrum of RuO2@FAU. The binding energy peak of O 1s in a range of 526.0 eV to 534.0 eV for RuO2@FAU was fitted with four pair peaks [29,30,31,32,33]. The peak at the binding energy of 529.7 eV was assigned to the lattice oxygen in zeolite and was noted as Oα2. Moreover, the peak at 531.9 eV was due to the oxygen of Al-OH-Si and was noted as Oγ. The peak at 530.9 eV corresponds to surface oxygen O22–, O2– or O− (denoted as Oβ) caused by structural defects [32,33]. The peak at 528.2 eV appeared which was assigned to lattice oxygen of the RuO2 crystal and was noted as Oα1. It again indicated that ruthenium oxide nanoparticles mainly exist in the form of RuO2.
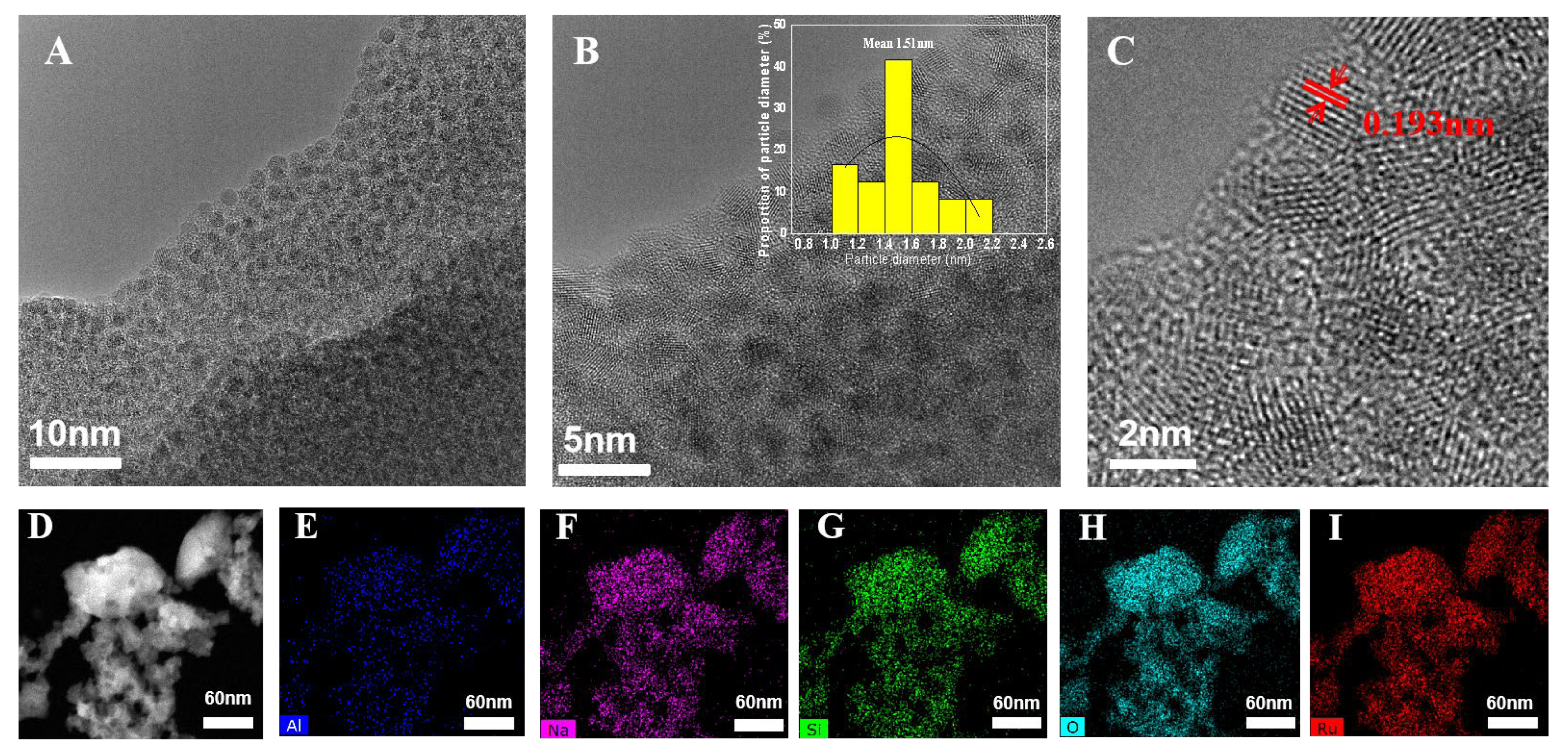
High-resolution transmission electron micrographs (HRTEM) (Figure 2A–I) clearly clarified that all visible ruthenium oxide nanoparticles dispersed uniformly in FAU zeolite. They had a very narrow size distribution around 1.5 ± 0.2 nm. More than 90% of RuO2 nanoparticles had a particle size smaller than 1.5 nm. The diameter of RuO2 nanoparticles was similar to that of α-cage in the FAU framework (1.3 nm) [18], indicating that RuO2 was successfully encapsulated within FAU zeolite channels. It was consistent with the absence of the RuO2 crystallization phase in XRD patterns. On the other hand, considering the structure of the FAU framework, the supercages are tetrahedrally connected by smaller 12-member ring channels (0.74 mm × 0.74 nm) [18]. RuO2 nanoparticles physically encapsulated in the supercages were not able to diffuse freely out of narrow channels. It greatly protected the active metallic sites against agglomeration during the catalytic reaction, which was a significant advantage when compared to the traditionally supported transition metal catalysts.
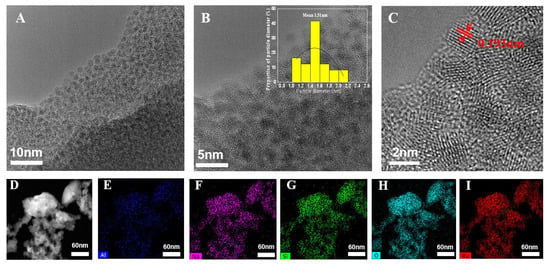
Figure 2.
(A–C) HRTEM images of RuO2@FAU catalyst, and (D–I) HADDF scanning transmission electron microscopy image and energy dispersive X-ray spectrometry (EDX) mapping of RuO2@FAU catalyst for Al, Na, Si, O, Ru elements, respectively.
2.2. Synthesis and Characterization of Modified RuO2@FAU Catalysts
The close proximity between the metal and acid sites inside the cavities or cages of the zeolites has been widely studied as many additional benefits are possible, for example, a shortened diffusion path, improved metal stability, or a synergistic effect. As the mechanism of the decarboxylation of L-lysine to cadaverine catalyzed by confined metal catalysts has not been revealed, it seems to be significantly meaningful to clarify the relationship between ruthenium oxide actives sites and the acid of zeolite, and especially their impact on catalytic performance. RuO2@FAU with different acidic properties were prepared by two different approaches: the ion exchange method, and the variation of Si/Al ratios.
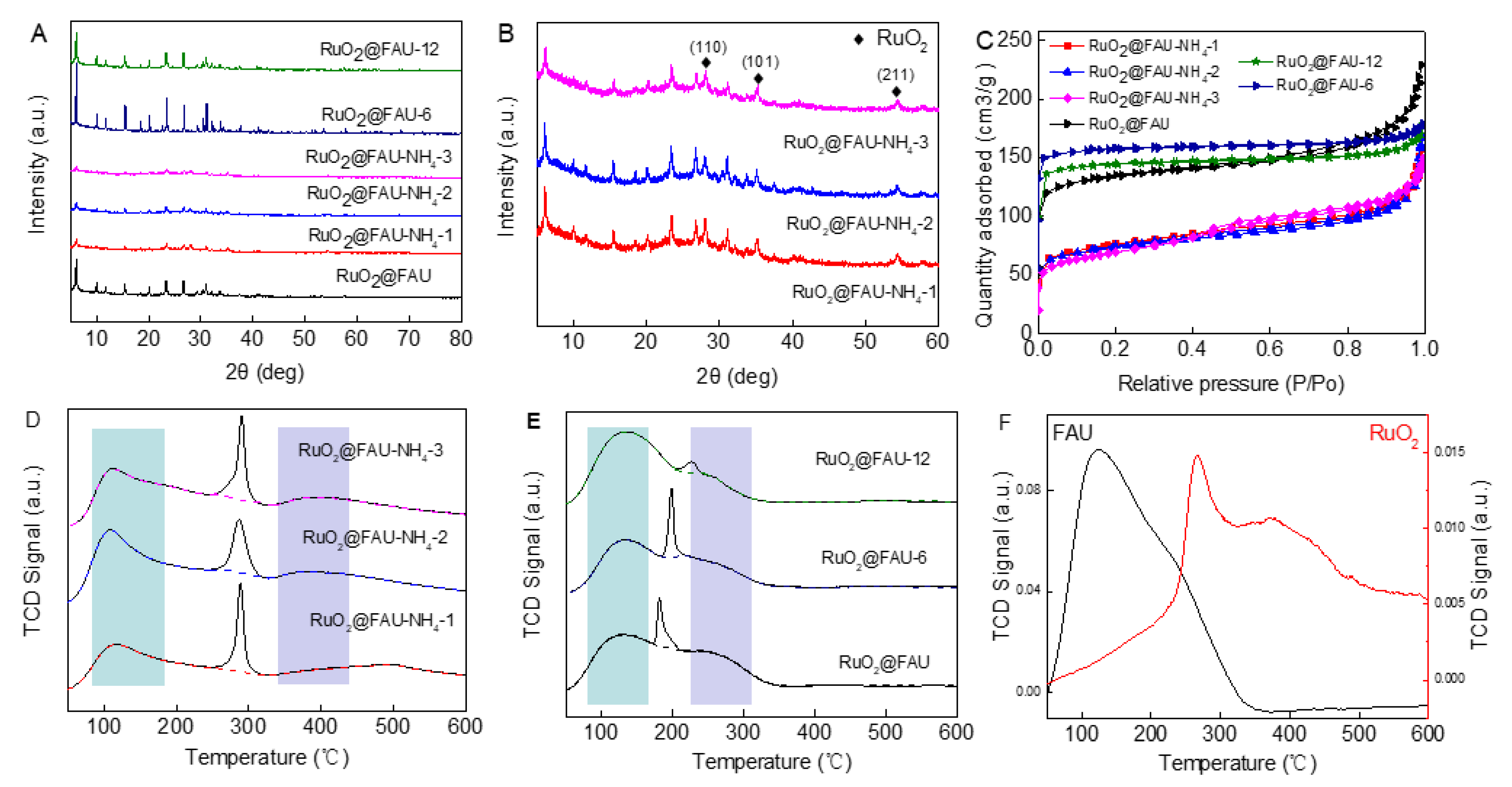
The modified catalysts were noted as RuO2@FAU-NH4-i, where i represented the exchange number, and RuO2@FAU-x, where x represented the Si/Al ratio. According to ICP results, RuO2@FAU-NH4-i and RuO2@FAU-x showed a different content of ruthenium (Table 1). It might be due to the uncontrollable encapsulation process of ruthenium oxide nanoparticles during crystallization. Figure 3A showed XRD patterns of RuO2@FAU-NH4-i and RuO2@FAU-x. All RuO2@FAU-NH4-i catalysts exhibited the typical diffraction peaks of FAU zeolite, but its diffraction peak intensity suffered an obvious decrease when compared to RuO2@FAU. It was generally demonstrated that factors affecting the lattice parameters in zeolites were complex, as the lattice parameters did not follow a fixed trend. It has been reported that the framework of FAU zeolite would undergo a distortion after ion exchange [34]. Marra et al. [35] and Li et al. [36] characterized the cation distribution in Y zeolite and concluded that cations would be relocated after ion exchange modification. All the above factors would lead to a decrease in diffraction peak intensity in RuO2@FAU. As shown in Figure 3B, new peaks at 28°, 35°, and 54° were observed in those modified catalysts, which could be attributed to (110), (101), and (211) crystal planes of RuO2, respectively [37,38,39,40]. It was suggested that ruthenium cations may relocate to form some occluded ruthenium species [41]. Moreover, RuO2@FAU-6 and RuO2@FAU-12 catalysts exhibited typical diffraction peaks of FAU zeolite, and no diffraction patterns of RuO2 were observed, indicating that RuO2 was successfully confined into the zeolite channel.
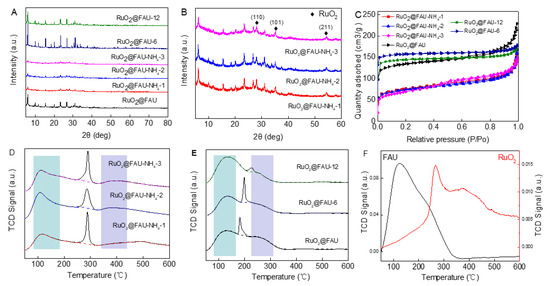
Figure 3.
Characterizations of different catalysts. (A) XRD patterns of all catalysts, (B) enlarged XRD patterns of RuO2@FAU-NH4-i catalysts between 5 and 60°, (C) Nitrogen adsorption and desorption isotherms curves of all catalysts, (D) NH3-TPD curves of RuO2@FAU-NH4-i catalysts, (E) NH3-TPD curves of RuO2@FAU-x catalysts, the light-blue rectangle in (D,E) represents weak acid sites, and the light-purple rectange represents medium strong acid sites, (F) NH3-TPD curves of RuO2 and FAU.
Nitrogen adsorption-desorption isotherms of RuO2@FAU-NH4-i and RuO2@FAU-x were shown in Figure 3C and a detailed textural property was given in Table 1. The adsorption-desorption isotherms of N2 were observed to be type I with all the catalysts, which represented a typical behavior for microporous materials. For RuO2@FAU-NH4-i catalysts, an obvious decrease was observed in surface areas (243~265 m2·g−1, entries 5~7 in Table 1) when compared with the intact RuO2@FAU catalyst (424 m2·g−1, entry 2 in Table 1). Differently, the mesoporous volume of those modified catalysts met a small rise from 0.104 cm3·g−1 to 0.160 cm3·g−1. The alteration of textural properties in RuO2@FAU-NH4-i catalysts might be assigned to the distorted crystalline structure after ion exchange modifications. For RuO2@FAU-x, the surface area and pore volume increased with Si/Al ratio. Ru@FAU-12 showed the largest surface area of 493 m2·g−1.
Ammonia temperature programmed desorption (NH3-TPD) curves of all the catalysts, FAU and RuO2 were described in Figure 3D–F. The peak located below 250 °C was assigned to weak acid sites. Peaks in the range of 250−400 °C were attributed to medium acid sites [35,36,37]. As shown in Figure 3D–F, a narrow and sharp peak was observed in the TPD curves of all samples except FAU. It indicated that this narrow peak was not caused by ammonia desorption. It has been reported that ruthenium oxide nanoparticles play a critical role in the temperature-programmed decomposition of ammonia molecules. This narrow peak might be mainly caused by the partial decomposition of ammonia by RuO2 during the NH3-TPD test [37]. The acid content of RuO2@FAU-NH4-i, RuO2@FAU-x and FAU were calculated according to the NH3-TPD curves and summarized in Table 1. Two ammonia desorption peaks were observed at 133 °C and 189 °C for RuO2@FAU-x catalysts, which were assigned to desorption at weak acid sites. It indicated that only weak acid sites existed in RuO2@FAU-x catalysts. It is well known that the acidity of zeolite is closely related to the Al content. With the Si/Al molar ratio increasing from 2 to 12, the total acid content of the samples decreased from 9.91 to 8.29 mmol/g. However, after ammonium ion exchange modification, desorption peaks at weak acid sites (189 °C) disappeared in RuO2@FAU-NH4-i catalysts, while those at medium strong acid sites were clearly observed, indicating that medium strong acid sites were formed in those modified catalysts. After NH4+ exchange, cations in FAU were converted from Na+ to H+. This led to an increase in acidity, resulting in a shift of the ammonia desorption peak to a higher temperature. RuO2@FAU-NH4-3 showed a maximum total acid content (13.56 mmol/g).
2.3. Enhanced Performance in Decarboxylation of L-Lysine to Cadaverine
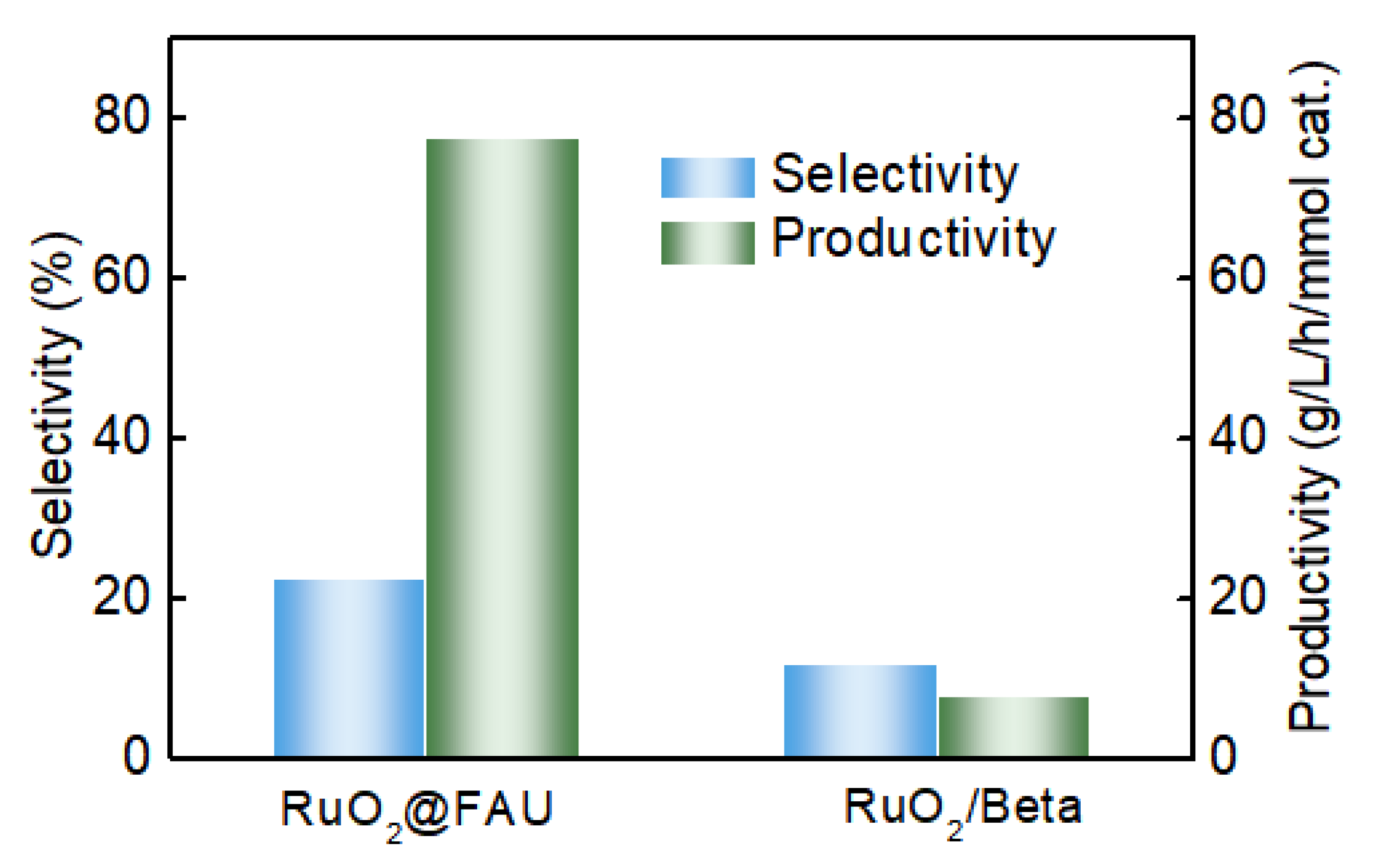
We have performed decarboxylation of L-lysine with RuO2@FAU as the catalyst. As shown in Figure 4, RuO2@FAU exhibited a similar activity (X = 48.2%) at 1 h to the traditionally supported ruthenium oxide catalysts RuO2/Beta (X = 46.9%) at 1.5 h. However, the selectivity of cadaverine with RuO2@FAU was 22.2%, which was almost twice that with RuO2/Beta. Meanwhile, the productivity of cadaverine was also significantly improved to 120.9 g/L/h/mmol cat. when compared with that of RuO2/Beta (7.8 g/L/h/mmol cat.). It indicated that confinement of ruthenium clusters into zeolite largely boosted the decarboxylation of L-lysine, especially with the improved productivity of cadaverine.
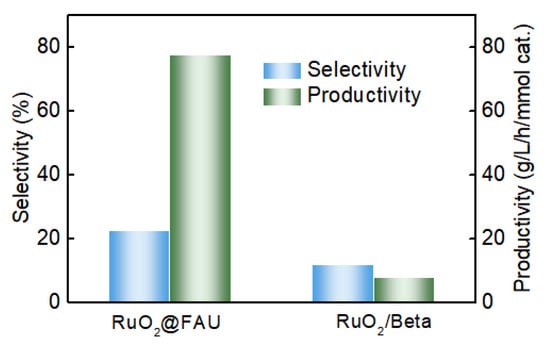
Figure 4.
Performance comparison of decarboxylation of L-lysine to cadaverine between RuO2@FAU with the traditional supported RuO2/Beta catalyst with similar activity. The reaction conditions are as follows: L-lysine (0.1 M), Ru catalysts (1.01 g), 200 °C, 800 rpm, 2 MPa H2.
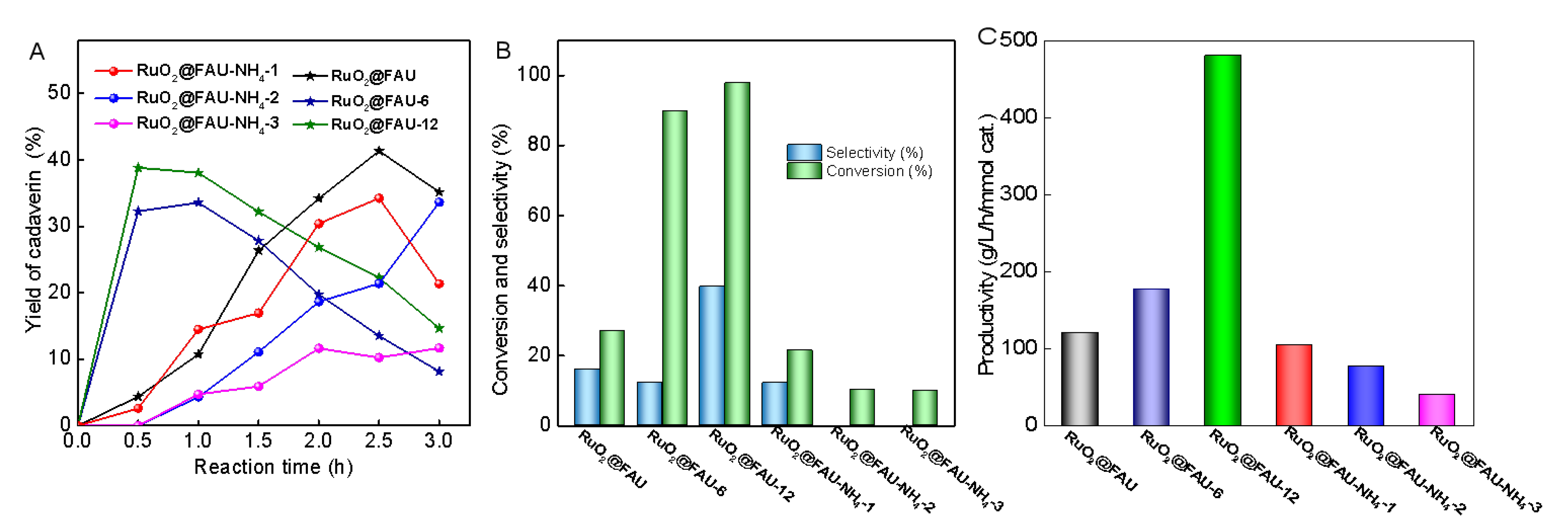
Modified catalysts, both RuO2@FAU-NH4-i and RuO2@FAU-x, were also evaluated in the decarboxylation of L-lysine to produce cadaverine. Time profiles of cadaverine yield over different catalysts were given in Figure 5A. Two different patterns were observed with RuO2@FAU-NH4-i and RuO2@FAU-x. For RuO2@FAU-6 and RuO2@FAU-12, the yield of cadaverine reached a maximum at 0.5 h and then began to decrease with reaction time. Cadaverine could further undergo deamination to give piperidine, resulting in a yield loss of cadaverine after 0.5 h. Differently, the yield of cadaverine increased with reaction time in a range of 0 to 3.0 h over RuO2@FAU-NH4-i, indicating that prolonging the reaction time seemed to be helpful for the synthesis of cadaverine. It was clear that RuO2@FAU showed a higher productivity rate of cadaverine than RuO2@FAU-NH4-i. As discussed above, the relocation of ruthenium nanoparticles after ammonium ion exchange occurred to form some occluded ruthenium species, which might be blamed for the slower productivity of cadaverine with RuO2@FAU-NH4-i. Notably, RuO2@FAU-x exhibited a higher cadaverine yield when compared to RuO2@FAU-NH4-i catalysts. To further clarify the effect of modification on the decarboxylation reaction, the initial activity of the different catalysts was further given in Figure 5B, together with the selectivity of cadaverine. The activity of catalysts suffered a decrease after ammonium ion exchange modification, with a conversion of less than 30%. The selectivity of cadaverine had a similar trend in RuO2@FAU-NH4-i catalysts. Differently, both the conversion and selectivity of cadaverine were greatly enhanced over RuO2@FAU-6 and RuO2@FAU-12 when compared to RuO2@FAU. For example, conversion was 27.2% at 0.5 h over RuO2@FAU, which greatly increased up to 89.8% and 97.8% for RuO2@FAU-6 and RuO2@FAU-12, respectively. The highest selectivity of cadaverine (39.7%) was obtained with RuO2@FAU-12. Additionally, the highest cadaverine productivity of 480.3 g/L/h/mmol cat. was obtained on RuO2@FAU-12, which is much higher than RuO2@FAU (120.9 g/L/h/mmol cat.), and RuO2@FAU-NH4-3 (40.1 g/L/h/mmol cat.). Productivity of cadaverine, defined as the number of cadaverine produced per molar catalyst per time, reflected the intrinsic reaction ability of catalysts. Although the selectivity of cadaverine was not greatly enhanced by confinement and modification, the significantly enhanced productivity of cadaverine in RuO2@FAU-12 brought new opportunities for the effective decarboxylation of L-lysine.
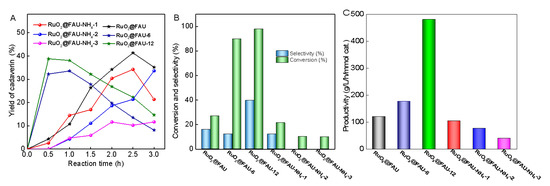
Figure 5.
Decarboxylation of L-lysine to cadaverine with different catalysts. (A) variation of cadaverine yield with reaction time (B) conversion and cadaverine selectivity at 0.5 h with different catalysts, and (C) productivity of cadaverine with different catalysts. Reaction conditions are as follows: L-lysine (0.1 M), Ru catalysts (0.101 g), 200 °C, 800 rpm, 2 MPa H2, pH 2.0.
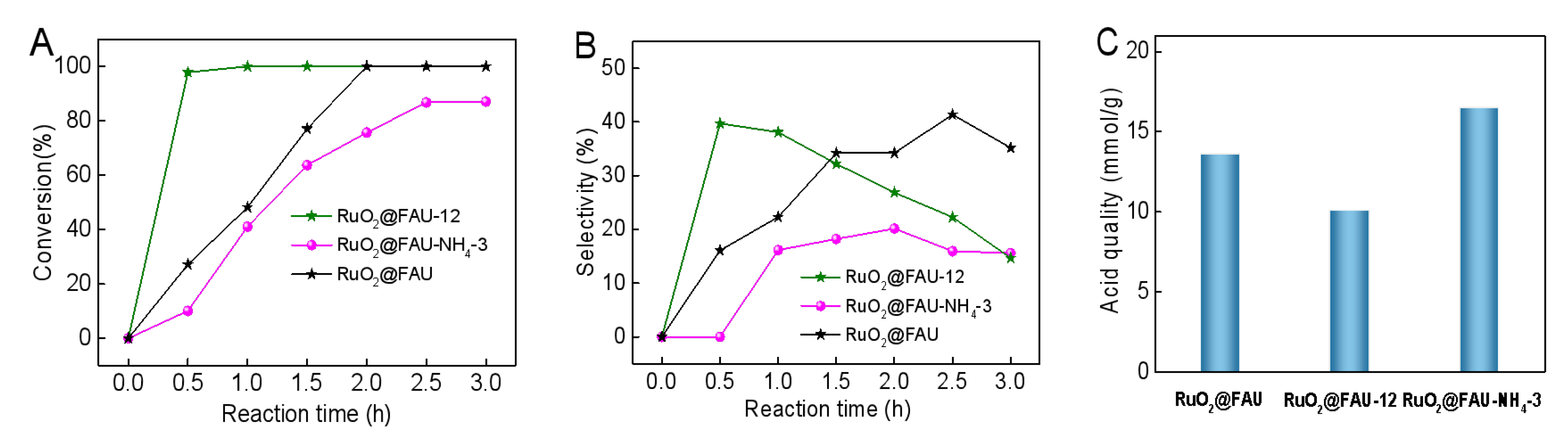
For either RuO2@FAU-NH4-i catalysts or RuO2@FAU-x catalysts, they both showed a change in acidity compared with RuO2@FAU. To further clearly illustrate the effect of acidic properties on catalytic performance, time profiles of conversion and cadaverine selectivity over RuO2@FAU, RuO2@FAU-12 and RuO2@FAU-NH4-3 were particularly selected for discussion in detail as shown in Figure 6A,B. The detailed acidic property was described in Figure 6C and Table 1. RuO2@FAU-12 outperformed the other two catalysts in activity, selectivity and productivity with much lower acid content. It seemed that the excessive acid content might be not helpful to cadaverine synthesis.
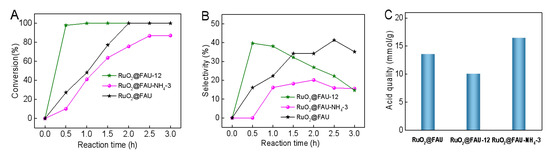
Figure 6.
Decarboxylation of L-lysine to cadaverine with RuO2@FAU, and RuO2@FAU-12 and RuO2@FAU-NH4-3 catalysts. (A) variation of conversion with reaction time, (B) variation of the selectivity of cadaverine with reaction time. (C) acid content comparison of RuO2@FAU, and RuO2@FAU-12 and RuO2@FAU-NH4-3.
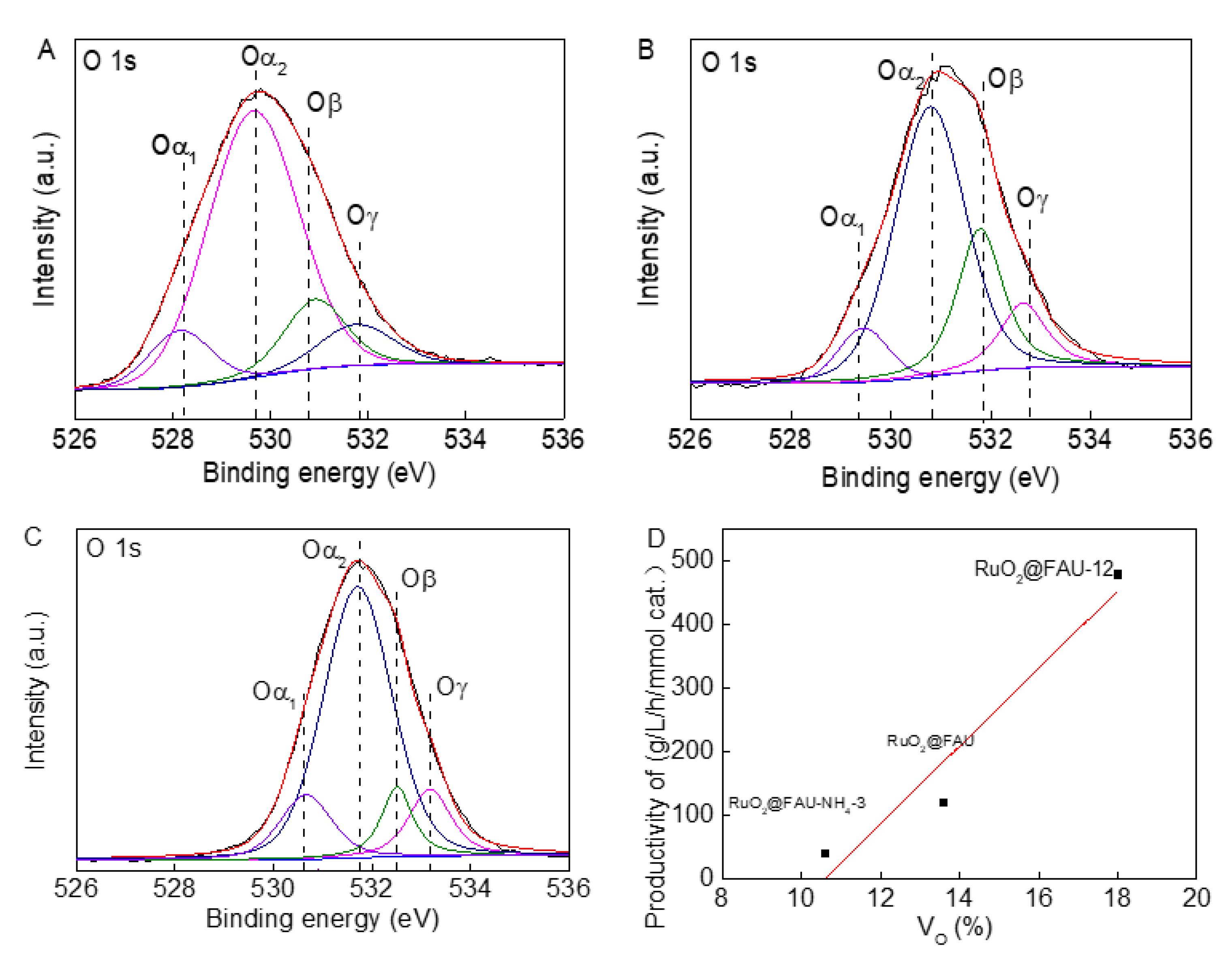
Our previous work showed that active surface oxygen vacancies in catalysts facilitated the adsorption of intermediate in the decarboxylation of L-lysine [13]; therefore, the relationship between the quantity of oxygen vacancies in catalysts and their catalytic performances were studied. The high-resolution O 1s spectra for RuO2@FAU, RuO2@FAU-12 and RuO2@FAU-NH4-3 catalyst were depicted in Figure 7A–C. The peak area of Oα1, Oα2, Oβ and Oγ were calculated. The density of oxygen vacancies (Ov) could be assumed as Oβ/(Oα + Oβ + Oγ). For RuO2@FAU, the peak of Oα2 shifted to 530.8 eV for RuO2@FAU-12 catalyst and to 531.7 eV for RuO2@FAU-NH4-3 catalyst, respectively. The shift in the binding energy for crystal oxygen of FAU in modified catalysts might be due to the distorted crystalline structure which has been described above in Figure 3A. According to the literature, 32–33 peaks at the binding energy of 530.9 eV for RuO2@FAU, 531.8 eV for RuO2@FAU-12, and 532.5 eV for RuO2@FAU-NH4-3 were assigned to surface oxygen vacancies. Then the cadaverine productivity as a function of oxygen vacancies in proportion was plotted as shown in Figure 7D. It showed that cadaverine productivity increased when there was oxygen vacancy. Undoubtedly, oxygen vacancies were significantly beneficial to the synthesis of cadaverine. It was supposed that oxygen vacancies interacted with the carbonyl oxygen atoms in L-lysine to weaken the C=O bond and finally, promote the target decarboxylation process [42].
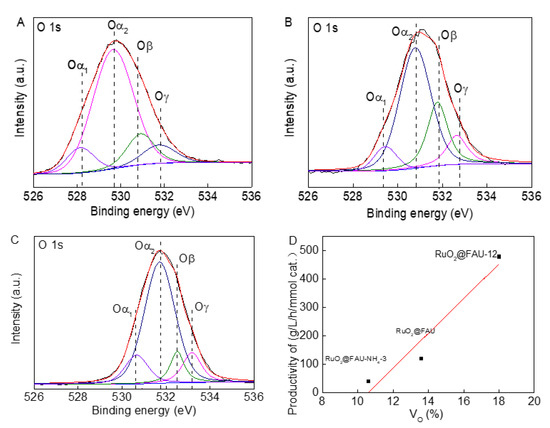
Figure 7.
High-resolution spectra of XPS of RuO2@FAU (A), RuO2@FAU-12 (B), RuO2@FAU-NH4-3 (C) for O 1s, and variation of cadaverine productivity with oxygen vacancies percentage in RuO2@FAU, RuO2@FAU-12, and RuO2@FAU-NH4-3 catalysts (D). Based on the XPS peak differentiating results for O 1s, the oxygen vacancies percentage was calculated by using area normalization method.
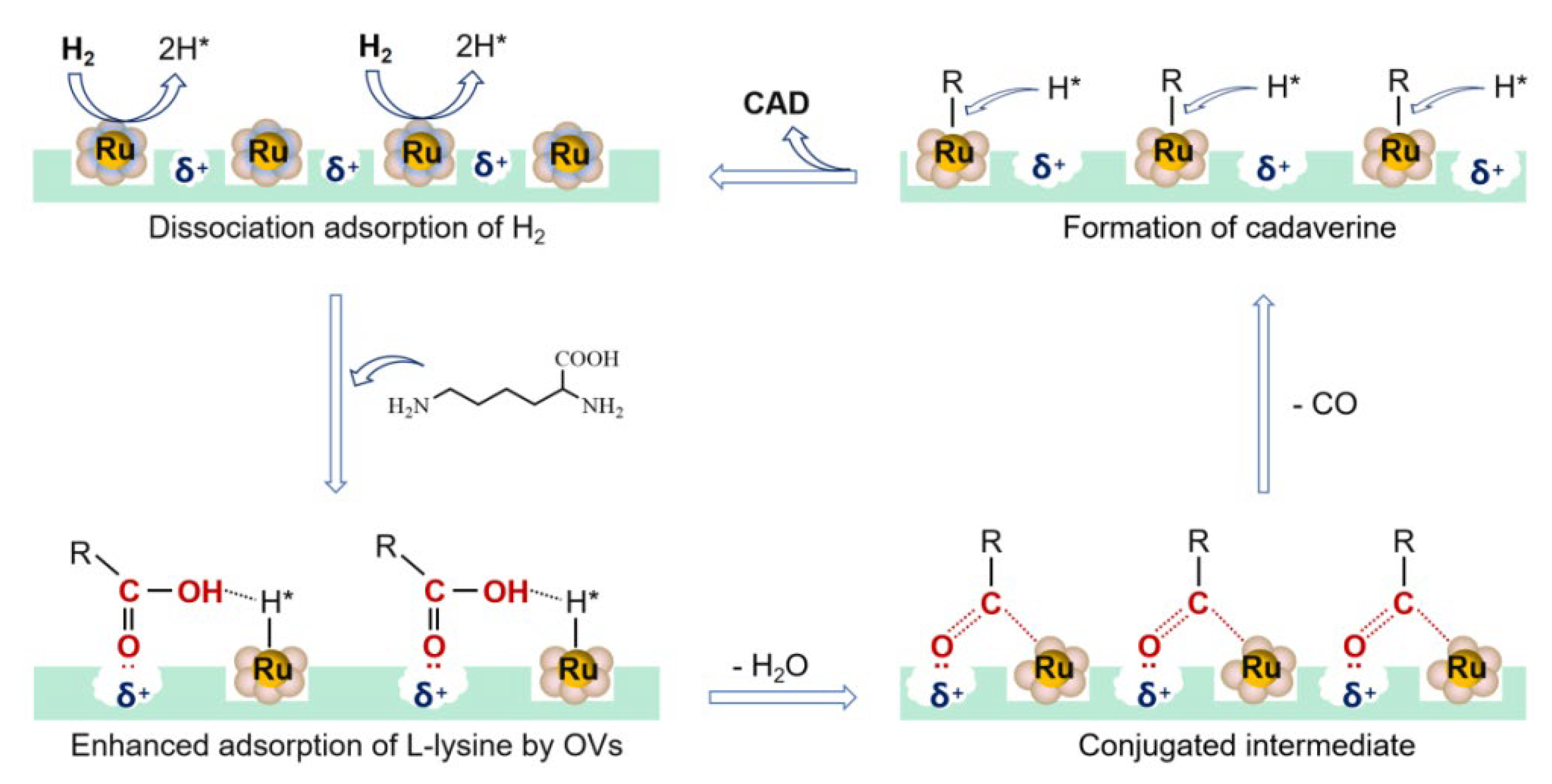
The mechanism of decarboxylation of L-lysine to cadaverine was proposed as shown in Figure 8. First, H2 dissociative adsorption on the polarized Ru–O bond releases the active H* species [43]. Two kinds of under-coordinated surface atoms, the bridging oxygen atoms and onefold under-coordinated Ru atoms, existed in RuO2. The bridging oxygen atoms were able to accept H atoms as a Brønsted base, which finally formed bridging O–H species and released active H* species [44,45,46,47]. Then L-lysine was adsorbed by positive oxygen vacancies in RuO2@FAU. The hydroxyl in the carboxyl group interacted with the active H* species through a hydrogen bond. After the dehydration process, the surface carboxylate species (Ru-COOR) with a conjugate structure was formed, where the character R represented NH2(CH2)4CH(NH2)– group. This conjugated chemical was believed to be the intermediate in the decarboxylation process [48,49]. Thirdly, the decarbonylation process occurred to form active R species while releasing one CO molecule. The alkylation product of CO (methane) was detected by in situ gas chromatography (GC). Finally, cadaverine was obtained with the attack of H* species.
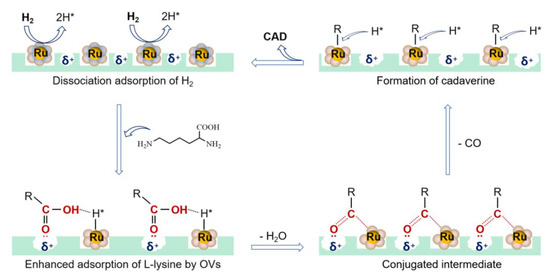
Figure 8.
Proposed mechanism of decarboxylation of L-lysine to cadaverine.
Finally, Table 2 described the performance comparison of various ruthenium oxide catalysts in the decarboxylation of L-lysine to cadaverine. As shown in Table 2, Ru/BaSO4 exhibited the highest selectivity for cadaverine at 62.0%. However, the concentration of L-lysine was merely 0.01 mol/L, which was too low for the industrial production of cadaverine on a large scale. For commercial Ru/C catalyst, the selectivity of cadaverine only reached 46.6%. The time needed for total conversion was even prolonged to 120 min. In comparison with commercial Ru/C catalyst, the selectivity of cadaverine had a certain improvement for the Ru-Mn/Beta catalyst. Nevertheless, the productivity of cadaverine was still not ideal. For the RuO2@FAU-12 catalyst in this study, ruthenium oxide was confined within the supercage of FAU zeolite. Compared with the traditionally supported ruthenium oxide catalysts, RuO2@FAU-12 showed excellent catalytic activity in the decarboxylation of L-lysine to cadaverine with a productivity of 480.3 g/L/h/mmol cat. which was almost three times that of the best-reported catalyst.
Table 2.
Performance of varied supported ruthenium oxide in decarboxylation of L-lysine to cadaverine.
3. Experimental Section
3.1. Chemicals
Ruthenium chloride hydrate (RuCl3·3H2O), sodium hydroxide (NaOH), sodium aluminates (NaAlO2), silica sol (25–30 wt.%), L-lysine monohydrochloride, and phosphoric acid (H3PO4) were obtained from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). They were used as accepted without any further modification.
3.2. Synthesis of RuO2@FAU
RuO2@FAU: The initial composition of the sample was SiO2:Al2O3:Na2O:H2O = 4:1.00:17.00:675. The designed Si/Al ratio was 2; 2.800 g NaOH and 0.332 g NaAlO2 were added to 25 mL of deionized water under stirring; 2.1 mL silica sol was poured into the above solution; 0.34 g RuCl3·3H2O was added to the mixture under stirring at 300 rpm. After stirring for 4 h, the above mixture was placed in a sealed Teflon recipient and crystallized at 100 °C for 12 h. The obtained sample was filtered and washed with deionized water until the filtrate was neutral. After drying at 80 °C for 24 h, we obtained a black powder and it was noted as RuO2@FAU. ICP results showed that the ruthenium content of RuO2@FAU was 1.48 wt.%.
3.3. Synthesis of RuO2@FAU with Different Si/Al Ratio
RuO2@FAU-6 and RuO2@FAU-12 have been synthesized by adjusting the raw material ratio Si/Al ratio of 6, and 12, respectively. ICP results showed that the ruthenium amount of RuO2@FAU-6 and RuO2@FAU-12 was 3.72 wt.%, 1.58 wt.%, respectively.
3.4. Modification of RuO2@FAU
We dissolved 0.5349 g ammonium chloride in 10 g water under stirring to obtain 1 mol/L NH4Cl solution; 1 g RuO2@FAU was added into the ammonium chloride solution under stirring at 70 °C. After stirring for 1 h, the solid powder was filtered and washed with 1 L distilled water. It was heated at 100 °C overnight and then calcinated at 400 °C for 2 h at a rate of 5 °C/min. The obtained sample was noted as RuO2@FAU-NH4-1. By changing the number of ammonium ion exchanges, we obtained RuO2@FAU-NH4-2 and RuO2@FAU-NH4-3 samples.
3.5. Characterizations
The structure of catalysts was studied by powder X-ray diffraction (XRD) patterns with Cu Kα radiation (40 kV, 100 mA, λ = 1.5406 Å) with a speed of 5°/min from 10° to 80°. N2 adsorption/desorption isotherm of samples at 77 K was investigated by a Micromeritics ASAP 2460 surface area and porosity analyzer. Before measurements, 100 mg samples were pretreated at 150 °C for 3 h. The specific surface areas of catalysts were calculated using the Brunauer–Emmett–Teller (BET) methods. The acid content of the catalysts was determined by NH3-TPD; 100 mg catalysts were pretreated at 300 °C for 1 h under Ar gas. Then the catalyst was cooled to 50 °C and exposed to NH3-Ar flow (30 mL/min) for 1 h, purged with Ar (30 mL/min) and heated to 600 °C at a rate of 10 °C/min. H2-TPR was carried out using an AutoChem II 2920 analyzer. The catalysts were pre-treated at 350 °C for 1 h under Ar gas. After cooling, the catalysts were purged with 5% H2/Ar (30 mL/min) and heated to 800 °C at a rate of 10 °C/min. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo ESCALAB 250 Xi spectrometer (ThermoFisher Scientific, Waltham, MA, USA) with Al Kα (1486.6 eV) radiation as the excitation source in an ultrahigh vacuum (6.710–8 Pa). The binding energy scale was corrected with respect to the C 1s line (284.4 eV) derived from adventitious carbon. The powder samples were pressed into self-supported disks loaded in the subchamber and evacuated for 4 h. The transmission electron microscopy (TEM) was carried out by Tecnai G2 F30/F20 (Yumpu, Diepoldsau, Switzerland).
3.6. Evaluation in Decarboxylation of L-Lysine to Cadaverine
The decarboxylation of L-lysine was performed in a high-pressure reactor with a 25 mL quartz liner. L-lysine (10 mL, 0.1 M) and catalyst were added to the reactor. After adjusting the pH of the mixture to 2 through the addition of H3PO4, the reactor was sealed and flushed with N2 six times and then H2 six times. Then the decarboxylation of L-lysine was performed at 200 °C with an H2 pressure of 2 MPa and a stirring speed of 800 r/min. The reaction was stopped by quenching in ice water.
Products were determined using LC-UV (Waters) equipped with a C18 column (100 Å, 5 µm, 4.6 mm × 250 mm) according to the method described in our previous work [13]. To be brief, 50 µL of the diluted solution and 50 µL of dansyl chloride (5 mg/mL in acetone) were successively added into a 4 mL centrifuge tube, and the mixture was heated by ultrasonic pretreatment for 10 min in the dark. The derivatized product was extracted by adding 1850 µL methanol into the tube. After the filtration product was analyzed. The conversion of L-lysine was calculated by (moles of consumed L-lysine)/(moles of initial L-lysine) and the selectivity of cadaverine was defined as (moles of cadaverine)/(moles of consumed L-lysine).
4. Conclusions
Ruthenium oxide nanoparticles (~1.5 nm) confined into supercages of FAU zeolites were successfully prepared using the in situ hydrothermal synthesis method. This new RuO2@FAU was used for the decarboxylation of L-lysine to cadaverine. After modification by ammonia exchange and different Si/Al ratios, the best catalyst (RuO2@FAU-12) had extraordinarily high cadaverine productivity of 480.3 g/L/h/mmol cat., which was 22.6 times that of traditionally supported ruthenium catalysts (21.2 g/L/h/mmol cat.). Moreover, the effect of encapsulated active metal centers and the acidic sites of zeolite that existed in catalysis was further investigated and it was found that the excessive acid content might be detrimental to the production of cadaverine.
Author Contributions
Conceptualization, Y.H. and Z.M.; methodology, Z.M. and Z.X.; validation, Z.M., Z.X. and S.Q.; formal analysis, Z.M., S.Q. and Z.X.; data curation, Z.M. and Z.X.; writing—original draft preparation, Z.M. and Z.X.; writing—review and editing, Z.M., Z.X. and S.Q.; project administration, Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by China Postdoctoral Science Foundation (N.O. 2021M690920).
Data Availability Statement
All the data generated or analyzed within the present investigation are included in this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yang, P.; Li, X.; Liu, H.; Li, Z.; Liu, J.; Zhuang, W.; Wu, J.; Ying, H. Thermodynamics, crystal structure, and characterization of a bio-based nylon 54 monomer. CrystEngComm 2019, 21, 7069–7077. [Google Scholar] [CrossRef]
- Qian, Z.G.; Xia, X.X.; Lee, S.Y. Metabolic engineering of Escherichia coli for the production of cadaverine: A five carbon diamine. Biotechnol. Bioeng. 2011, 108, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Scott, E.L.; Zeeland, T.A.; Sanders, J.P.M. The use of l-lysine decarboxylase as a means to separate amino acids by electrodialysis. Green Chem. 2011, 13, 624–630. [Google Scholar] [CrossRef]
- Kwak, D.H.; Lim, H.G.; Yang, J.; Seo, S.W.; Jung, G.Y. Synthetic redesign of Escherichia coli for cadaverine production from galactose. Biotechnol. Biofuels 2017, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Kind, S.; Wittmann, C. Bio-based production of the platform chemical 1,5-diaminopentane. Appl. Microbial. Biot. 2011, 91, 1287–1296. [Google Scholar] [CrossRef]
- Moon, Y.M.; Yang, S.Y.; Choi, T.R.; Jung, H.R.; Song, H.S.; Han, Y.H.; Park, H.Y.; Bhatia, S.K.; Gurav, R.; Park, K.; et al. Enhanced production of cadaverine by the addition of hexadecyltrimethylammonium bromide to whole cell system with regeneration of pyridoxal-5′-phosphate and ATP. Enzym. Microb. Technol. 2019, 127, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhao, Y.; Ji, X.; Yao, J.; Busk, P.K.; Lange, L.; Huang, Y.; Zhang, S. Advances in bio-nylon 5X: Discovery of new lysine decarboxylases for the high-level production of cadaverine. Green Chem. 2020, 22, 8656–8668. [Google Scholar] [CrossRef]
- Schouwer, F.; Claes, L.; Claes, N.; Bals, S.; Degreve, J.; De Vos, D.E. Pd-catalyzed decarboxylation of glutamic acid and pyroglutamic acid to bio-based 2-pyrrolidone. Green Chem. 2015, 17, 2263–2270. [Google Scholar] [CrossRef]
- Verduyckt, J.; Van Hoof, M.; Schouwer, F.; Wolberg, M.; Kurttepeli, M.; Eloy, P.; Gaigneaux, E.M.; Bals, S.; Kirschhock, C.E.A.; De Vos, D.E. PdPb-catalyzed decarboxylation of proline to pyrrolidine: Highly selective formation of a biobased amine in water. ACS Catal. 2016, 6, 7303–7310. [Google Scholar] [CrossRef] [Green Version]
- Verduyckt, J.; Coeck, R.; De Vos, D.E. Ru-Catalyzed Hydrogenation–Decarbonylation of Amino Acids to Bio-based Primary Amines. ACS Sustain. Chem. Eng. 2017, 5, 3290–3295. [Google Scholar] [CrossRef]
- Xie, S.; Jia, C.; Wang, Z.; Ong, S.S.G.; Zhu, M.J.; Lin, H. Mechanistic Insight into Selective Deoxygenation of l-Lysine to Produce Biobased Amines. ACS Sustain. Chem. Eng. 2020, 8, 11805–11817. [Google Scholar] [CrossRef]
- Lv, X.; Ma, Z.; Li, X.; Zhang, Y.; Huang, Y.; Li, T. Highly efficient decarboxylation of l-lysine to cadaverine catalyzed by supported ruthenium oxide. Catal. Commun. 2021, 158, 106339. [Google Scholar] [CrossRef]
- Ma, Z.; Xin, Z.; Qin, S.; Huang, Y. Mn-Doped Highly Dispersed RuO2 Catalyst with Abundant Oxygen Vacancies for Efficient Decarboxylation of l-Lysine to Cadaverine. ACS Sustain. Chem. Eng. 2021, 9, 13480–13490. [Google Scholar] [CrossRef]
- Ji, S.; Chen, Y.; Zhao, S.; Chen, W.; Shi, L.; Wang, Y.; Dong, J.; Li, Z.; Li, F.; Chen, C.; et al. Atomically Dispersed Ruthenium Species Inside Metal-Organic Frameworks: Combining the High Activity of Atomic Sites and the Molecular Sieving Effect of MOFs. Angew. Chem. Int. Ed. 2019, 58, 4271–4275. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, R.; Pan, X.; Wang, C.; Fan, M.; Zhu, Y.; Wang, Y.; Peng, J. Catalyst design strategies towards highly shape-selective HZSM-5 for para-xylene through toluene alkylation. Green Energy Environ. 2020, 5, 385–393. [Google Scholar] [CrossRef]
- Zhan, B.Z.; Iglesia, E. RuO2 Clusters within LTA Zeolite Cages: Consequences of Encapsulation on Catalytic Reactivity and Selectivity. Angew. Chem. 2007, 119, 3771–3774. [Google Scholar] [CrossRef]
- Altwasser, S.; Glaser, R.; Lo, A.S.; Liu, P.H.; Chao, K.J. Incorporation of RuO2 nanoparticles into MFI-type zeolites. Micropor. Mesopor. Mater. 2006, 89, 109–122. [Google Scholar] [CrossRef]
- Zhan, B.; White, M.A.; Sham, T.; Pincock, J.A.; Doucet, R.J.; Rao, K.V.R.; Robertson, K.N.; Cameron, T.S. Zeolite-Confined Nano-RuO2: A Green, Selective, and Efficient Catalyst for Aerobic Alcohol Oxidation. J. Am. Chem. Soc. 2003, 125, 2195–2199. [Google Scholar] [CrossRef]
- Yang, J.; He, Y.; He, J.; Liu, Y.; Geng, H.; Chen, S.; Lin, L.; Liu, M.; Chen, T.; Jiang, Q.; et al. Enhanced catalytic performance through in situ encapsulation of ultrafine Ru clusters within a high-aluminum zeolite. ACS Catal. 2022, 12, 1847–1856. [Google Scholar] [CrossRef]
- Xiong, P.; He, P.; Qu, Y.X.; Wang, L.G.; Cao, Y.; Xu, S.; Chen, J.Q.; Ammar, M.; Li, H.Q. The adsorption properties of NaY zeolite for separation of ethylene glycol and 1,2-butanediol: Experiment and molecular modelling. Green Energy Environ. 2021, 6, 102–113. [Google Scholar] [CrossRef]
- Devadas, A.; Baranton, S.; Coutanceau, C. Green synthesis and modification of RuO2 materials for the oxygen evolution reaction. Front. Energy Res. 2020, 8, 571704. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Zhou, W.; Zhang, X.; Zhang, B.; Yu, Y. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 2022, 61, e202202604. [Google Scholar]
- Chen, L.; Li, Y.; Zhang, X.; Zhang, Q.; Wang, T.; Ma, L. Mechanistic insights into the effects of support on the reaction pathway for aqueous-phase hydrogenation of carboxylic acid over the supported Ru catalysts. Appl. Catal. A Gen. 2014, 478, 117–128. [Google Scholar] [CrossRef]
- Lv, L.; Wang, S.; Ding, Y.; Zhang, L.; Gao, Y.; Wang, S. Mechanistic insights into the contribution of Lewis acidity to brominated VOCs combustion over titanium oxide supported Ru catalyst. Chemosphere 2021, 263, 128112. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Browne, M.P.; Nolan, H.; Duesberg, G.S.; Colavita, P.E.; Lyons, M.E.G. Low-overpotential high-activity mixed manganese and ruthenium oxide electrocatalysts for oxygen evolution reaction in alkaline media. ACS Catal. 2016, 6, 2408–2415. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Ji, C.; Sun, Y.; Wang, Y.; Zhao, Z.; Lv, Z. Surface basicity induced RuO2 modification on nanosized Li-rich cathode Li1.26Fe0.22Mn0.52O2 with superior electrochemical performance. J. Electrochem. Soc. 2016, 163, A2040. [Google Scholar] [CrossRef]
- Qadir, K.; Joo, S.H.; Mun, B.S.; Butcher, D.R.; Renzas, J.R.; Aksoy, F.; Liu, Z.; Somorjai, G.A.; Park, J.Y. Intrinsic relation between catalytic activity of CO oxidation on Ru nanoparticles and Ru oxides uncovered with ambient pressure XPS. Nano Lett. 2012, 12, 5761–5768. [Google Scholar] [CrossRef]
- Ou, G.; Xu, Y.; Wen, B.; Lin, R.; Ge, B.; Tang, Y.; Liang, Y.; Yang, C.; Huang, K.; Zu, D.; et al. Tuning defects in oxides at room temperature by lithium reduction. Nat. Commun. 2018, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Wang, S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.; Liu, X. Insights into the influence of CeO2 crystal facet on CO2 hydrogenation to methanol over Pd/CeO2 catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Gao, X.; Chen, J.; Sun, X.; Wu, B.; Li, B.; Ning, Z.; Li, J.; Wang, N. Ru/RuO2 nanoparticle composites with N-doped reduced graphene oxide as electrocatalysts for hydrogen and oxygen evolution. ACS Appl. Nano Mater. 2020, 3, 12269–12277. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, Q.; Xiao, Z.; Li, X.; Huo, J.; Wang, S.; Dai, L. Plasma-Engraved Co3O4 Nanosheets with Oxygen Vacancies and High Surface Area for the Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2016, 55, 5277–5281. [Google Scholar] [CrossRef]
- Tong, Y.; Chen, P.; Zhang, M.; Zhou, T.; Zhang, L.; Chu, W.; Wu, C.; Xie, Y. Oxygen vacancies confined in nickel molybdenum oxide porous nanosheets for promoted electrocatalytic urea oxidation. ACS Catal. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Weitkamp, J.; Ernst, S.; Hunger, M.; Roser, T.; Huber, S.; Schubert, U.A.; Thomasson, P.; Knozinger, H. Solid state ion exchange of alkali metal cations into zeolite Y: Physicochemical characterization and catalytic tests. Stud. Surf. Sci. Catal. 1996, 101, 731–740. [Google Scholar]
- Marra, G.L.; Fitch, A.N.; Zecchina, A.; Ricchiardi, G.; Salvalaggio, M.; Bordiga, S.; Lamberti, C. Cation location in dehydrated Na–Rb–Y zeolite: An XRD and IR study. J. Phys. Chem. B 1997, 101, 10653–10660. [Google Scholar] [CrossRef]
- Li, J.; Davis, R.J. Raman spectroscopy and dioxygen adsorption on Cs-loaded zeolite catalysts for butene isomerization. J. Phys. Chem. B 2005, 109, 7141–7148. [Google Scholar] [CrossRef]
- Ristic, M.; Marcius, M.; Petrovic, Z.; Ivanda, M.; Music, S. Formation and characterization of ribbon-like RuO2/Ru fibers. Mater. Lett. 2015, 156, 142–145. [Google Scholar] [CrossRef]
- López, N.; Gómez-Segura, J.; Marín, R.P.; Pérez-Ramírez, J. Mechanism of HCl oxidation (Deacon process) over RuO2. J. Catal. 2008, 255, 29–39. [Google Scholar] [CrossRef]
- Anjum, S.M.; Riazunnisa, K. Fine ultra-small ruthenium oxide nanoparticle synthesis by using Catharanthus roseus and Moringa oleifera leaf extracts and their efficacy towards in vitro assays, antimicrobial activity and catalytic: Adsorption kinetic studies using methylene blue dye. J. Clust. Sci. 2022, 33, 1103–1117. [Google Scholar] [CrossRef]
- Bepari, S.; Li, X.; Abrokwah, R.; Mohammad, N.; Arslan, M.; Kuila, D. Co-Ru catalysts with different composite oxide supports for Fischer–Tropsch studies in 3D-printed stainless steel microreactors. Appl. Catal. A Gen. 2020, 608, 117838. [Google Scholar] [CrossRef]
- Prasanth, K.P.; Pillai, R.S.; Peter, S.A.; Bajaj, H.C.; Jasra, R.V.; Chung, H.D.; Kim, T.H.; Song, S.D. Hydrogen uptake in palladium and ruthenium exchanged zeolite X. J. Alloy. Compd. 2008, 466, 439–446. [Google Scholar] [CrossRef]
- Cao, X.; Long, F.; Zhai, Q.; Liu, P.; Xu, J.; Jiang, J. Enhancement of fatty acids hydrodeoxygenation selectivity to diesel-range alkanes over the supported Ni-MoOx catalyst and elucidation of the active phase. Renew. Energy 2020, 162, 2113–2125. [Google Scholar] [CrossRef]
- Wang, J.; Fan, C.; Sun, Q.; Reuter, K.; Jacobi, K.; Scheffler, M.; Ertl, G. Surface coordination chemistry: Dihydrogen versus hydride complexes on RuO2(110). Angew. Chem. Int. Ed. 2003, 42, 2151–2154. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.D.; Seitsonen, A.P.; Over, H. The atomic geometry of oxygen-rich Ru(0001) surfaces: Coexistence of (1 × 1)O and RuO2(110) domains. Surf. Sci. 2000, 465, 1–8. [Google Scholar] [CrossRef]
- Over, H.; Seitsonen, A.P.; Lundgren, E.; Wiklund, M.; Andersen, J.N. Spectroscopic characterization of catalytically active surface sites of a metallic oxide. Chem. Phys. Lett. 2001, 342, 467–472. [Google Scholar] [CrossRef]
- Henrich, V.E.; Cox, P.A. The Surface Science of Metal Oxides; Cambridge University Press: Cambridge, NY, USA, 1994. [Google Scholar] [CrossRef]
- Sun, Q.; Reuter, K.; Scheffler, M. Hydrogen adsorption on RuO2(110): Density-functional calculations. Phys. Rev. B 2004, 70, 235402. [Google Scholar] [CrossRef]
- Liang, J.; Chen, T.; Liu, J.; Zhang, Q.; Peng, W.; Li, Y.; Zhang, F.; Fan, X. Chemoselective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over Ni/MoO2@ Mo2CTx catalyst with extraordinary synergic effect. Chem. Eng. J. 2020, 391, 123472. [Google Scholar] [CrossRef]
- Feng, W.; Yu, M.; Wang, L.; Miao, Y.; Shakouri, M.; Ran, J.; Hu, Y.; Li, Z.; Huang, R.; Lu, Y.; et al. Insights into bimetallic oxide synergy during carbon dioxide hydrogenation to methanol and dimethyl ether over GaZrOx oxide catalysts. ACS Catal. 2021, 11, 4704–4711. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).