
Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants



Abstract
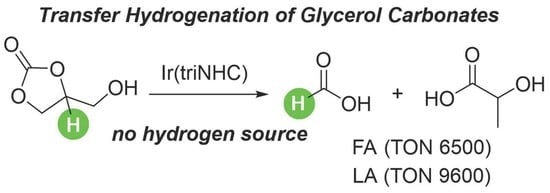
1. Introduction
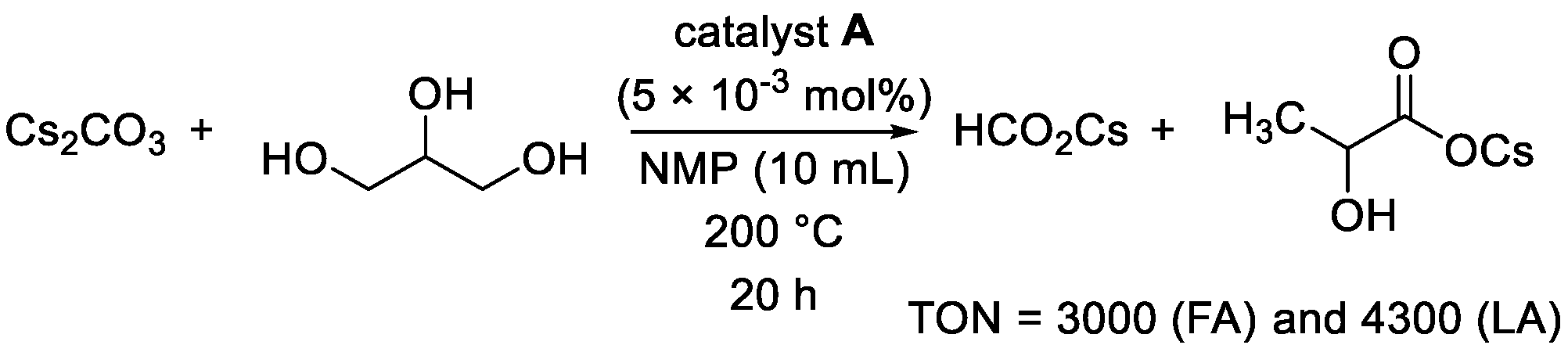
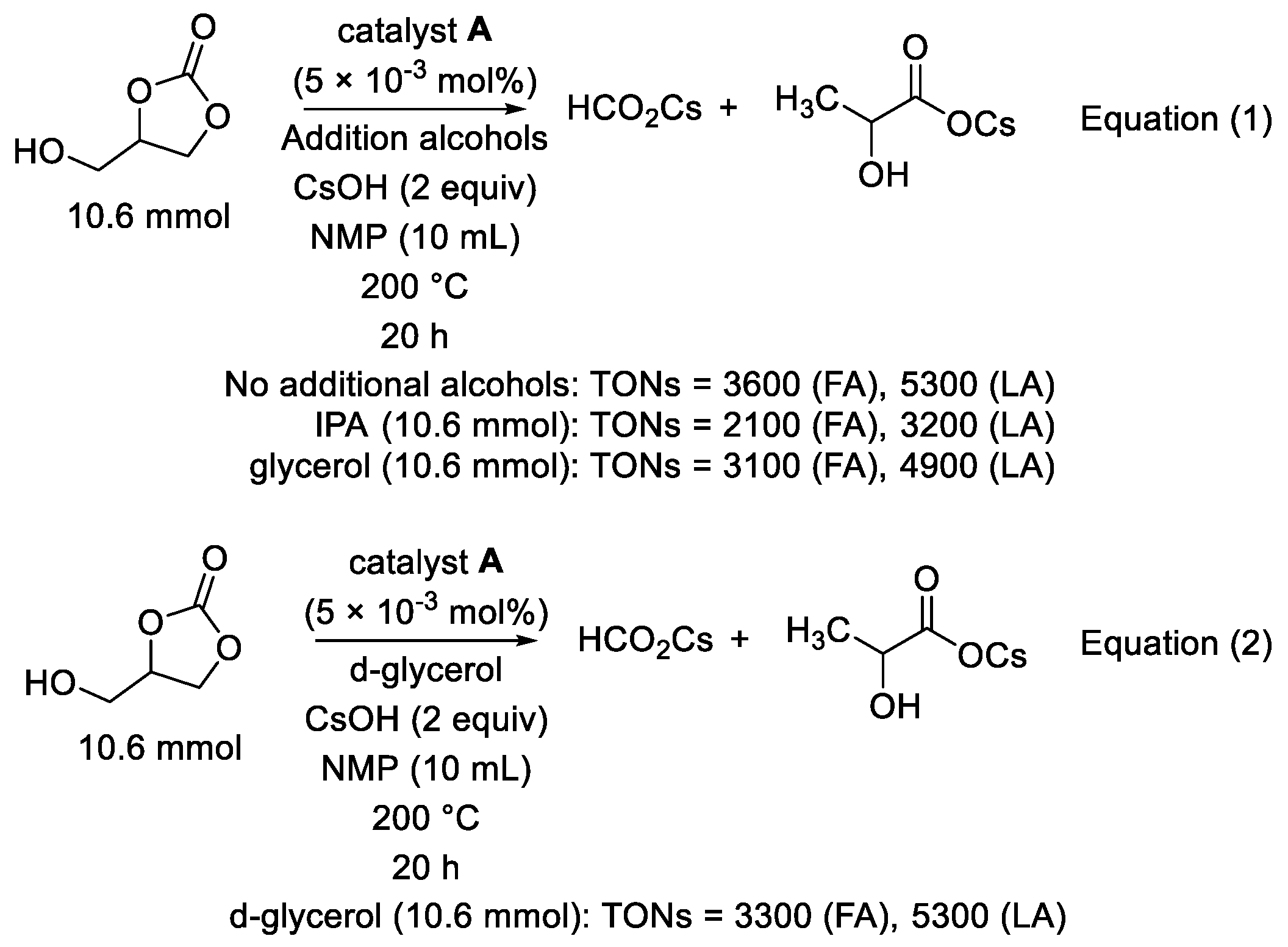
2. Results and Discussion

3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous hydrogenation of carbon-dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- Jessop, P.G.; Joó, F.; Tai, C.C. Recent advances in the homogeneous hydrogenation of carbon dioxide. Coord. Chem. Rev. 2004, 248, 2425–2442. [Google Scholar] [CrossRef]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The use of carbon dioxide (CO2) as a building block in organic synthesis from an industrial perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef]
- Krall, E.M.; Klein, T.W.; Andersen, R.J.; Nett, A.J.; Glasgow, R.W.; Reader, D.S.; Dauphinais, B.C.; Mc Ilrath, S.P.; Fischer, A.A.; Carney, M.J.; et al. Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(II) PNN pincer complexes. Chem. Commun. 2014, 50, 4884–4887. [Google Scholar] [CrossRef]
- Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Catalytic hydrogenation of cyclic carbonates: A practical approach from CO2 and epoxides to methanol and diols. Angew. Chem. Int. Ed. 2012, 51, 13041–13045. [Google Scholar] [CrossRef]
- Li, Y.; Junge, K.; Beller, M. Improving the efficiency of the hydrogenation of carbonates and carbon dioxide to methanol. ChemCatChem 2013, 5, 1072–1074. [Google Scholar] [CrossRef]
- Wu, X.; Ji, L.; Ji, Y.; Elageed, E.H.M.; Gao, G. Hydrogenation of ethylene carbonate catalyzed by lutidine-bridged N-heterocyclic carbene ligands and ruthenium precursors. Catal. Commun. 2016, 85, 57–60. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese catalyzed hydrogenation of organic carbonates to methanol and alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef] [PubMed]
- Zubar, V.; Levedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepelgy, O.; Rueping, M. Hydrogenation of CO2-derived carbonates and polycarbonates to methanol and diols by metal-ligand cooperative manganese catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; Hölscher, M.; Leitner, W. Catalytic hydrogenation of cyclic carbonates using manganese complexes. Angew. Chem. Int. Ed. 2018, 57, 13449–13453. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, P.; Gangwar, M.K.; Sundararaju, B. Well-defined Cp*Co(III)-catalyzed hydrogenation of carbonates and polycarbonates. ChemCatChem 2021, 13, 934–939. [Google Scholar] [CrossRef]
- Kim, S.H.; Hong, S.H. Transfer hydrogenation of organic formates and cyclic carbonates: An alternative route to methanol from carbon dioxide. ACS Catal. 2014, 4, 3630–3636. [Google Scholar] [CrossRef]
- Liu, X.; de Vries, J.G.; Werner, T. Transfer hydrogenation of cyclic carbonates and polycarbonate to methanol and diols by iron pincer catalysts. Green Chem. 2019, 21, 5248–5255. [Google Scholar] [CrossRef]
- Szewczyk, M.; Magre, M.; Zubar, V.; Rueping, M. Reduction of cyclic and linear organic carbonates using a readily available magnesium catalyst. ACS Catal. 2019, 9, 11634–11639. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Menoud, F.; Dyson, P.J. Synthesis of methanol and diols from CO2 via cyclic carbonates under metal-free, ambient pressure, and solvent-free conditions. ACS Sustain. Chem. Eng. 2018, 6, 12119–12123. [Google Scholar] [CrossRef]
- Feghali, E.; Cantat, T. Room temperature organocatalyzed reductive depolymerization of waste polyethers, polyesters, and polycarbonates. ChemSusChem 2015, 8, 980–984. [Google Scholar] [CrossRef]
- Zassinovich, G.; Mestroni, G.; Gladiali, S. Asymmetric hydrogen transfer reactions promoted by homogeneous transition metal catalysis. Chem. Rev. 1992, 92, 1051–1069. [Google Scholar] [CrossRef]
- Díaz-Álvarez, A.E.; Cadierno, V. Glycerol: A promising green solvent and reducing agent for metal-catalyzed transfer hydrogenation reactions and nanoparticles formation. Appl. Sci. 2013, 3, 55–69. [Google Scholar] [CrossRef]
- Crabtree, R.H. Transfer hydrogenation with glycerol as H-donor: Catalyst activation, deactivation and homogeneity. ACS Sustain. Chem. Eng. 2019, 7, 15845–15853. [Google Scholar] [CrossRef]
- Cheong, Y.-J.; Sung, K.; Park, S.; Jung, J.; Jang, H.-Y. Valorization of chemical wates: Ir(biscarbene)-catalyzed transfer hydrogenation of inorganic carbonates using glycerol. ACS Sustain. Chem. Eng. 2020, 8, 6972–6978. [Google Scholar] [CrossRef]
- Sung, K.; Lee, M.-h.; Cheong, Y.-J.; Jang, H.-Y. Ir(triscarbene)-catalyzed sustainable transfer hydrogenation of levulinic acid to γ-valerolactone. Appl. Organomet. Chem. 2020, 35, e6105. [Google Scholar] [CrossRef]
- Cheong, Y.-J.; Sung, K.; Kim, J.-A.; Kim, Y.K.; Yoon, W.; Yun, H.; Jang, H.-Y. Iridium(NHC)-catalyzed sustainable transfer hydrogenation of CO2 and inorganic carbonates. Catalysts 2021, 11, 695. [Google Scholar] [CrossRef]
- Christy, S.; Noschese, A.; Lomelí-Rodrigeuz, M.; Greeves, N.; Lopez-Sanchez, J.A. Recent progress in the synthesis and applications of glycerol carbonate. Curr. Opin. Green Sustain. Chem. 2018, 14, 99–107. [Google Scholar] [CrossRef]
- Szöri, M.; Giri, B.R.; Wang, Z.; Dawood, A.E.; Viskolcz, B.; Farooq, A. Glycerol carbonate as a fuel additive for a sustainable future. Sustain. Energy Fuels 2018, 2, 2171–2178. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Gartner, F.; Junge, H.; Beller, M. Catalytic generation of hydrogen from formic acid and its derivatives: Useful hydrogen storage materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source-recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material-development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef]
- Bottari, G.; Barta, K. Lactic acid and hydrogen from glycerol via acceptorless dehydrogenation using homogeneous catalysts. Recycl. Catal. 2015, 2, 70–77. [Google Scholar] [CrossRef][Green Version]
- Frey, G.D.; Rentzsch, C.F.; von Preysing, D.; Scherg, T.; Mühlhofer, M.; Herdtweck, E.; Herrmann, W.A. Rhodium and iridium complexes of N-heterocyclic carbenes: Structural investigations and their catalytic properties in the borylation reaction. J. Organomet. Chem. 2006, 691, 5725–5738. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Campos, J.; Manas, M.G.; Crabtree, R.H. Efficient selective and atom economic catalytic conversion of glycerol to lactic acid. Nat. Commun. 2014, 5, 5084. [Google Scholar] [CrossRef] [PubMed]
- Cheong, Y.-J.; Sung, K.; Kim, J.-A.; Kim, Y.K.; Jang, H.-Y. Highly efficient iridium-catalyzed production of hydrogen and lactate from glycerol: Rapid hydrogen evolution by bimetallic iridium catalysts. Eur. J. Inorg. Chem. 2020, 2020, 4064–4068. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Mercado, B.Q.; Crabtree, R.H.; Hazari, N. Selective conversionof glycerol to lactic acid with iron pincer precatalysts. Chem. Commun. 2015, 51, 16201–16204. [Google Scholar] [CrossRef]
- Lu, Z.; Cherepakhin, V.; Demianets, I.; Lauridsen, P.J.; Williams, T.J. Iridium-based hydride transfer catalysts: From hydrogen storage to fine chemicals. Chem. Commun. 2018, 54, 7711–7724. [Google Scholar] [CrossRef]
- Truscott, B.J.; Kruger, H.; Webb, P.B.; Bühl, M.; Nolan, S.P. The mechanism of CO2 insertion into iridium(I) hydroxide and alkoxide bonds: A kinetics and computational study. Chem. Eur. J. 2015, 21, 6930–6935. [Google Scholar] [CrossRef]
- Vummaleti, S.V.C.; Talarico, G.; Nolan, S.P.; Cavallo, L.; Poater, A. Mechanism of CO2 fixation by IrI-X bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 2015, 4653–4657. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Catalyst (mol%) | Base (Equiv.) | Solvent | Formate (TON) | Lactate (TON) |
|---|---|---|---|---|---|
| 1 | A (2.5 × 10−3) | CsOH⋅H2O (2) | NMP | 6500 | 9600 |
| 2 | A (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 3600 | 5300 |
| 3 | A (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 980 | 1900 |
| 4 | A (5.0 × 10−3) | CsOH⋅H2O (2) | H2O | 21 | 89 |
| 5 | A (5.0 × 10−3) | KOH (2) | NMP | 12 | 65 |
| 6 | A (5.0 × 10−3) | NaOH (2) | NMP | 160 | 130 |
| 7 | A (5.0 × 10−3) | CsOH⋅H2O (3) | NMP | 930 | 4800 |
| 8 | A (5.0 × 10−3) | -- | NMP | -- | -- |
| 9 | -- | CsOH⋅H2O (2) | NMP | -- | -- |
| 10 | B (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2400 | 3600 |
| 11 | B (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 5200 | 7400 |
| 12 | C (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2600 | 4200 |
| 13 | D (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2100 | 3400 |
| 14 | E (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 580 | 2400 |
| 15 | F (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 310 | 1700 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheong, Y.-J.; Lee, M.-h.; Byeon, H.; Park, J.; Yu, S.; Jang, H.-Y. Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts 2022, 12, 656. https://doi.org/10.3390/catal12060656
Cheong Y-J, Lee M-h, Byeon H, Park J, Yu S, Jang H-Y. Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts. 2022; 12(6):656. https://doi.org/10.3390/catal12060656
Chicago/Turabian StyleCheong, Yeon-Joo, Mi-hyun Lee, Heemin Byeon, Jiyong Park, Sungju Yu, and Hye-Young Jang. 2022. "Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants" Catalysts 12, no. 6: 656. https://doi.org/10.3390/catal12060656
APA StyleCheong, Y.-J., Lee, M.-h., Byeon, H., Park, J., Yu, S., & Jang, H.-Y. (2022). Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts, 12(6), 656. https://doi.org/10.3390/catal12060656