
Dimethyl Ether Oxidation over Copper Ferrite Catalysts



Abstract
:
1. Introduction
2. Results
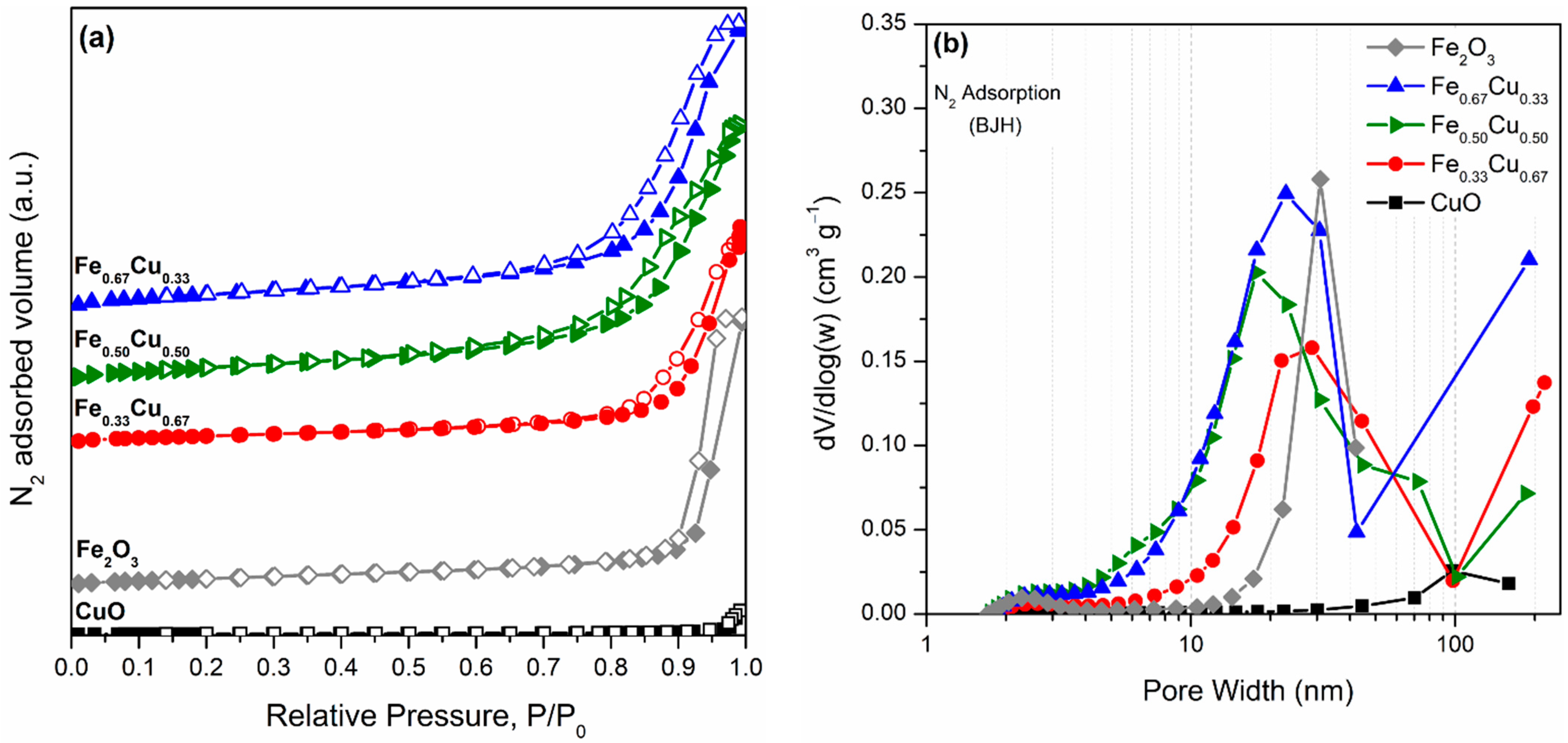
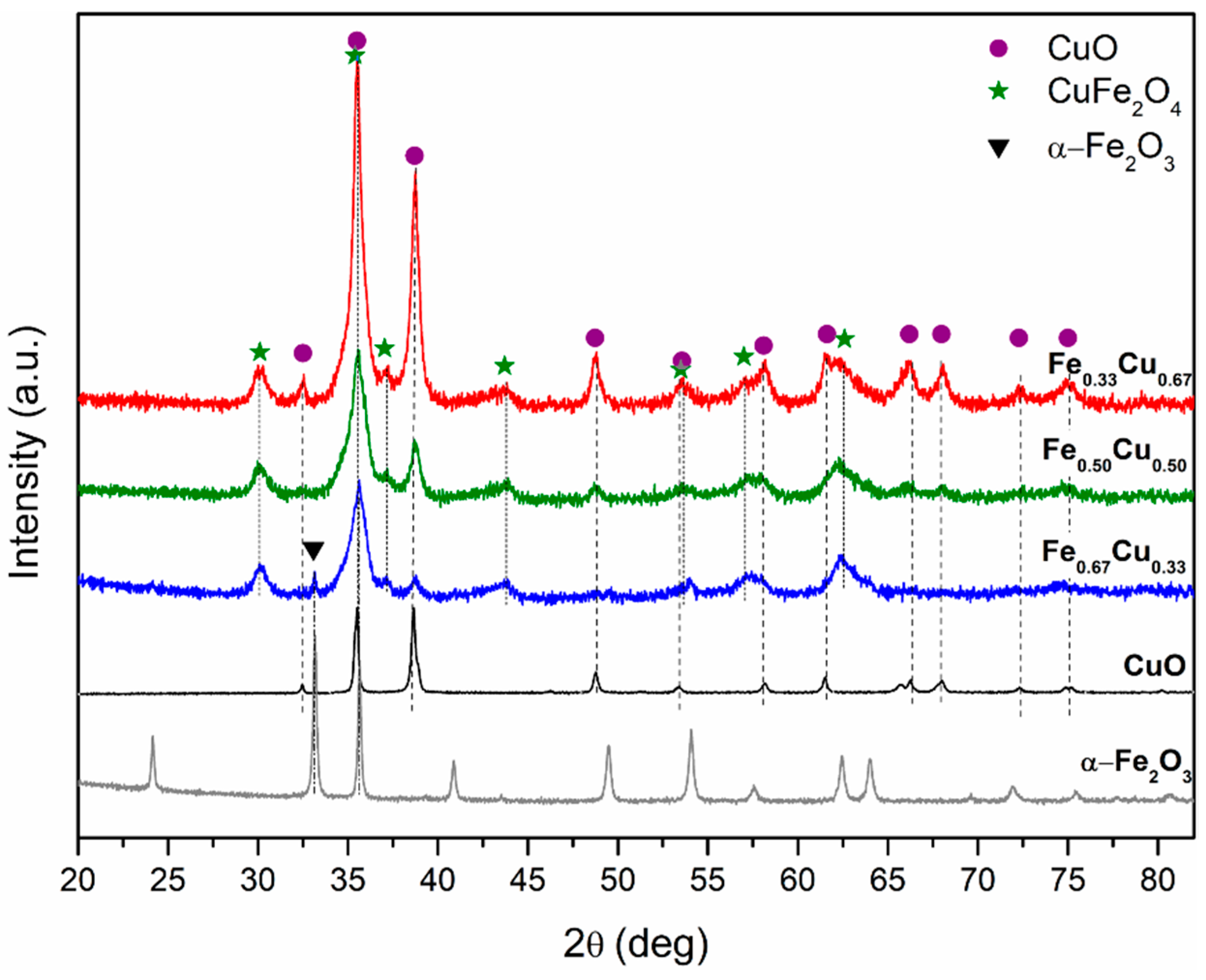
2.1. Textural and Structural Properties of FexCu1−x Oxide Catalysts
2.2. Reducibility of FexCu1−x Oxide Catalysts
2.3. XPS Measurements
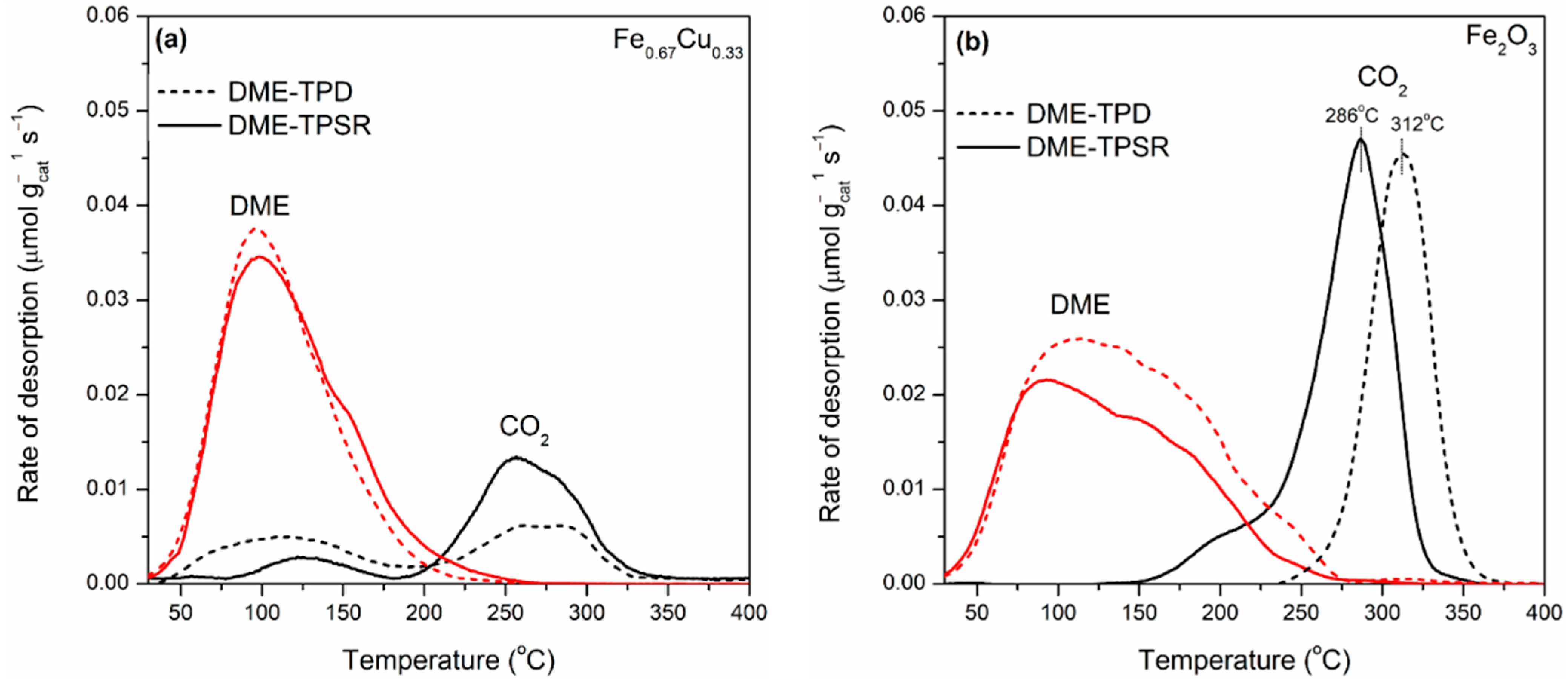
2.4. Temperature-Programmed Desorption and Surface Reaction of DME over FexCu1−x Catalysts (DME-TPD/TPSR)
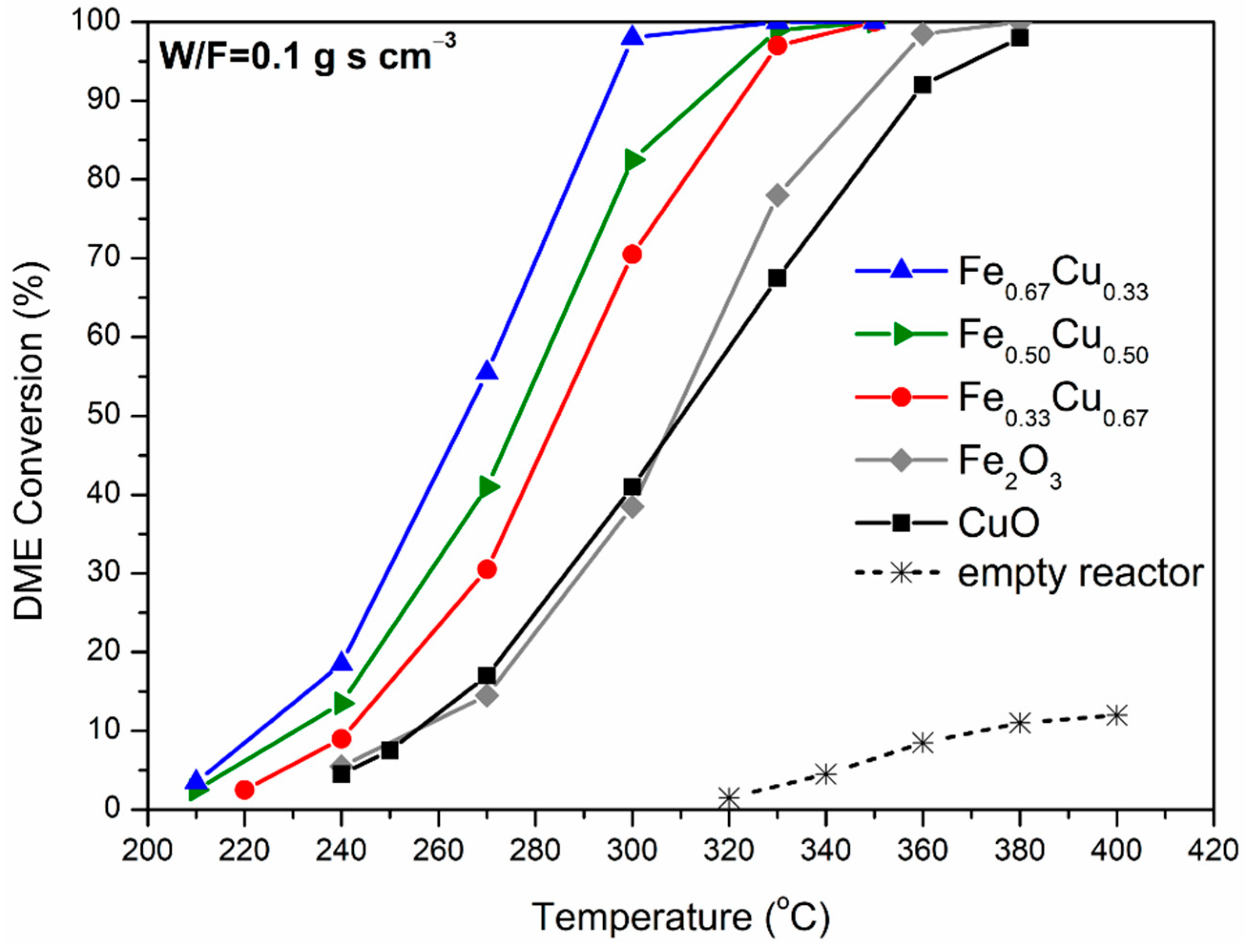
2.5. DME Oxidation Activity
3. Materials and Methods
3.1. Catalysts Synthesis
3.2. Catalyst Characterization
3.3. Catalyst Evaluation Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Generating Hydrogen-Rich Fuel-Cell Feeds from Dimethyl Ether (DME) Using Cu/Zn Supported on Various Solid-Acid Substrates. Appl. Catal. A Gen. 2006, 309, 210–223. [Google Scholar] [CrossRef]
- Han, D.; Yin, H.; Qian, E.; Ye, L.; Liu, D. Pyrolysis and Catalysis of Dimethyl Ether in a Flow Reactor. Fuel 2020, 263, 116700. [Google Scholar] [CrossRef]
- Arcoumanis, C.; Bae, C.; Crookes, R.; Kinoshita, E. The Potential of Di-Methyl Ether (DME) as an Alternative Fuel for Compression-Ignition Engines: A Review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Mihai, O.; Fathali, A.; Auvray, X.; Olsson, L. DME, Propane and CO: The Oxidation, Steam Reforming and WGS over Pt/Al2O3. The Effect of Aging and Presence of Water. Appl. Catal. B Environ. 2014, 160–161, 480–491. [Google Scholar] [CrossRef]
- Cheng, G.; Yu, L.; He, B.; Sun, M.; Zhang, B.; Ye, W.; Lan, B. Catalytic Combustion of Dimethyl Ether over α-MnO2 Nanostructures with Different Morphologies. Appl. Surf. Sci. 2017, 409, 223–231. [Google Scholar] [CrossRef]
- Sun, M.; Yu, L.; Ye, F.; Diao, G.; Yu, Q.; Hao, Z.; Zheng, Y.; Yuan, L. Transition Metal Doped Cryptomelane-Type Manganese Oxide for Low-Temperature Catalytic Combustion of Dimethyl Ether. Chem. Eng. J. 2013, 220, 320–327. [Google Scholar] [CrossRef]
- Tabakova, T.; Kolentsova, E.; Dimitrov, D.; Ivanov, K.; Manzoli, M.; Venezia, A.M.; Karakirova, Y.; Petrova, P.; Nihtianova, D.; Avdeev, G. CO and VOCs Catalytic Oxidation Over Alumina Supported Cu–Mn Catalysts: Effect of Au or Ag Deposition. Top. Catal. 2017, 60, 110–122. [Google Scholar] [CrossRef]
- Huang, H.; Xu, Y.; Feng, Q.; Leung, D.Y.C. Low Temperature Catalytic Oxidation of Volatile Organic Compounds: A Review. Catal. Sci. Technol. 2015, 5, 2649–2669. [Google Scholar] [CrossRef]
- Santos, V.P.; Carabineiro, S.A.C.; Tavares, P.B.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. Oxidation of CO, Ethanol and Toluene over TiO2 Supported Noble Metal Catalysts. Appl. Catal. B Environ. 2010, 99, 198–205. [Google Scholar] [CrossRef]
- Scirè, S.; Liotta, L.F. Supported Gold Catalysts for the Total Oxidation of Volatile Organic Compounds. Appl. Catal. B Environ. 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Papaefthimiou, P.; Ioannides, T.; Verykios, X.E. Performance of Doped Pt/TiO2 (W6+) Catalysts for Combustion of Volatile Organic Compounds (VOCs). Appl. Catal. B Environ. 1998, 15, 75–92. [Google Scholar] [CrossRef]
- Liotta, L.F. Catalytic Oxidation of Volatile Organic Compounds on Supported Noble Metals. Appl. Catal. B Environ. 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Ishikawa, A.; Iglesia, E. Bifunctional Pathways Mediated by Pt Clusters and Al2O3 in the Catalytic Combustion of Dimethyl Ether. Chem. Commun. 2007, 28, 2992–2993. [Google Scholar] [CrossRef]
- Solymosi, F.; Cserényi, J.; Ovári, L. A comparative study of the complete oxidation of dimethyl ether on supported group VIII metals. Catal. Lett. 1997, 44, 89–93. [Google Scholar] [CrossRef]
- Idakiev, V.; Dimitrov, D.; Tabakova, T.; Ivanov, K.; Yuan, Z.-Y.; Su, B.-L. Catalytic Abatement of CO and Volatile Organic Compounds in Waste Gases by Gold Catalysts Supported on Ceria-Modified Mesoporous Titania and Zirconia. Chin. J. Catal. 2015, 36, 579–587. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Liu, Z.-Q.; Haruta, M.; Shen, W. Low-Temperature Oxidation of CO Catalysed by Co3O4 Nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef]
- Krämer, M.; Schmidt, T.; Stöwe, K.; Maier, W.F. Structural and Catalytic Aspects of Sol–Gel Derived Copper Manganese Oxides as Low-Temperature CO Oxidation Catalyst. Appl. Catal. A Gen. 2006, 302, 257–263. [Google Scholar] [CrossRef]
- Chen, X.; Carabineiro, S.A.C.; Bastos, S.S.T.; Tavares, P.B.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Exotemplated Copper, Cobalt, Iron, Lanthanum and Nickel Oxides for Catalytic Oxidation of Ethyl Acetate. J. Environ. Chem. Eng. 2013, 1, 795–804. [Google Scholar] [CrossRef]
- Delimaris, D.; Ioannides, T. VOC Oxidation over CuO–CeO2 Catalysts Prepared by a Combustion Method. Appl. Catal. B Environ. 2009, 89, 295–302. [Google Scholar] [CrossRef]
- Soltan, W.B.; Sun, J.; Wang, W.; Song, Z.; Zhao, X.; Mao, Y.; Zhang, Z. Discovering the Key Role of MnO2 and CeO2 Particles in the Fe2O3 Catalysts for Enhancing the Catalytic Oxidation of VOC: Synergistic Effect of the Lattice Oxygen Species and Surface-Adsorbed Oxygen. Sci. Total Environ. 2022, 819, 152844. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Jirsak, T.; Pérez, M.; Chaturvedi, S.; Kuhn, M.; González, L.; Maiti, A. Studies on the Behavior of Mixed-Metal Oxides and Desulfurization: Reaction of H2S and SO2 with Cr2O3 (0001), MgO(100), and CrxMg1-xO(100). J. Am. Chem. Soc. 2000, 122, 12362–12370. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; Di Carlo, G.; Pantaleo, G.; Deganello, G.; Boreave, A.; Giroir-Fendler, A. Catalytic Removal of Toluene over Co3O4–CeO2 Mixed Oxide Catalysts: Comparison with Pt/Al2O3. Catal. Lett. 2009, 127, 270–276. [Google Scholar] [CrossRef]
- Shah, P.M.; Burnett, J.W.H.; Morgan, D.J.; Davies, T.E.; Taylor, S.H. Ceria–Zirconia Mixed Metal Oxides Prepared via Mechanochemical Grinding of Carbonates for the Total Oxidation of Propane and Naphthalene. Catalysts 2019, 9, 475. [Google Scholar] [CrossRef] [Green Version]
- Jirátová, K.; Kovanda, F.; Ludvíková, J.; Balabánová, J.; Klempa, J. Total Oxidation of Ethanol over Layered Double Hydroxide-Related Mixed Oxide Catalysts: Effect of Cation Composition. Catal. Today 2016, 277, 61–67. [Google Scholar] [CrossRef]
- Neha; Prasad, R.; Singh, S.V. Catalytic Abatement of CO, HCs and Soot Emissions over Spinel-Based Catalysts from Diesel Engines: An Overview. J. Environ. Chem. Eng. 2020, 8, 103627. [Google Scholar] [CrossRef]
- Davari, E.; Ivey, D.G. Bifunctional Electrocatalysts for Zn–Air Batteries. Sustain. Energy Fuels 2018, 2, 39–67. [Google Scholar] [CrossRef]
- Manos, D.; Miserli, K.; Konstantinou, I. Perovskite and Spinel Catalysts for Sulfate Radical-Based Advanced Oxidation of Organic Pollutants in Water and Wastewater Systems. Catalysts 2020, 10, 1299. [Google Scholar] [CrossRef]
- Zhao, Q.; Yan, Z.; Chen, C.; Chen, J. Spinels: Controlled Preparation, Oxygen Reduction/Evolution Reaction Application, and Beyond. Chem. Rev. 2017, 117, 10121–10211. [Google Scholar] [CrossRef]
- Ren, L.; Zhong, Y.; Xu, J.; Chen, J.; Zou, T.; Liao, X.-L.; Chen, Z.-F.; Yu, L. Nano Fe3-xCuxO4 as the Heterogeneous Catalyst in an Advanced Oxidation Process for Excellent Peroxymonosulfate Activation toward Climbazole Degradation. Chem. Eng. J. 2022, 439, 135553. [Google Scholar] [CrossRef]
- Yan, K.; Wu, X.; An, X.; Xie, X. Facile Synthesis and Catalytic Property of Spinel Ferrites by a Template Method. J. Alloy Compd. 2013, 552, 405–408. [Google Scholar] [CrossRef]
- Ren, Y.; Lin, L.; Ma, J.; Yang, J.; Feng, J.; Fan, Z. Sulfate Radicals Induced from Peroxymonosulfate by Magnetic Ferrospinel MFe2O4 (M = Co, Cu, Mn, and Zn) as Heterogeneous Catalysts in the Water. Appl. Catal. B Environ. 2015, 165, 572–578. [Google Scholar] [CrossRef]
- Manikandan, A.; Sridhar, R.; Arul Antony, S.; Ramakrishna, S. A Simple Aloe Vera Plant-Extracted Microwave and Conventional Combustion Synthesis: Morphological, Optical, Magnetic and Catalytic Properties of CoFe2O4 Nanostructures. J. Mol. Struct. 2014, 1076, 188–200. [Google Scholar] [CrossRef]
- Liu, P.; He, H.; Wei, G.; Liang, X.; Qi, F.; Tan, F.; Tan, W.; Zhu, J.; Zhu, R. Effect of Mn Substitution on the Promoted Formaldehyde Oxidation over Spinel Ferrite: Catalyst Characterization, Performance and Reaction Mechanism. Appl. Catal. B Environ. 2016, 182, 476–484. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Mountapmbeme Kouotou, P.; El Kasmi, A.; Tchoua Ngamou, P.H.; Kohse-Höinghaus, K.; Vieker, H.; Beyer, A.; Gölzhäuser, A. Low-Temperature Deep Oxidation of Olefins and DME over Cobalt Ferrite. Proc. Combust. Inst. 2015, 35, 2207–2214. [Google Scholar] [CrossRef]
- Djinović, P.; Ristić, A.; Žumbar, T.; Dasireddy, V.D.B.C.; Rangus, M.; Dražić, G.; Popova, M.; Likozar, B.; Zabukovec Logar, N.; Novak Tušar, N. Synergistic Effect of CuO Nanocrystals and Cu-Oxo-Fe Clusters on Silica Support in Promotion of Total Catalytic Oxidation of Toluene as a Model Volatile Organic Air Pollutant. Appl. Catal. B Environ. 2020, 268, 118749. [Google Scholar] [CrossRef]
- Tu, Y.-J.; Chang, C.-K.; You, C.-F. Combustion of Isopropyl Alcohol Using a Green Manufactured CuFe2O4. J. Hazard. Mater. 2012, 229–230, 258–264. [Google Scholar] [CrossRef]
- Amini, E.; Rezaei, M. Preparation of Mesoporous Fe-Cu Mixed Metal Oxide Nanopowder as Active and Stable Catalyst for Low-Temperature CO Oxidation. Chin. J. Catal. 2015, 36, 1711–1718. [Google Scholar] [CrossRef]
- Popescu, I.; Boudjemaa, A.; Helaili, N.; Bessekhouad, Y.; Tudorache, M.; Bachari, K.; Marcu, I.-C. Study of the Electrical and Catalytic Properties of Spinels with CuFe2−xMnxO4 Composition (X = 0, 0.4, 0.8, 1.6 and 2). Appl. Catal. A Gen. 2015, 504, 29–36. [Google Scholar] [CrossRef]
- Liu, H.; Liu, L.; Wei, L.; Chu, B.; Qin, Z.; Jin, G.; Tong, Z.; Dong, L.; Li, B. Preparation of Three-Dimensionally Ordered Macroporous MFe2O4 (M = Co, Ni, Cu) Spinel Catalyst and Its Simultaneous Catalytic Application in CO Oxidation and NO + CO Reaction. Fuel 2020, 272, 117738. [Google Scholar] [CrossRef]
- Rezlescu, N.; Rezlescu, E.; Popa, P.D.; Popovici, E.; Doroftei, C.; Ignat, M. Preparation and Characterization of Spinel-Type MeFe2O4 (Me = Cu, Cd, Ni and Zn) for Catalyst Applications. Mater. Chem. Phys. 2013, 137, 922–927. [Google Scholar] [CrossRef]
- Tsoncheva, T.; Manova, E.; Velinov, N.; Paneva, D.; Popova, M.; Kunev, B.; Tenchev, K.; Mitov, I. Thermally Synthesized Nanosized Copper Ferrites as Catalysts for Environment Protection. Catal. Commun. 2010, 12, 105–109. [Google Scholar] [CrossRef]
- Yeste, M.P.; Vidal, H.; García-Cabeza, A.L.; Hernández-Garrido, J.C.; Guerra, F.M.; Cifredo, G.A.; González-Leal, J.M.; Gatica, J.M. Low Temperature Prepared Copper-Iron Mixed Oxides for the Selective CO Oxidation in the Presence of Hydrogen. Appl. Catal. A Gen. 2018, 552, 58–69. [Google Scholar] [CrossRef]
- Luo, M.-F.; Zhong, Y.-J.; Yuan, X.-X.; Zheng, X.-M. TPR and TPD Studies of CuOCeO2 Catalysts for Low Temperature CO Oxidation. Appl. Catal. A Gen. 1997, 162, 121–131. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Song, L.; Wang, Y.; He, C.; Wang, Z.; Cui, L. High and Stable Catalytic Activity of Ag/Fe 2 O 3 Catalysts Derived from MOFs for CO Oxidation. Mol. Catal. 2018, 447, 80–89. [Google Scholar] [CrossRef]
- Perezalonso, F.; Meliancabrera, I.; Lopezgranados, M.; Kapteijn, F.; Fierro, J. Synergy of FexCe1−xO2 Mixed Oxides for N2O Decomposition. J. Catal. 2006, 239, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Espinós, J.P.; Morales, J.; Barranco, A.; Caballero, A.; Holgado, J.P.; González-Elipe, A.R. Interface Effects for Cu, CuO, and Cu2O Deposited on SiO2 and ZrO2. XPS Determination of the Valence State of Copper in Cu/SiO2 and Cu/ZrO2 Catalysts. J. Phys. Chem. B 2002, 106, 6921–6929. [Google Scholar] [CrossRef]
- Kundakovic, L.; Flytzani-Stephanopoulos, M. Reduction Characteristics of Copper Oxide in Cerium and Zirconium Oxide Systems. Appl. Catal. A Gen. 1998, 171, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Hayes, P. Analysis of XPS Spectra of Fe2+ and Fe3+ Ions in Oxide Materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Weiss, W.; Ranke, W. Surface Chemistry and Catalysis on Well-Defined Epitaxial Iron-Oxide Layers. Prog. Surf. Sci. 2002, 70, 1–151. [Google Scholar] [CrossRef] [Green Version]
- Wen, N.; Su, Y.; Deng, W.; Zhou, H.; Zhao, B. Selective Catalytic Reduction of NO with C3H6 over CuFe-Containing Catalysts Derived from Layered Double Hydroxides. Fuel 2021, 283, 119296. [Google Scholar] [CrossRef]
- Dong, X.; Ren, B.; Sun, Z.; Li, C.; Zhang, X.; Kong, M.; Zheng, S.; Dionysiou, D.D. Monodispersed CuFe2O4 Nanoparticles Anchored on Natural Kaolinite as Highly Efficient Peroxymonosulfate Catalyst for Bisphenol A Degradation. Appl. Catal. B Environ. 2019, 253, 206–217. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Chen, X.; Martynyuk, O.; Bogdanchikova, N.; Avalos-Borja, M.; Pestryakov, A.; Tavares, P.B.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Gold Supported on Metal Oxides for Volatile Organic Compounds Total Oxidation. Catal. Today 2015, 244, 103–114. [Google Scholar] [CrossRef]
- Saqer, S.M.; Kondarides, D.I.; Verykios, X.E. Catalytic Oxidation of Toluene over Binary Mixtures of Copper, Manganese and Cerium Oxides Supported on γ-Al2O3. Appl. Catal. B Environ. 2011, 103, 275–286. [Google Scholar] [CrossRef]
- Durán, F.G.; Barbero, B.P.; Cadús, L.E.; Rojas, C.; Centeno, M.A.; Odriozola, J.A. Manganese and Iron Oxides as Combustion Catalysts of Volatile Organic Compounds. Appl. Catal. B Environ. 2009, 92, 194–201. [Google Scholar] [CrossRef]
- Lutterotti, L.; Bortolotti, M.; Ischia, G.; Lonardelli, I.; Wenk, H.-R. Rietveld Texture Analysis from Diffraction Images. Z. Kristallogr. Suppl. 2007, 26, 125–130. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m2 g−1) | Pore Volume (cm3 g−1) | Pore Diameter (nm) | Phase Composition | dcryst (nm) | H2 Consumption (mmol gcat−1) |
|---|---|---|---|---|---|---|
| CuO | 1.5 | 0.013 | 9 | 100% (CuO) | 27.3 | 12.3 (12.6) * |
| Fe0.33Cu0.67 | 17 | 0.121 | 24 | 49.9% (CuFe2O4), 50.1% (CuO) | 14 (CuFe2O4), 17.7 (CuO) | 14.9 |
| Fe0.50Cu0.50 | 31 | 0.147 | 17 | 77.3% (CuFe2O4), 22.7% (CuO) | 10.4 (CuFe2O4), 16.6 (CuO) | 12.4 |
| Fe0.67Cu0.33 | 33 | 0.163 | 19 | 90.4% (CuFe2O4), 4.3% (CuO), 5.3% (Fe2O3) | 12.4 (CuFe2O4), 20.9 (CuO), 62.4 (Fe2O3) | 11.3 |
| Fe2O3 | 17 | 0.147 | 34 | 100% (α-Fe2O3) | 39.8 | 13.3 (18.8) ** |
| Catalyst | Fe/(Fe + Cu) Atomic Ratio | BE (eV) | ||||||
|---|---|---|---|---|---|---|---|---|
| Nominal | XPS | Cu 2p | Fe 2p | O 1s | Olatt./Oads. | |||
| 2p3/2 | 2p1/2 | 2p3/2 | 3p1/2 | |||||
| CuO | 0 | 0 | 933.8 | 953.7 | - | 529.5 | 1.96 | |
| Fe0.33Cu0.67 | 0.33 | 0.31 | 933.8 | 953.7 | 710.9 | 724.4 | 529.5 | 1.13 |
| Fe0.50Cu0.50 | 0.50 | 0.44 | 933.7 | 953.5 | 710.7 | 724.4 | 529.9 | 10.23 |
| Fe0.67Cu0.33 | 0.67 | 0.61 | 933.7 | 953.7 | 710.8 | 724.3 | 530.0 | 9.24 |
| Fe2O3 | 1 | 1 | - | 711.0 | 724.4 | 529.9 | 8.15 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyrnioti, M.; Ioannides, T. Dimethyl Ether Oxidation over Copper Ferrite Catalysts. Catalysts 2022, 12, 604. https://doi.org/10.3390/catal12060604
Smyrnioti M, Ioannides T. Dimethyl Ether Oxidation over Copper Ferrite Catalysts. Catalysts. 2022; 12(6):604. https://doi.org/10.3390/catal12060604
Chicago/Turabian StyleSmyrnioti, Maria, and Theophilos Ioannides. 2022. "Dimethyl Ether Oxidation over Copper Ferrite Catalysts" Catalysts 12, no. 6: 604. https://doi.org/10.3390/catal12060604
APA StyleSmyrnioti, M., & Ioannides, T. (2022). Dimethyl Ether Oxidation over Copper Ferrite Catalysts. Catalysts, 12(6), 604. https://doi.org/10.3390/catal12060604