
Detailed Kinetic Modeling of CO2-Based Fischer–Tropsch Synthesis



Abstract
:1. Introduction
- (1)
- (2)
- (3)
- Chain-length-dependent kinetic parameters [38];
- (4)
2. Modeling
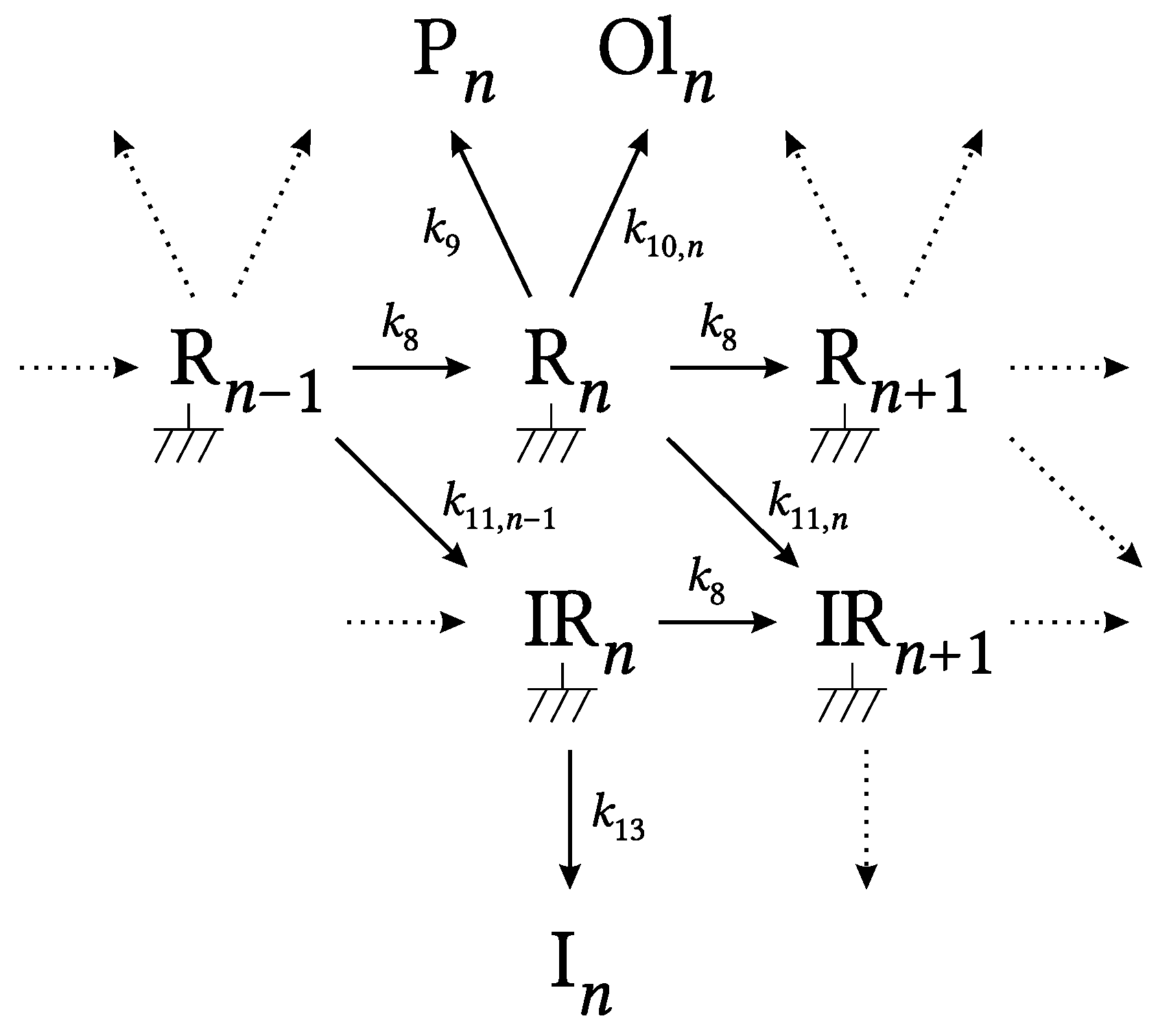
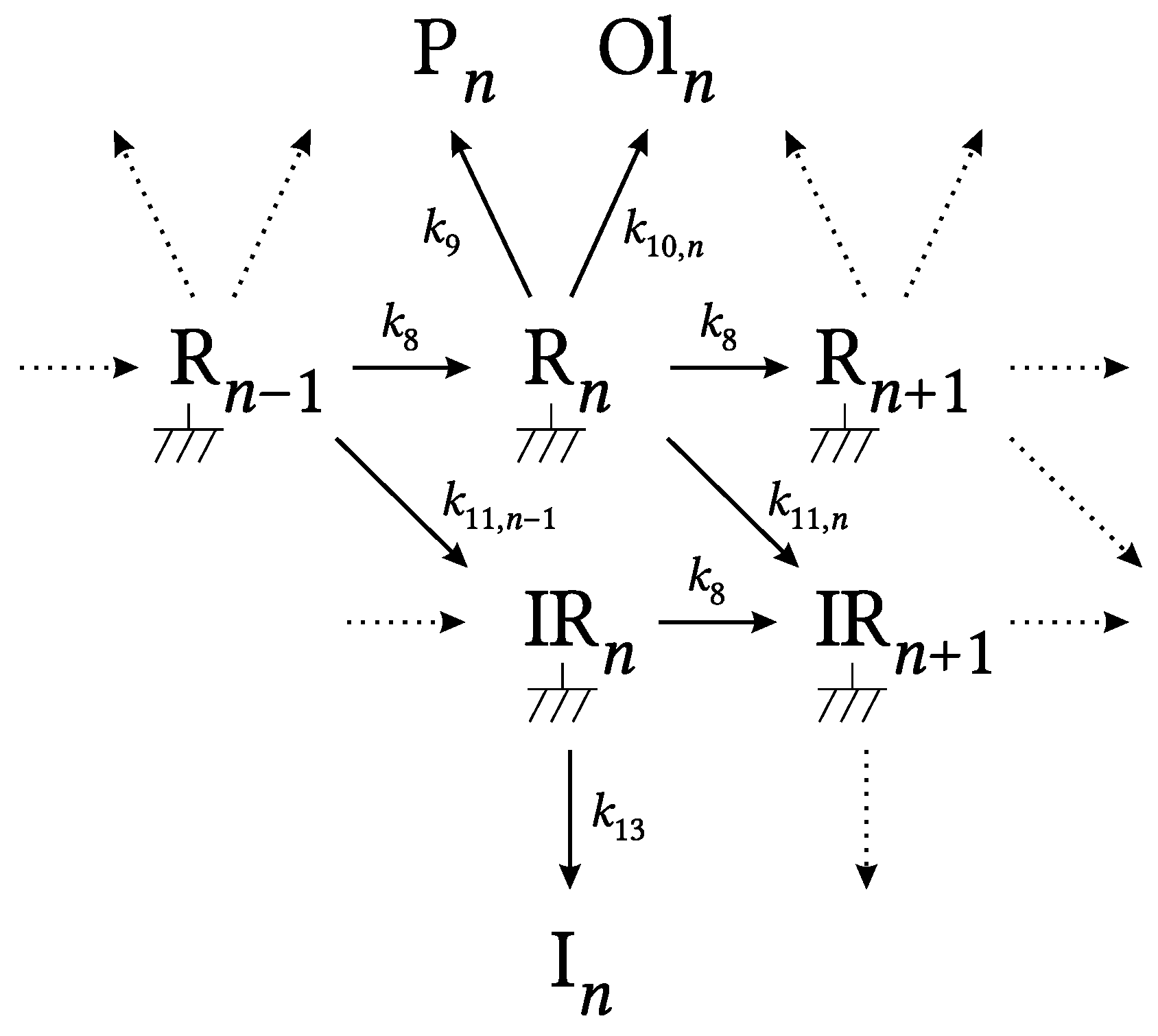
2.1. Kinetic Model
2.2. Reactor Model
2.3. Parameter Regression
3. Results and Discussion
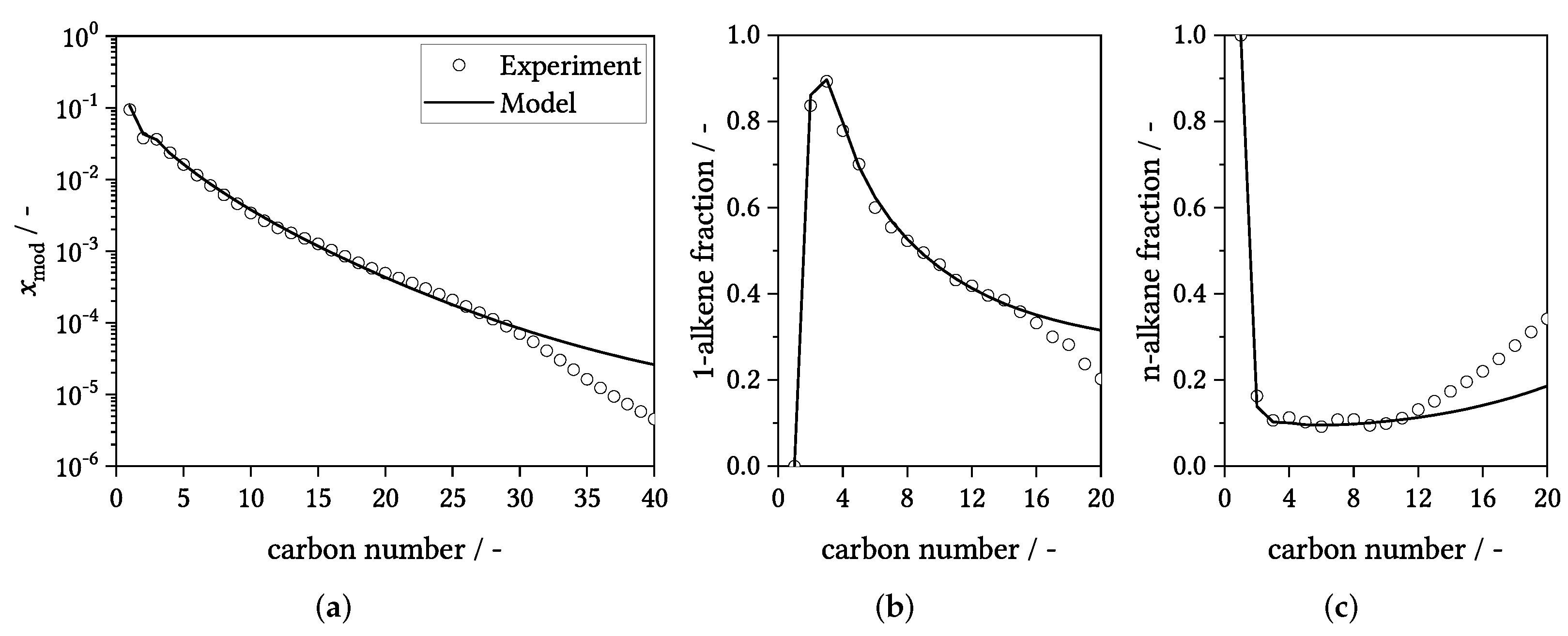
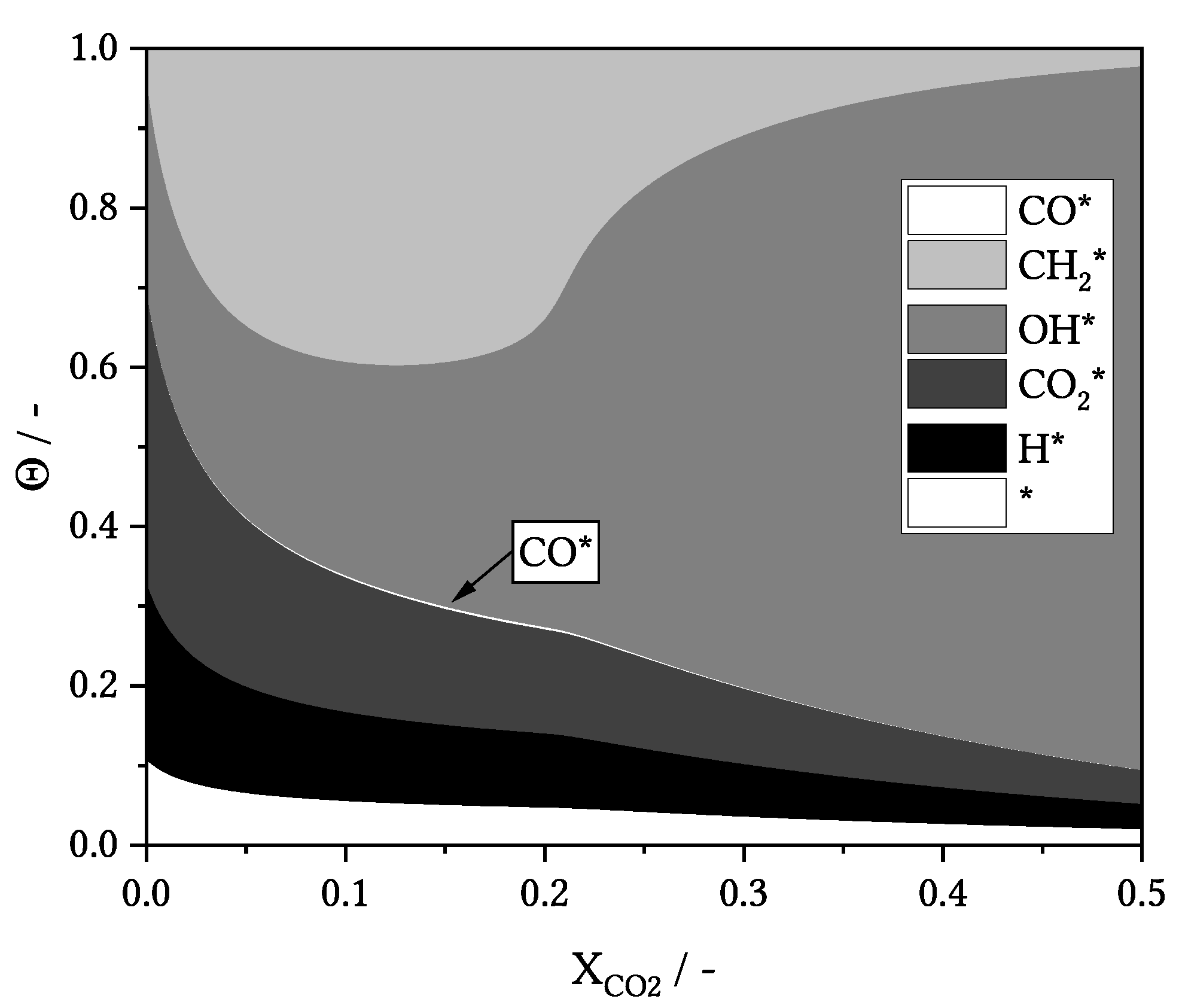
3.1. General Results and Model Details
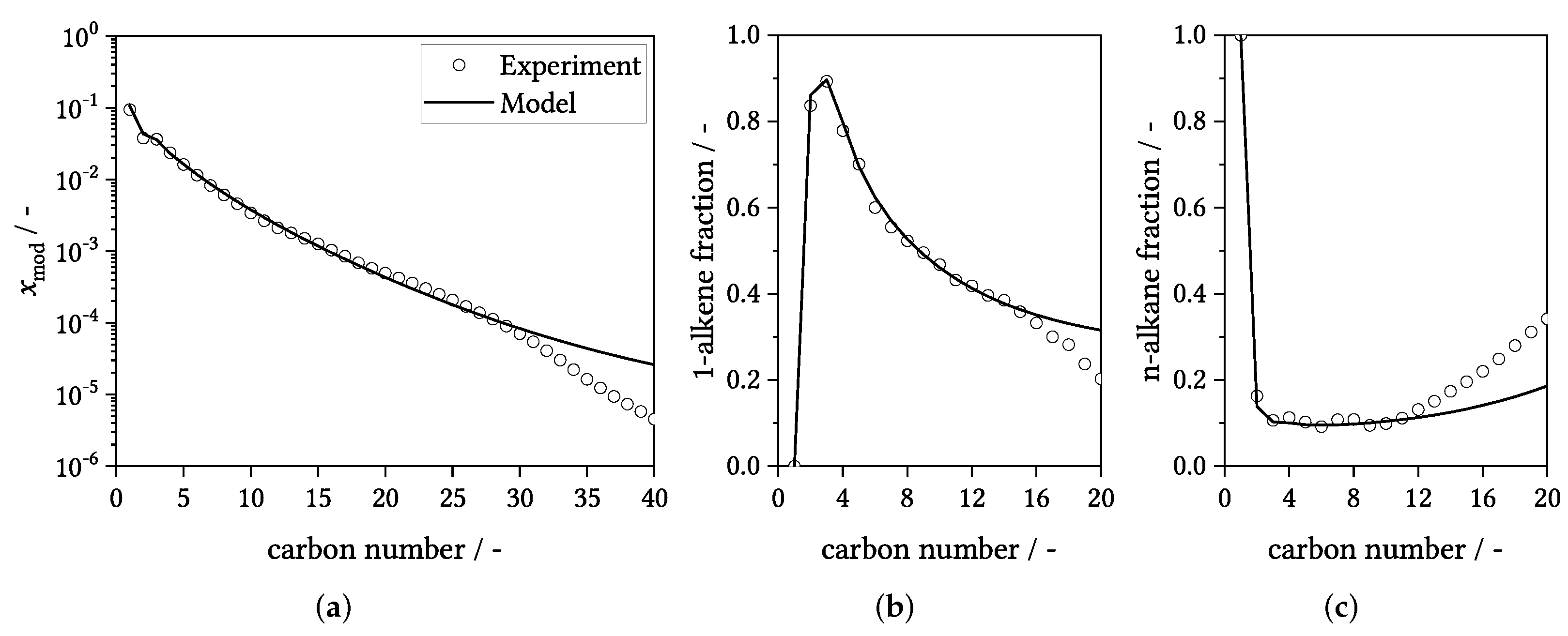
3.2. Parameter Estimates
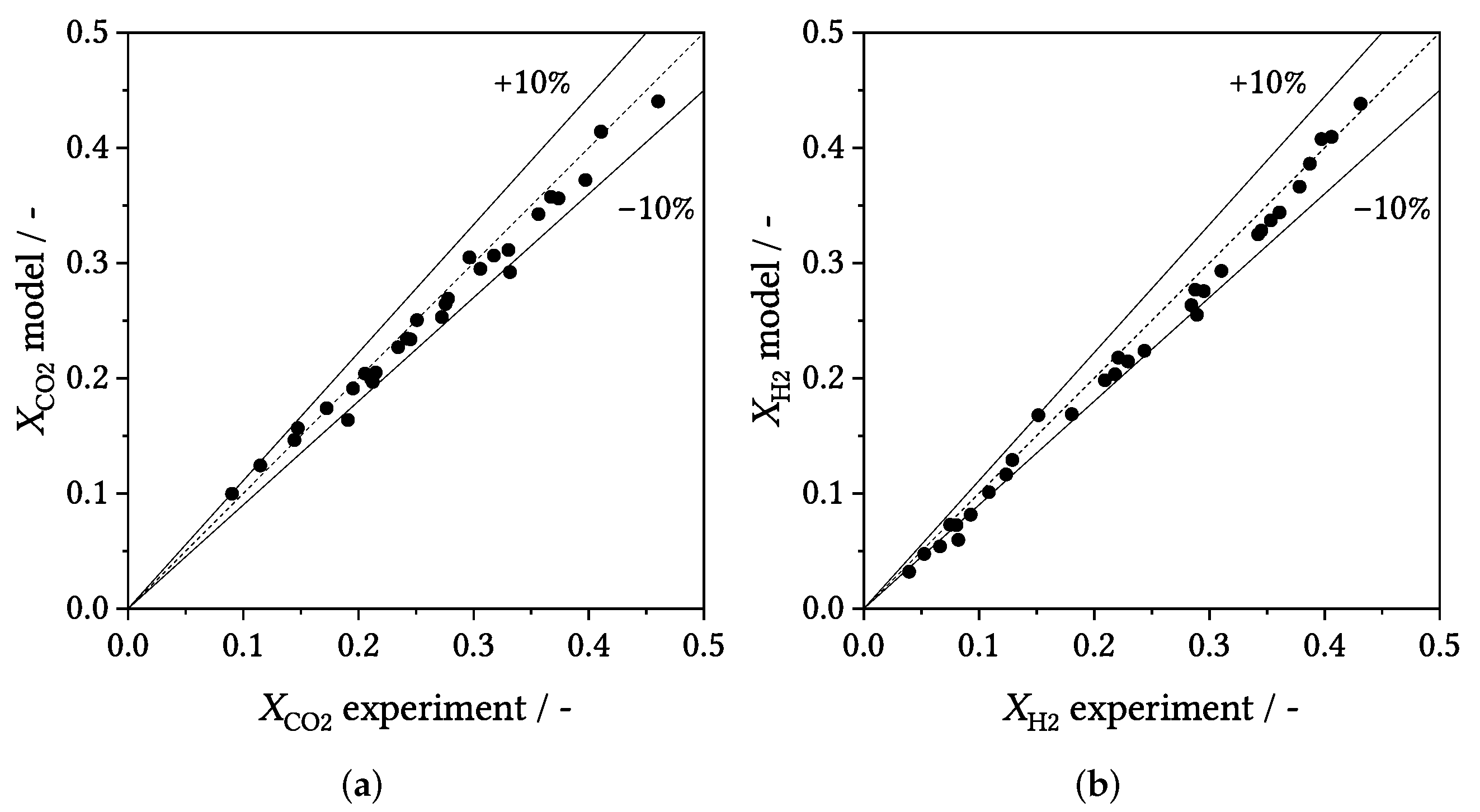
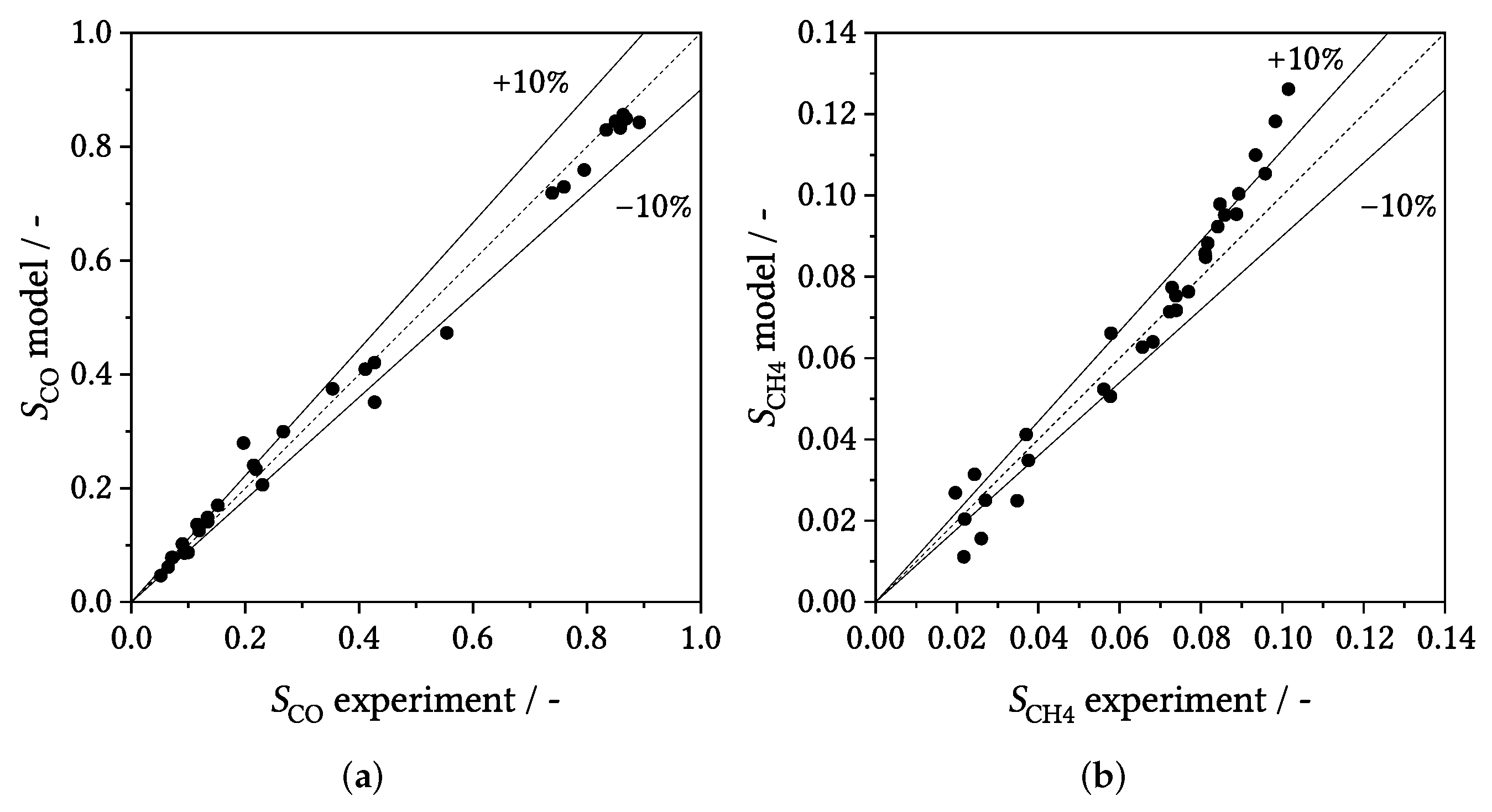
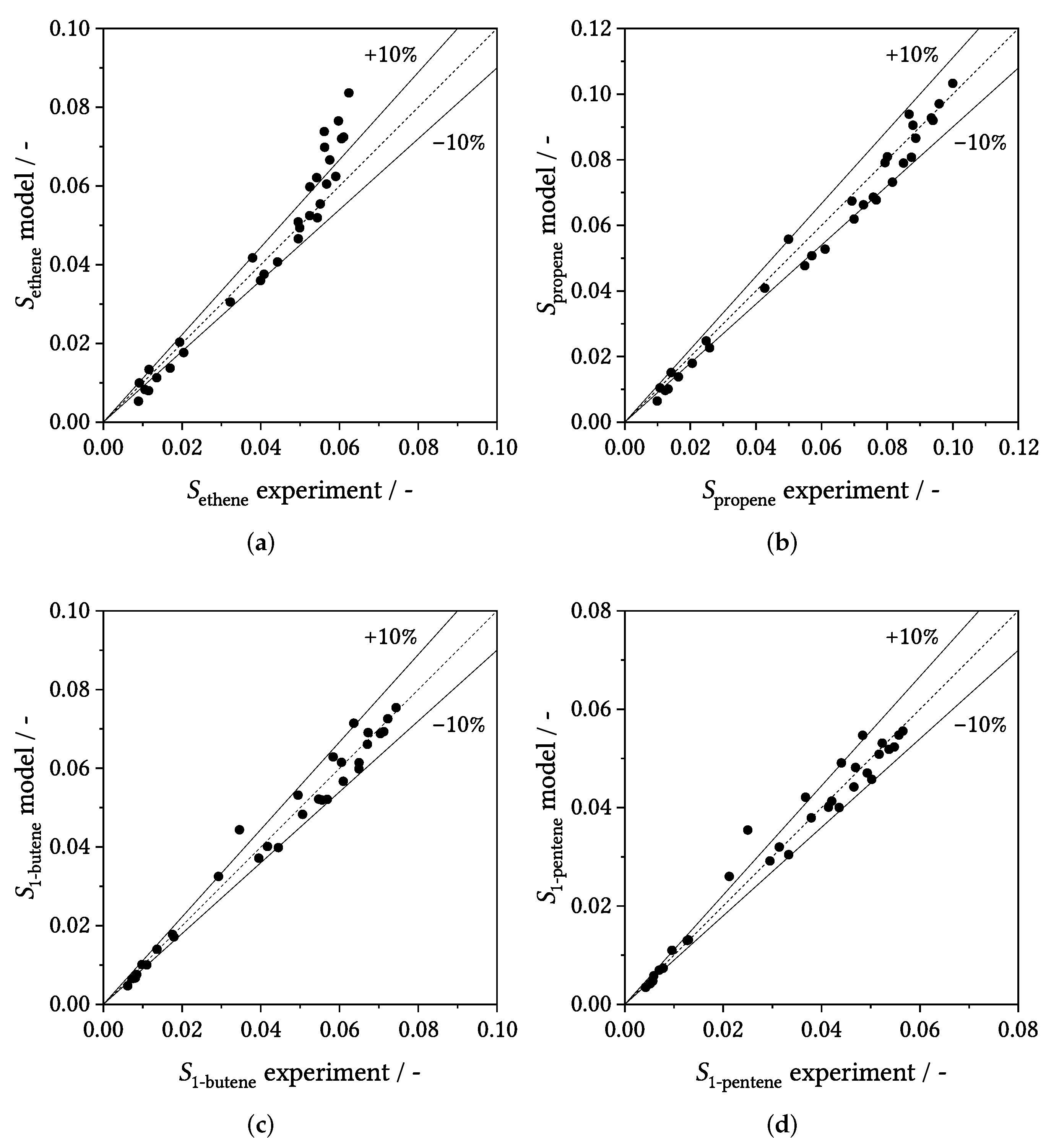
3.3. Parity Plots
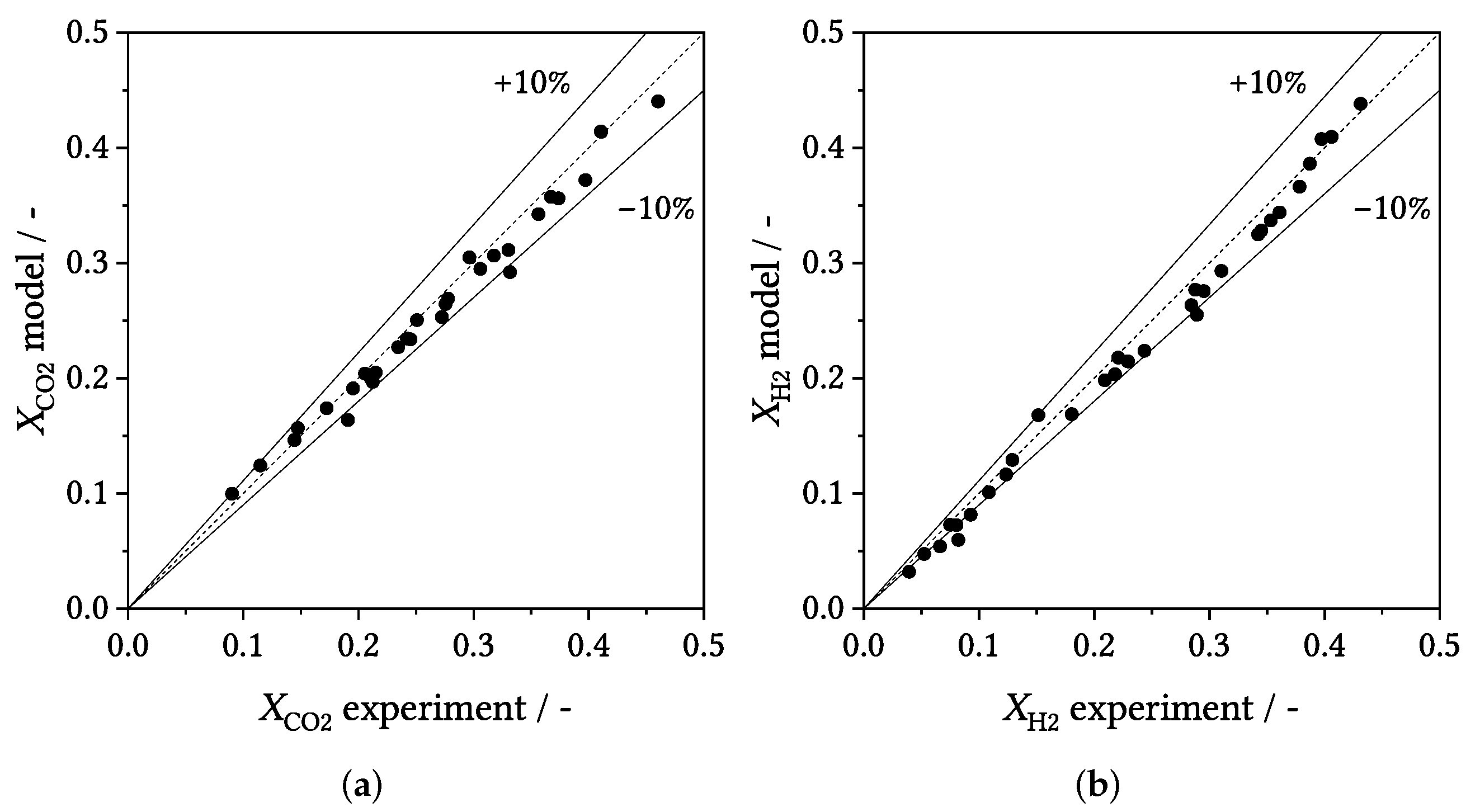
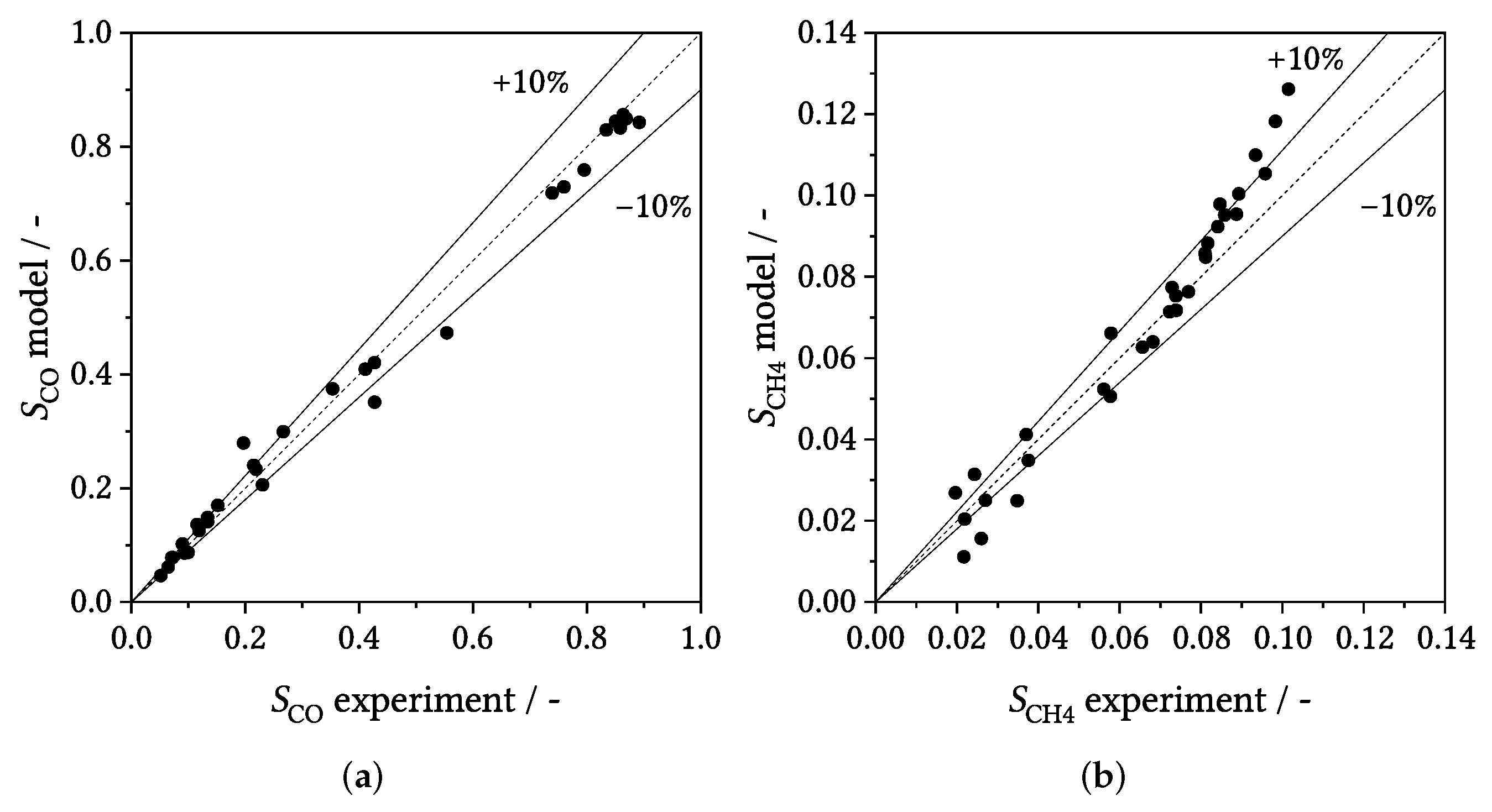
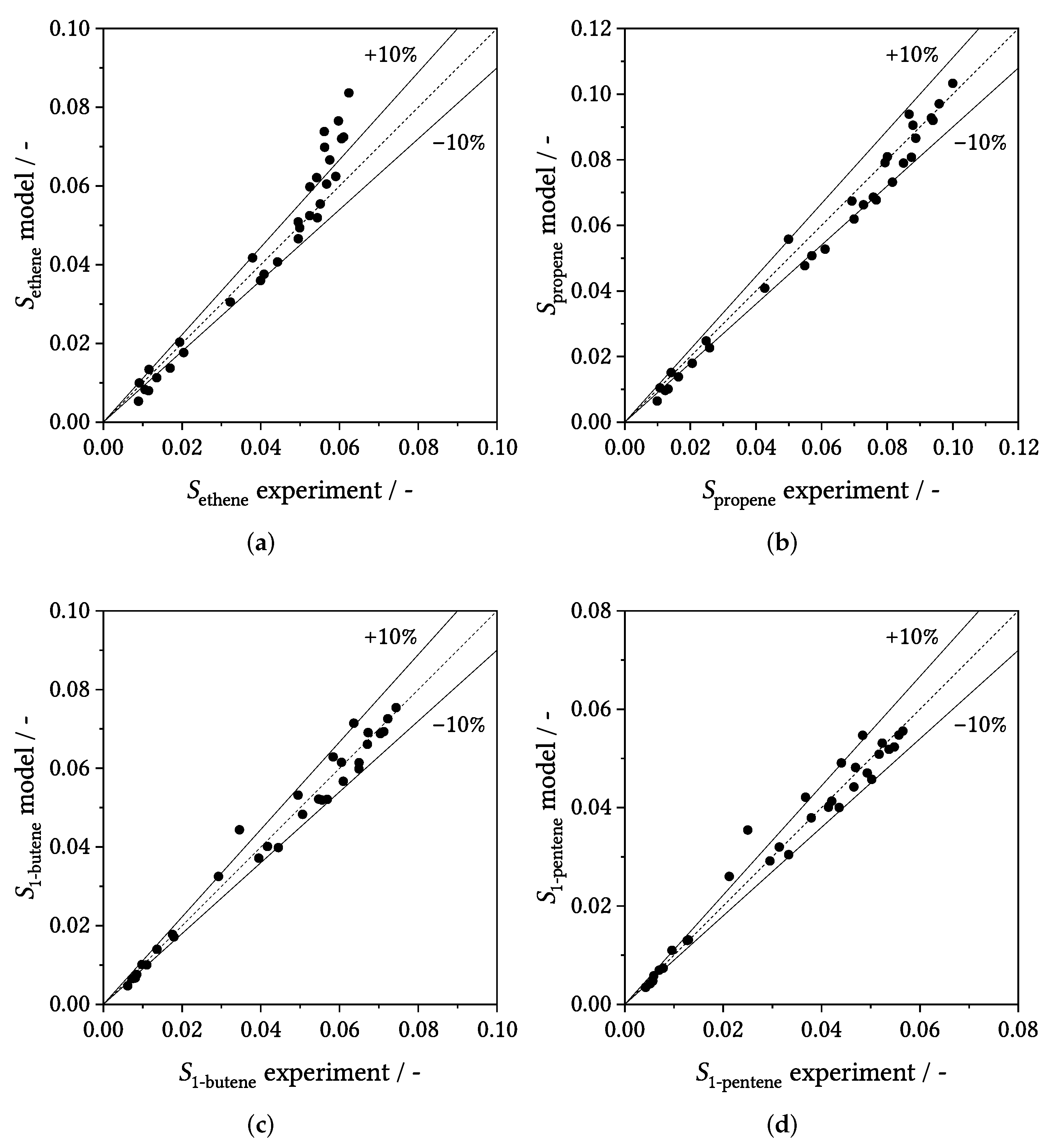
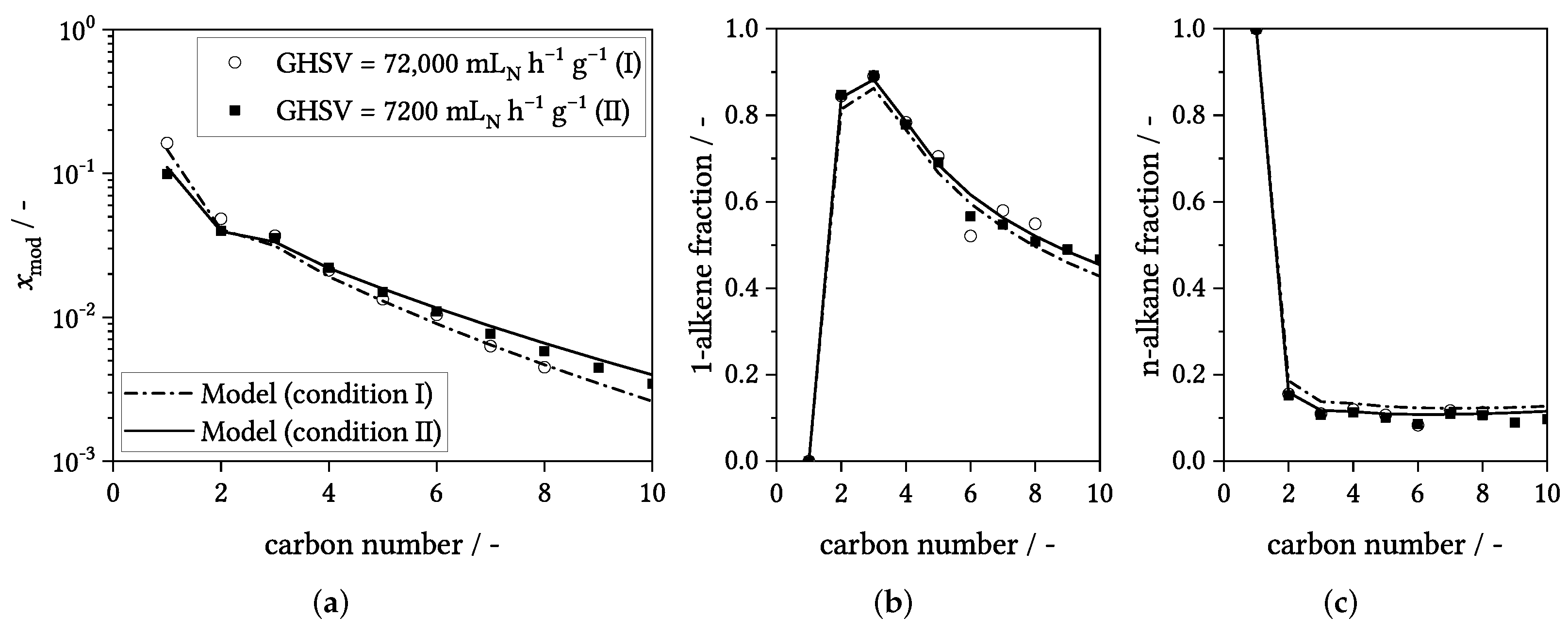
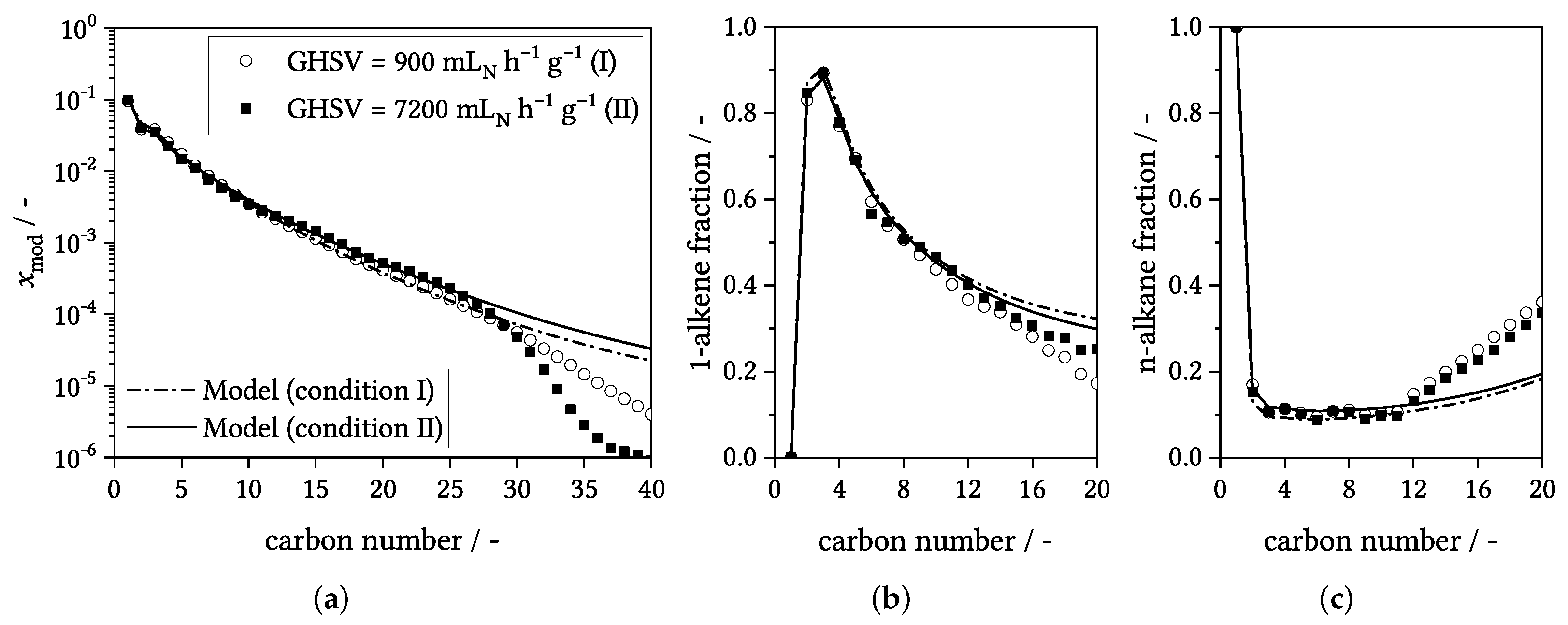
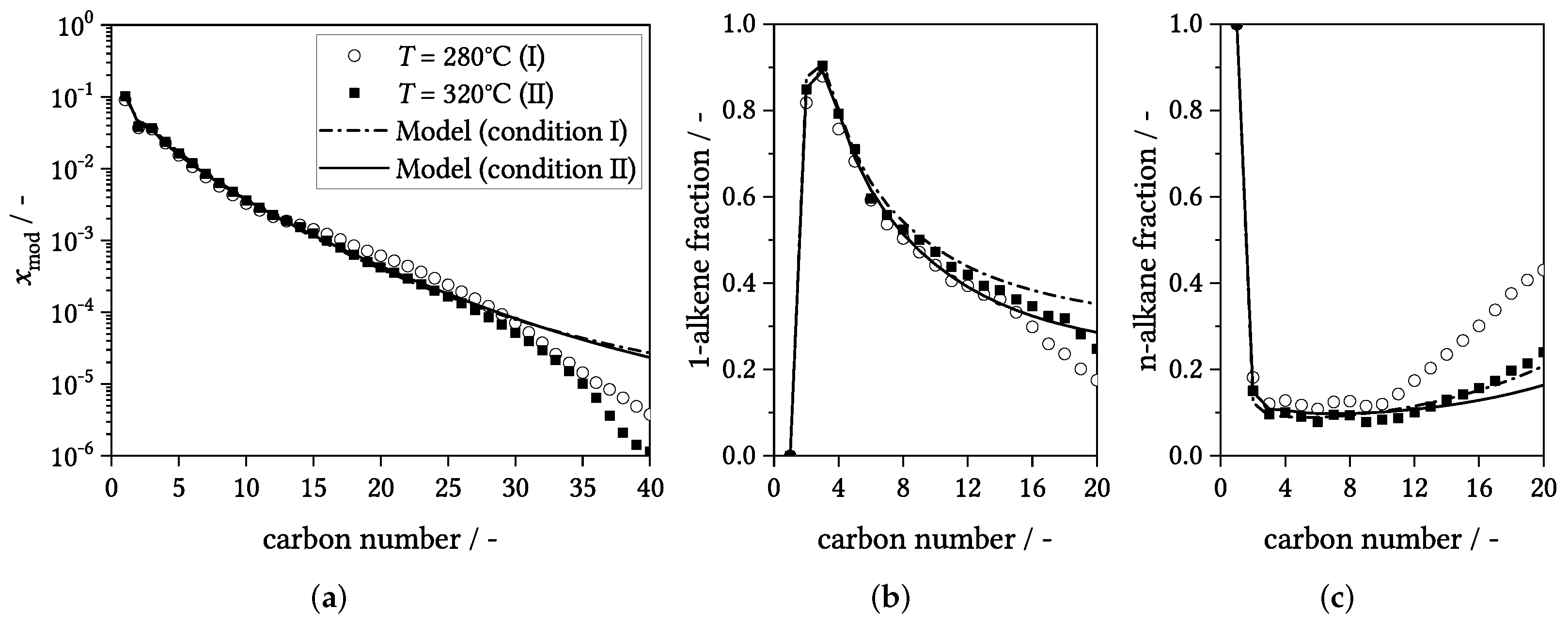
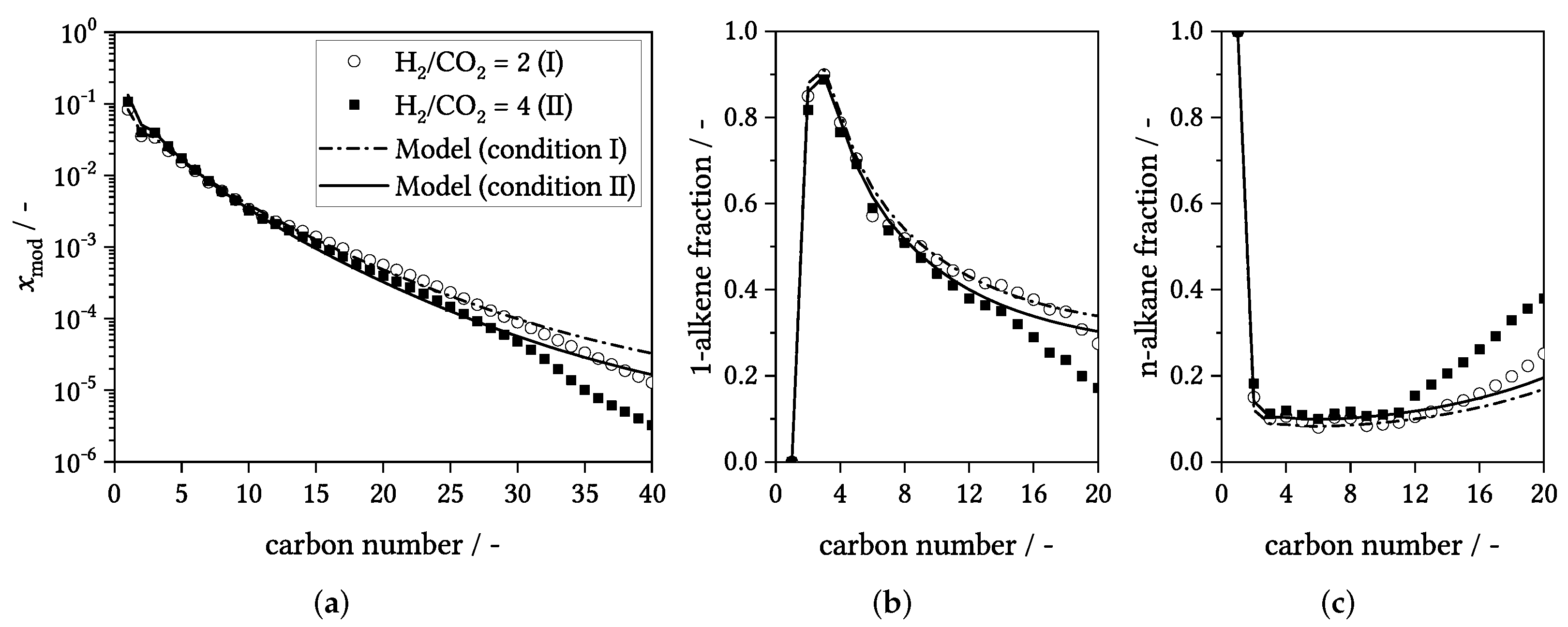
3.4. Influence of Process Conditions on Product Distribution
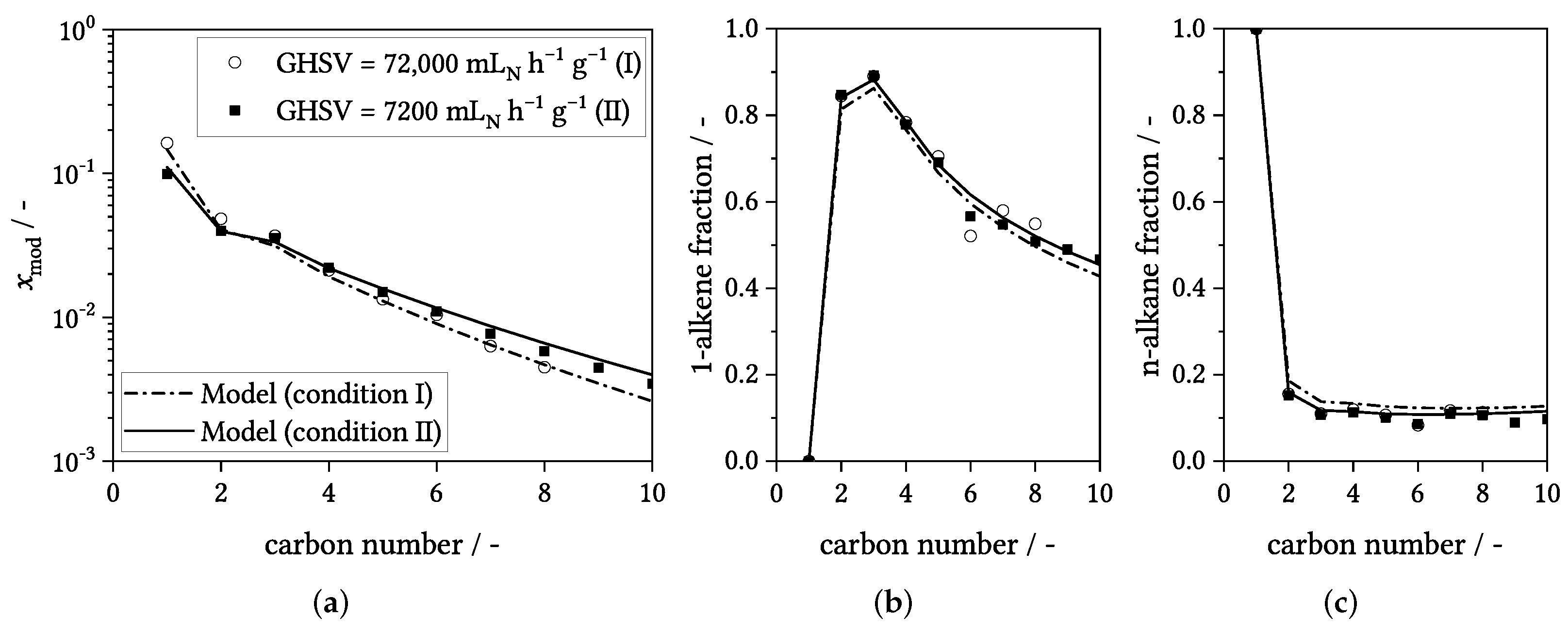
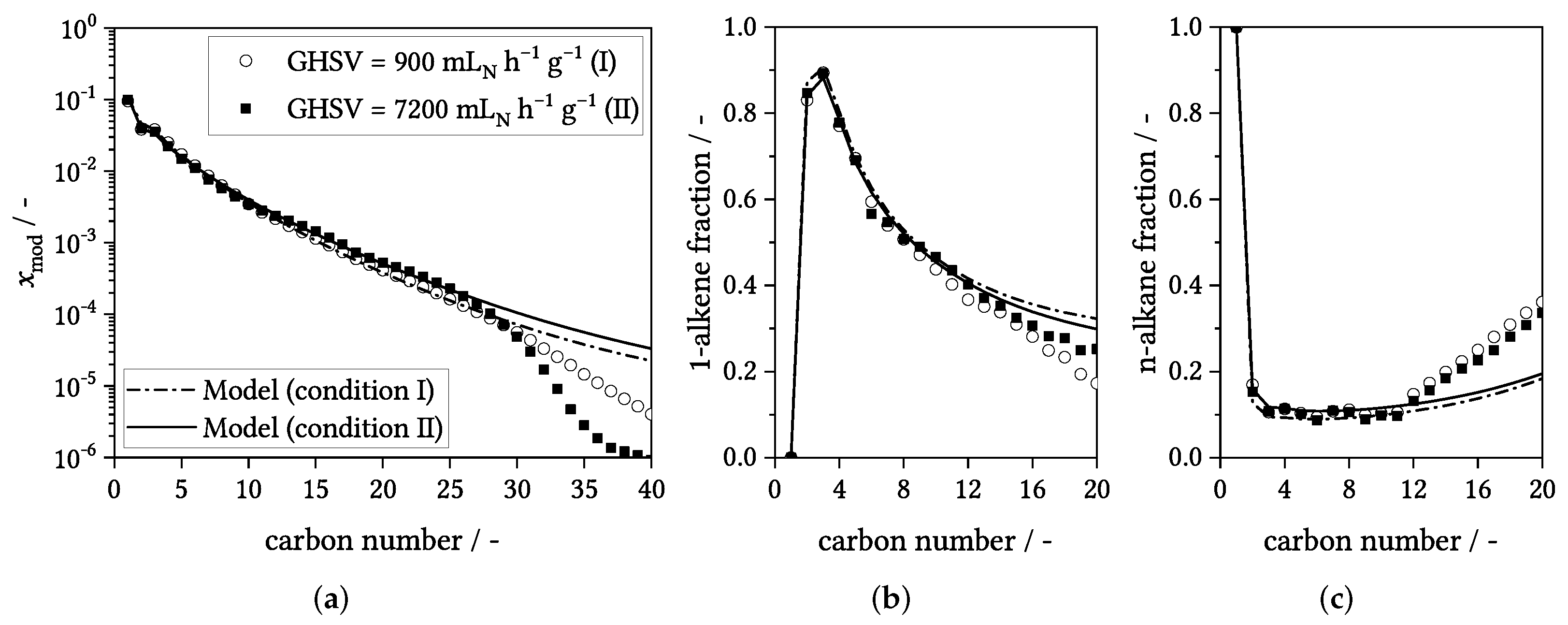
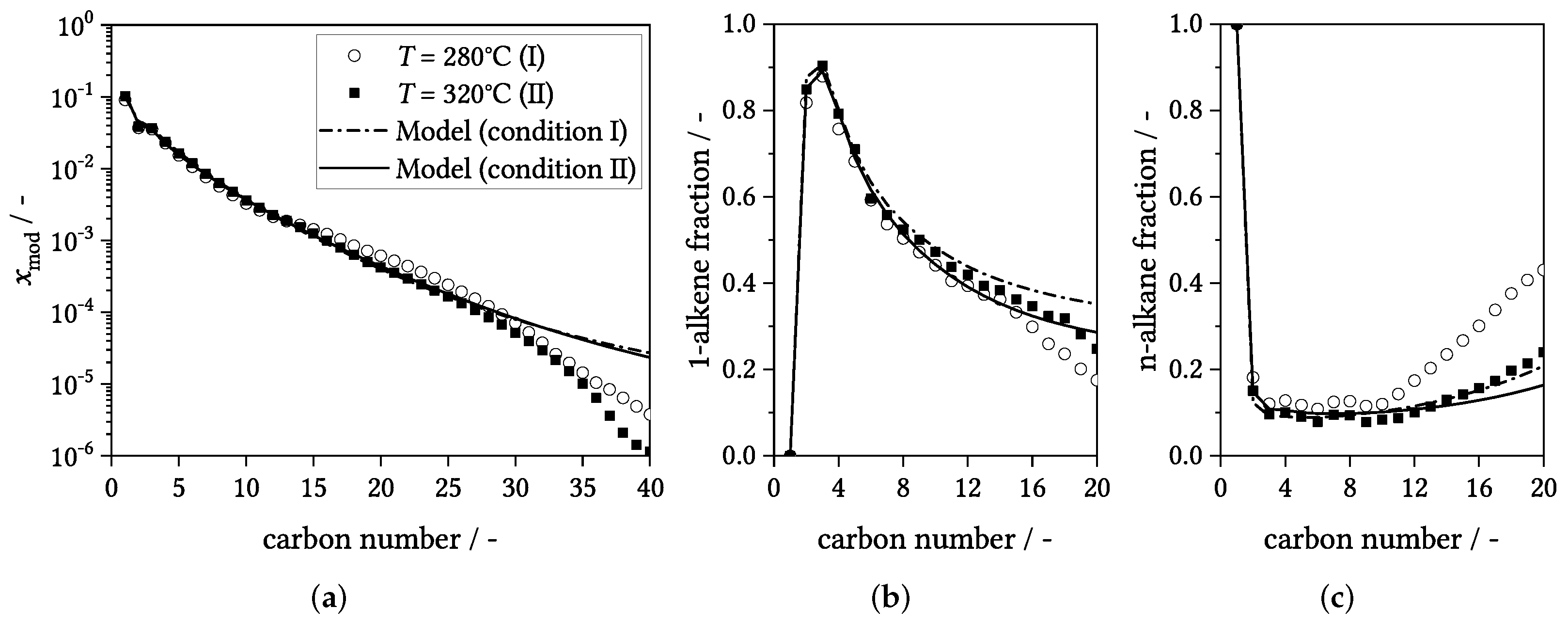
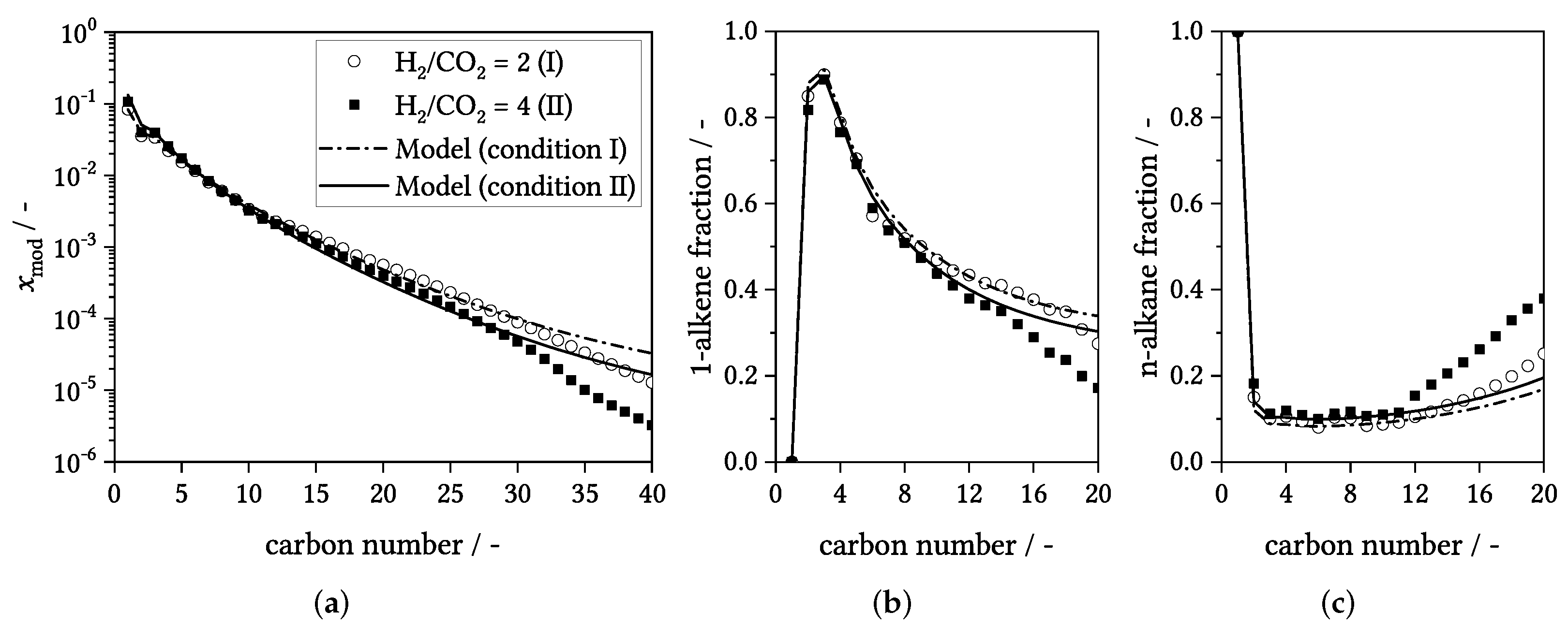
4. Experimental
4.1. Kinetic Experiments and Product Analysis
4.2. Data Analysis and Definitions
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASF | Anderson–Schulz–Flory |
| CSTR | continuously stirred tank reactor |
| DFT | density functional theory |
| EPMA | electron probe microanalysis |
| FID | flame ionization detector |
| FT | Fischer–Tropsch |
| FTS | Fischer–Tropsch synthesis |
| GC | gas chromatograph |
| GHSV | gas hourly space velocity |
| ICP-OES | inductively coupled plasma optical emission spectrometry |
| LHHW | Langmuir–Hinshelwood–Hougen–Watson |
| PFR | plug flow reactor |
| RDS | rate-determining step |
| RWGS | reverse water gas shift |
| VLE | vapor–liquid–equilibrium |
| Symbols | |
| chain growth probability | |
| activation energy (kJ mol−1) | |
| F | objective value of minimization function (Equation (6)) |
| chain-length dependence parameter (kJ mol−1) | |
| fraction of free surface sites | |
| surface coverage fraction | |
| iso-alkene | |
| IR | adsorbed iso-alkyl species |
| K | equilibrium constant () |
| k | reaction rate constant (mol g−1 h−1/mol g−1 h−1 bar−1) |
| m | mass (kg) |
| n | number of carbon atoms |
| molar flow rate (mol h−1) | |
| stoichiometric coefficient | |
| 1-alkene (-olefin) | |
| n-alkane (n-paraffin) | |
| p | pressure (bar) |
| partial pressure (bar) | |
| R | universal gas constant (8.314 J mol−1 K−1) |
| R | adsorbed alkyl species |
| r | reaction rate (mol h−1 g−1) |
| S | carbon based selectivity |
| T | temperature (°C/K) |
| volumetric flow rate (mL min−1) | |
| modified mole fraction | |
| X | conversion |
| ∗ | free surface site |
| Subscripts and Superscripts | |
| a,b | subreaction index |
| cat | catalyst |
| exp | experimental value |
| eq | equilibrium |
| i | species/component index |
| j | reaction index |
| k | surface species index |
| l | experiment index |
| in | reactor inlet |
| n | number of carbon atoms |
| N | normal conditions (0 °C and 1 atm) |
| out | reactor outlet |
| ref | reference temperature (here: 300 °C) |
| sim | simulated value |
| + | forward reaction |
| − | backward reaction |
| * | adsorbed species |
References
- Martinelli, M.; Gnanamani, M.K.; LeViness, S.; Jacobs, G.; Shafer, W.D. An overview of Fischer–Tropsch Synthesis: XtL processes, catalysts and reactors. Appl. Catal. A Gen. 2020, 608, 117740. [Google Scholar] [CrossRef]
- Panzone, C.; Philippe, R.; Chappaz, A.; Fongarland, P.; Bengaouer, A. Power-to-Liquid catalytic CO2 valorization into fuels and chemicals: Focus on the Fischer–Tropsch route. J. CO2 Util. 2020, 38, 314–347. [Google Scholar] [CrossRef]
- Ye, R.P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [Green Version]
- Ra, E.C.; Kim, K.Y.; Kim, E.H.; Lee, H.; An, K.; Lee, J.S. Recycling Carbon Dioxide through Catalytic Hydrogenation: Recent Key Developments and Perspectives. ACS Catal. 2020, 10, 11318–11345. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Sai Prasad, P.S.; Bae, J.W.; Jun, K.W.; Lee, K.W. Fischer–Tropsch Synthesis by Carbon Dioxide Hydrogenation on Fe-Based Catalysts. Catal. Surv. Asia 2008, 12, 170–183. [Google Scholar] [CrossRef]
- Van Der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and Selectivity of the Fischer–Tropsch Synthesis: A Literature Review. Catal. Rev. Sci. Eng. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Basha, O.M.; Sehabiague, L.; Abdel-Wahab, A.; Morsi, B.I. Fischer–Tropsch Synthesis in Slurry Bubble Column Reactors: Experimental Investigations and Modeling—A Review. Int. J. Chem. React. Eng. 2015, 13, 201–288. [Google Scholar] [CrossRef]
- Riedel, T.; Schaub, G.; Jun, K.W.; Lee, K.W. Kinetics of CO2 Hydrogenation on a K-Promoted Fe Catalyst. Ind. Eng. Chem. Res. 2001, 40, 1355–1363. [Google Scholar] [CrossRef]
- Meiri, N.; Dinburg, Y.; Amoyal, M.; Koukouliev, V.; Nehemya, R.V.; Landau, M.V.; Herskowitz, M. Novel process and catalytic materials for converting CO2 and H2 containing mixtures to liquid fuels and chemicals. Faraday Discuss. 2015, 183, 197–215. [Google Scholar] [CrossRef]
- Meiri, N.; Radus, R.; Herskowitz, M. Simulation of novel process of CO2 conversion to liquid fuels. J. CO2 Util. 2017, 17, 284–289. [Google Scholar] [CrossRef]
- Pour, A.N.; Housaindokht, M.R. A new kinetic model for direct CO2 hydrogenation to higher hydrocarbons on a precipitated iron catalyst: Effect of catalyst particle size. J. Energy Chem. 2017, 26, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Panzone, C.; Philippe, R.; Nikitine, C.; Vanoye, L.; Bengaouer, A.; Chappaz, A.; Fongarland, P. Catalytic and Kinetic Study of the CO2 Hydrogenation Reaction over a Fe-K/Al2O3 Catalyst toward Liquid and Gaseous Hydrocarbon Production. Ind. Eng. Chem. Res. 2021, 60, 16635–16652. [Google Scholar] [CrossRef]
- Brübach, L.; Hodonj, D.; Pfeifer, P. Kinetic Analysis of CO2 Hydrogenation to Long-Chain Hydrocarbons on a Supported Iron Catalyst. Ind. Eng. Chem. Res. 2022, 61, 1644–1654. [Google Scholar] [CrossRef]
- Panzone, C.; Philippe, R.; Nikitine, C.; Bengaouer, A.; Chappaz, A.; Fongarland, P. Development and Validation of a Detailed Microkinetic Model for the CO2 Hydrogenation Reaction toward Hydrocarbons over an Fe-K/Al2O3 Catalyst. Ind. Eng. Chem. Res. 2022, 61, 4514–4533. [Google Scholar] [CrossRef]
- Schulz, H. Principles of Fischer–Tropsch synthesis—Constraints on essential reactions ruling FT-selectivity. Catal. Today 2013, 214, 140–151. [Google Scholar] [CrossRef]
- Claeys, M.; van Steen, E. Chapter 8—Basic studies. In Fischer–Tropsch Technology; Studies in Surface Science and Catalysis; Steynberg, A., Dry, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 152, pp. 601–680. [Google Scholar] [CrossRef]
- Chakrabarti, D.; Prasad, V.; Klerk, A.D. Mechanism of the Fischer–Tropsch Process. In Fischer–Tropsch Synthesis, Catalysts, and Catalysis; Chemical Industries; Davis, B.H., Occelli, M.L., Eds.; CRC Press: Bosa Roca, FL, USA, 2016; Volume 118, pp. 183–222. [Google Scholar] [CrossRef]
- Davis, B.H. Fischer–Tropsch Synthesis: Reaction mechanisms for iron catalysts. Catal. Today 2009, 141, 25–33. [Google Scholar] [CrossRef]
- Fischer, F.; Tropsch, H. Über die Herstellung synthetischer Ölgemische (Synthol) durch Aufbau aus Kohlenoxyd und Wasserstoff. Brennst.-Chem. 1923, 4, 276–285. [Google Scholar]
- Fischer, F.; Tropsch, H. Über die direkte Synthese von Erdöl–Kohlenwasserstoffen bei gewöhnlichem Druck. Ber. Dtsch. Chem. Ges. A/B 1926, 59, 832–836. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer–Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Ozbek, M.O.; Niemantsverdriet, J.W. Elementary reactions of CO and H2 on C-terminated chi-Fe5C2(001) surfaces. J. Catal. 2014, 317, 158–166. [Google Scholar] [CrossRef]
- Pham, T.H.; Duan, X.; Qian, G.; Zhou, X.; Chen, D. CO Activation Pathways of Fischer–Tropsch Synthesis on chi-Fe5C2 (510): Direct versus Hydrogen-Assisted CO Dissociation. J. Phys. Chem. C 2014, 118, 10170–10176. [Google Scholar] [CrossRef]
- Petersen, M.A.; van Rensburg, W.J. CO Dissociation at Vacancy Sites on Hägg Iron Carbide: Direct Versus Hydrogen-Assisted Routes Investigated with DFT. Top. Catal. 2015, 58, 665–674. [Google Scholar] [CrossRef]
- Lox, E.S.; Froment, G.F. Kinetics of the Fischer–Tropsch reaction on a precipitated promoted iron catalyst. 2. Kinetic modeling. Ind. Eng. Chem. Res. 1993, 32, 71–82. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y.; Chang, J.; Wang, Y.N.; Bai, L.; Xu, Y.Y.; Xiang, H.W.; Li, Y.W.; Zhong, B. Detailed Kinetics of Fischer–Tropsch Synthesis on an Industrial Fe-Mn Catalyst. Ind. Eng. Chem. Res. 2003, 42, 5066–5090. [Google Scholar] [CrossRef]
- Teng, B.T.; Chang, J.; Zhang, C.H.; Cao, D.B.; Yang, J.; Liu, Y.; Guo, X.H.; Xiang, H.W.; Li, Y.W. A comprehensive kinetics model of Fischer–Tropsch synthesis over an industrial Fe–Mn catalyst. Appl. Catal. A 2006, 301, 39–50. [Google Scholar] [CrossRef]
- Lozano-Blanco, G.; Thybaut, J.W.; Surla, K.; Galtier, P.; Marin, G.B. Single-Event Microkinetic Model for Fischer–Tropsch Synthesis on Iron-Based Catalysts. Ind. Eng. Chem. Res. 2008, 47, 5879–5891. [Google Scholar] [CrossRef]
- Zhou, L.; Froment, G.F.; Yang, Y.; Li, Y. Advanced fundamental modeling of the kinetics of Fischer–Tropsch synthesis. AIChE J. 2016, 62, 1668–1682. [Google Scholar] [CrossRef]
- van der Laan, G.P.; Beenackers, A.A.C.M. Hydrocarbon Selectivity Model for the Gas–Solid Fischer–Tropsch Synthesis on Precipitated Iron Catalysts. Ind. Eng. Chem. Res. 1999, 38, 1277–1290. [Google Scholar] [CrossRef]
- Schulz, H.; Claeys, M. Kinetic modelling of Fischer–Tropsch product distributions. Appl. Catal. A Gen. 1999, 186, 91–107. [Google Scholar] [CrossRef]
- Nowicki, L.; Ledakowicz, S.; Bukur, D.B. Hydrocarbon selectivity model for the slurry phase Fischer–Tropsch synthesis on precipitated iron catalysts. Chem. Eng. Sci. 2001, 56, 1175–1180. [Google Scholar] [CrossRef]
- Olewski, T.; Todic, B.; Nowicki, L.; Nikacevic, N.; Bukur, D.B. Hydrocarbon selectivity models for iron-based Fischer–Tropsch catalyst. Chem. Eng. Res. Des. 2015, 95, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Huff, G.A.; Satterfield, C.N. Evidence for two chain growth probabilities on iron catalysts in the Fischer–Tropsch synthesis. J. Catal. 1984, 85, 370–379. [Google Scholar] [CrossRef]
- König, L.; Gaube, J. Fischer–Tropsch-Synthese. Neuere Untersuchungen und Entwicklungen. Chem. Ing. Tech. 1983, 55, 14–22. [Google Scholar] [CrossRef]
- Gaube, J.; Klein, H.F. Studies on the reaction mechanism of the Fischer–Tropsch synthesis on iron and cobalt. J. Mol. Catal. 2008, 283, 60–68. [Google Scholar] [CrossRef]
- Botes, F.G. Proposal of a New Product Characterization Model for the Iron-Based Low-Temperature Fischer–Tropsch Synthesis. Energy Fuels 2007, 21, 1379–1389. [Google Scholar] [CrossRef]
- Hanlon, R.T.; Satterfield, C.N. Reactions of selected 1-olefins and ethanol added during the Fischer–Tropsch synthesis. Energy Fuels 1988, 2, 196–204. [Google Scholar] [CrossRef]
- Tau, L.M.; Dabbagh, H.A.; Davis, B.H. Fischer–Tropsch synthesis: Carbon-14 tracer study of alkene incorporation. Energy Fuels 1990, 4, 94–99. [Google Scholar] [CrossRef]
- Botes, F.G.; Govender, N.S. Secondary Reactions of Ethylene As Studied in a Laboratory-Scale Recycle Slurry Reactor. Energy Fuels 2007, 21, 3095–3101. [Google Scholar] [CrossRef]
- Han, S.J.; Hwang, S.M.; Park, H.G.; Zhang, C.; Jun, K.W.; Kim, S.K. Identification of active sites for CO2 hydrogenation in Fe catalysts by first-principles microkinetic modelling. J. Mater. Chem. A 2020, 8, 13014–13023. [Google Scholar] [CrossRef]
- Nie, X.; Wang, H.; Janik, M.J.; Guo, X.; Song, C. Computational Investigation of Fe-Cu Bimetallic Catalysts for CO2 Hydrogenation. J. Phys. Chem. C 2016, 120, 9364–9373. [Google Scholar] [CrossRef]
- Nie, X.; Wang, H.; Janik, M.J.; Chen, Y.; Guo, X.; Song, C. Mechanistic Insight into C-C Coupling over Fe-Cu Bimetallic Catalysts in CO2 Hydrogenation. J. Phys. Chem. C 2017, 121, 13164–13174. [Google Scholar] [CrossRef]
- Schulz, H.; Erich, E.; Gorre, H.; van Steen, E. Regularities of selectivity as a key for discriminating FT-surface reactions and formation of the dynamic system. Catal. Lett. 1990, 7, 157–167. [Google Scholar] [CrossRef]
- Visconti, C.G.; Tronconi, E.; Lietti, L.; Zennaro, R.; Forzatti, P. Development of a complete kinetic model for the Fischer–Tropsch synthesis over Co/Al2O3 catalysts. Chem. Eng. Sci. 2007, 62, 5338–5343. [Google Scholar] [CrossRef]
- Bates, D.M.; Watts, D.G. Nonlinear Regression Analysis and Its Applications; Wiley: New York, NY, USA, 1988. [Google Scholar] [CrossRef] [Green Version]
- Visconti, C.G.; Tronconi, E.; Lietti, L.; Forzatti, P.; Rossini, S.; Zennaro, R. Detailed Kinetics of the Fischer–Tropsch Synthesis on Cobalt Catalysts Based on H-Assisted CO Activation. Top. Catal. 2011, 54, 786–800. [Google Scholar] [CrossRef]
- Donnelly, T.J.; Satterfield, C.N. Product distributions of the Fischer–Tropsch synthesis on precipitated iron catalysts. Appl. Catal. 1989, 52, 93–114. [Google Scholar] [CrossRef]
- Pour, A.N.; Zamani, Y.; Tavasoli, A.; Kamali Shahri, S.M.; Taheri, S.A. Study on products distribution of iron and iron–zeolite catalysts in Fischer–Tropsch synthesis. Fuel 2008, 87, 2004–2012. [Google Scholar] [CrossRef]
- Botes, F.G.; Niemantsverdriet, J.W.; van de Loosdrecht, J. A comparison of cobalt and iron based slurry phase Fischer–Tropsch synthesis. Catal. Today 2013, 215, 112–120. [Google Scholar] [CrossRef]
- Choi, P.H.; Jun, K.W.; Lee, S.J.; Choi, M.J.; Lee, K.W. Hydrogenation of carbon dioxide over alumina supported Fe-K catalysts. Catal. Lett. 1996, 40, 115–118. [Google Scholar] [CrossRef]
- Landau, M.V.; Meiri, N.; Utsis, N.; Vidruk Nehemya, R.; Herskowitz, M. Conversion of CO2, CO, and H2 in CO2 Hydrogenation to Fungible Liquid Fuels on Fe-Based Catalysts. Ind. Eng. Chem. Res. 2017, 56, 13334–13355. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Description | Reaction Step | Equilibrium/Kinetic Expression | Notes |
|---|---|---|---|---|
| 1 | adsorption/desorption | 2H* | ||
| 2 | adsorption/desorption | + * * | ||
| 3 | adsorption/desorption | CO CO* | ||
| 4 | adsorption/desorption | + 2∗ * + H* | ||
| 5 1 | dissociation | * + H* * + * | ||
| 6 2 | Monomer formation | * + 3H* * + * + 2∗ | ||
| 7 | Chain initiation | CH2* + H* R1* + ∗ | ||
| 8 | Chain growth | * | ||
| 9a | Methane formation | * + H* + 2∗ | ||
| 9b | n-Alkane formation | |||
| 10a | Ethene formation | * + H* | ||
| 10b | 1-Alkene fomation | + H* | ||
| 11a | Propyl branching | * + * + ∗ | ||
| 11b | Chain branching | * + ∗ | ||
| 12 | Branched chain growth | + * + ∗ | 4 | |
| 13 | iso-Alkene termination | + H* | 4 |
| No. | Elementary Step | Equilibrium/Kinetic Expression |
|---|---|---|
| 5a | * + ∗ * + O* | |
| 5b | O* + H* OH* + ∗ | |
| 5 | * + H* * + * |
| No. | Elementary Step | Equilibrium/Kinetic Expression | Notes |
|---|---|---|---|
| 6a | CO* + H* * + ∗ | ||
| 6b | HCO* + H* HCOH*+ ∗ | RDS | |
| 6c | HCOH* + ∗ | ||
| 6d | CH* + H* + ∗ | ||
| 6 | CO* + 3H* + OH* + 2∗ |
| Parameter | Value | Unit | Parameter | Value | Unit |
|---|---|---|---|---|---|
| 5.82 × 10−1 | 1.97 × 103 | ||||
| 1.37 | 9.57 × 102 | ||||
| 1.43 × 103 | 1.79 × 103 | ||||
| 9.21 × 103 | 2.18 × 102 | ||||
| 3.49 × 102 | 2.49 × 103 | ||||
| 1.45 × 101 | 7.67 × 102 | ||||
| 2.21 × 101 | 4.52 × 10−1 | ||||
| 2.35 × 102 | 2.89 | ||||
| 4.10 × 101 | 1.51 × 102 | ||||
| 1.64× 10−1 | 1.99 × 102 | ||||
| 1.37 × 103 | 5.97 | ||||
| 2.59× 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brübach, L.; Hodonj, D.; Biffar, L.; Pfeifer, P. Detailed Kinetic Modeling of CO2-Based Fischer–Tropsch Synthesis. Catalysts 2022, 12, 630. https://doi.org/10.3390/catal12060630
Brübach L, Hodonj D, Biffar L, Pfeifer P. Detailed Kinetic Modeling of CO2-Based Fischer–Tropsch Synthesis. Catalysts. 2022; 12(6):630. https://doi.org/10.3390/catal12060630
Chicago/Turabian StyleBrübach, Lucas, Daniel Hodonj, Linus Biffar, and Peter Pfeifer. 2022. "Detailed Kinetic Modeling of CO2-Based Fischer–Tropsch Synthesis" Catalysts 12, no. 6: 630. https://doi.org/10.3390/catal12060630
APA StyleBrübach, L., Hodonj, D., Biffar, L., & Pfeifer, P. (2022). Detailed Kinetic Modeling of CO2-Based Fischer–Tropsch Synthesis. Catalysts, 12(6), 630. https://doi.org/10.3390/catal12060630