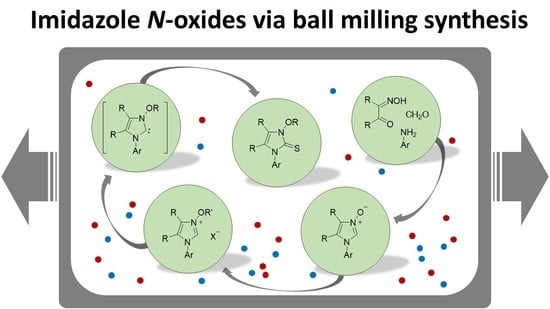
3.2. Methods
3.2.1. Synthesis of Imidazole N-Oxides 1a–1m
Method A: α-Hydroxyiminoketone 7a or 7b (1.1 mmol) and formaldimine 8 (1.0 mmol) were dissolved in glacial acetic acid (3 mL) and the mixture was stirred for 12 h at room temperature. After the concentrated aq. HCl (0.5 mL) was added dropwise, the solvent was evaporated under reduced pressure and the residue was dissolved in MeOH (5 mL). Then, excess solid NaHCO3 (1.0 g) was added and stirring was continued for 30 min. In order to remove NaCl the mixture was diluted with CH2Cl2 (10 mL), filtered, and the solvents were removed. Crude products 1c–1i were filtered through a short silica gel plug (AcOEt gradient AcOEt/MeOH 1:1). If not stated otherwise, the resulting product was triturated with Et2O, the precipitated imidazole N-oxide 1 was filtered, washed with additional portions of Et2O (2 × 5 mL) and air-dried.
Method B: α-Hydroxyiminoketone 7a or 7b (1.1 mmol), formaldimine 8 (1.0 mmol), a small amount of glacial acetic acid (0.1 mL, 0.175 mmol), and a zirconium ball (∅ 5.0 mm) were placed in the zirconium mechanochemical vial, and the mixture was ball-milled for 0.5 h at 25 Hz. The resulting material was dissolved in MeOH (5 mL), concentrated aq. HCl (0.5 mL) was added followed by solid NaHCO3 (1.0 g) and the mixture was magnetically stirred for 20 min. After the mixture was diluted with CH2Cl2 (10 mL) and filtered, the solvents were removed in vacuo. Crude N(1)-aryl imidazole N(3)-oxides 1c–1i were filtered through a short silica gel plug (AcOEt gradient AcOEt/MeOH 1:1). If not stated otherwise, the resulting material was triturated with Et2O, the precipitated imidazole N-oxide 1 was filtered and washed with additional portions of Et2O (2 × 5 mL).
Method C: α-Hydroxyiminoketone 7a or 7b (1.1 mmol), paraformaldehyde (30 mg, 1.2 mmol), the appropriate amine (1.0 mmol), glacial acetic acid (0.1 mL, 0.175 mmol), and a zirconium ball (∅ 5.0 mm) were placed in the zirconium mechanochemical vial and the mixture was ball-milled for 45 min at 25 Hz. Product 1 was isolated following the general work-up described for Method B.
1-Benzyl-4,5-dimethylimidazole 3-oxide (
1a):
Method A, 131 mg (65%), colorless crystals, m.p. 192−195 °C (ref. [
32], m.p. 199−201 °C);
Method B, 186 mg (92%);
Method C, 133 mg (68%).
1H NMR (600 MHz, CDCl
3):
δ 2.03, 2.14 (2 s, 6 H, 2 Me), 4.93 (s, 2 H, CH
2), 7.02–7.05, 7.26–7.33 (2 m, 2 H, 3 H), 7.74 (s, 1 H, C(2)H).
1-Benzyl-4,5-diphenylimidazole 3-oxide (
1b):
Method A, 202 mg (62%), colorless crystals, m.p. 183−185 °C (ref. [
32], m.p. 176−178 °C);
Method B, 300 mg (92%);
Method C, 270 mg (83%).
1H NMR (600 MHz, CDCl
3):
δ 4.98 (s, 2 H, CH
2), 7.08–7.10, 7.21–7.25, 7.28–7.32, 7.34–7.37, 7.38–7.42, 7.43–7.46, 7.56–7.59 (7 m, 2 H, 2 H, 3 H, 3 H, 2 H, 1 H, 2 H), 8.01 (s, 1 H, C(2)H).
1-Phenyl-4,5-dimethylimidazole 3-oxide (1c): Method A, 171 mg (91%), colorless crystals, m.p. 183−185 °C (CH2Cl2/i-Pr2O); Method B, 148 mg (79%); Method C, 132 mg (70%). 1H NMR (600 MHz, CDCl3): δ 2.09, 2.25 (2 s, 3 H each, 2 Me), 7.23–7.26, 7.45–7.51 (2 m, 2 H, 3 H), 7.91 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 7.5, 9.6, 121.7, 124.5, 126.0*, 127.7, 129.4, 130.0*, 135.2; *higher intensity. IR (neat): ν 3049, 2929, 1595, 1491, 1379, 1350, 1330, 1200, 816, 764, 697 cm–1. ESI-MS (m/z): 211 (20, [M+Na]+), 189 (100, [M+H]+). Elemental analysis for C11H12N2O (188.2): calculated, C 70.19, H 6.43, N 14.88; found, C 70.19, H 6.38, N 15.04.
1,4,5-Triphenylimidazole 3-oxide (1d): Method A, 299 mg (96%), colorless crystals, m.p. 208−210 °C (CH2Cl2/i-Pr2O); Method B, 298 mg (96%); Method C, 243 mg (78%). 1H NMR (600 MHz, CDCl3): δ 6.98–7.01, 7.09–7.12, 7.15–7.19, 7.23–7.36, 7.52–7.55 (5 m, 2 H, 2 H, 2 H, 7 H, 2 H), 8.27 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 125.9, 126.4, 126.6, 126.9, 127.1, 128.3, 128.6, 128.7, 129.0, 129.1, 129.7, 130.2, 130.6, 131.3, 134.9. IR (neat): ν 3071, 1506, 1364, 1223, 1163, 842, 760, 697 cm–1. ESI-MS (m/z): 335 (50, [M+Na]+), 313 (100, [M+H]+). Elemental analysis for C21H16N2O · 1.3H2O (336.1): calculated, C 74.98, H 5.59, N 8.33; found, C 75.12, H 5.35, N 8.68.
1-(4-Methoxyphenyl)-4,5-dimethylimidazole 3-oxide (1e): Method A, 107 mg (49%), colorless crystals, m.p. 82−84 °C (CH2Cl2/i-Pr2O); Method B, 124 mg (57%); Method C, 172 mg (79%). 1H NMR (600 MHz, CDCl3): δ 1.98, 2.16 (2 s, 3 H each, 2 Me), 3.78 (s, 3 H, OMe), 6.90–6.93, 7.10–7.12 (2 m, 2 H each), 7.85 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 7.4, 9.3, 55.6, 114.9, 122.1, 124.7, 127.0, 127.3, 127.5, 160.1. IR (neat): ν 3269, 3116, 3053, 2967, 2930, 2840, 1510, 1297, 1252, 1178, 1111, 1029, 846, cm–1. ESI-MS (m/z): 219 (100, [M+H]+). Elemental analysis for C12H14N2O2 · 2H2O (254.1): calculated, C 56.68, H 7.14, N 11.02; found, C 56.21, H 6.87, N 11.48.
1-(4-Methoxyphenyl)-4,5-diphenylimidazole 3-oxide (1f): Method A, 233 mg (68%), colorless crystals, m.p. 165−167 °C (CH2Cl2/i-Pr2O); Method B, 202 mg (59%); Method C, 239 mg (70%). 1H NMR (600 MHz, CDCl3): δ 3.78 (s, 3 H, Ome), 6.82–6.85, 6.99–7.06, 7.18–7.22, 7.24–7.32, 7.55–7.57 (5 m, 2 H, 4 H, 2 H, 4 H, 2 H), 8.16 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 55.6, 126.5, 114.7, 127.3, 128.2, 128.4, 128.6, 128.9, 130.1, 130.6, 126.8, 127.0, 127.3, 127.7, 131.1, 159.8. IR (neat): ν 3049, 2937, 1505, 1439, 1368, 1293, 1215, 1025, 835, 760, 693 cm–1. ESI-MS (m/z): 365 (15, [M+Na]+), 343 (100, [M+H]+). Elemental analysis for C22H18N2O2 · 1.5H2O (369.2): calculated, C 71.53, H 5.73, N 7.58; found, C 71.16, H 5.42, N 7.46.
1-(4-Bromophenyl)-4,5-dimethylimidazole 3-oxide (1g): Method A, 164 mg (42%), colorless crystals, m.p. 166−168 °C (CH2Cl2/i-Pr2O); Method B, 152 mg (39%); Method C, 250 mg (64%). 1H NMR (600 MHz, CDCl3): δ 2.07, 2.22 (2 s, 3 H each, 2 Me), 7.11–7.13, 7.61–7.63 (2 m, 2 H each), 7.88 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 7.4, 9.5, 121.8, 123.4, 125.3, 127.5, 127.6, 133.1, 133.8. IR (neat): v 3100, 2926, 1485, 1381, 1351, 1202, 1073, 941, 840, 807 cm–1. ESI-MS (m/z): 291 (95, [M{81Br}+Na]+), 289 (49, [M{79Br}+Na]+), 269 (95, [M{81Br}+H]+), 267 (100, [M{79Br}+H]+). Elemental analysis for C11H11BrN2O · 0.75H2O (279.5): calculated, C 47.08, H 4.49, N 9.98; found, C 47.00, H 4.11, N 10.37.
1-(4-Fluorophenyl)-4,5-dimethylimidazole 3-oxide (1h): Thick brown oil; Method A, 169 mg (82%); Method B, 161 mg (78%); Method C, 179 mg (87%). 1H NMR (600 MHz, CDCl3): δ 2.07, 2.25 (2 s, 3 H each, 2 Me), 7.17–7.22, 7.29–7.32 (2 m, 2 H each), 8.26 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 7.5, 9.5, 117.1 (d, 2JC–F = 23.3 Hz), 122.2, 125.6, 127.4, 128.1 (d, 3JC–F = 8.9 Hz), 130.8 (d, 4JC–F = 3.2 Hz), 162.9 (d, 1JC–F = 250.8 Hz). 19F NMR (565 MHz, CDCl3): δ −110.6 (mc). IR (neat): ν 3058, 3071, 2930, 1677, 1506, 1387, 1223, 1160, 1101, 831 cm–1. ESI-MS (m/z): 207 (100, [M+H]+). Elemental analysis for C11H11FN2O (206.1): calculated, C 64.07, H 5.38, N 13.58; found, C 64.09, H 5.40, N 13.52.
1-(4-Fluorophenyl)-4,5-diphenylimidazole 3-oxide (1i): Method A, 112 mg (34%), colorless crystals, m.p. 252−254 °C (Et2O); Method B, 191 mg (58%); Method C, 224 mg (68%). 1H NMR (600 MHz, CD3OD): δ 7.13–7.19, 7.25–7.28, 7.31–7.39, 7.49–7.51 (4 m, 4 H, 2 H, 6 H, 2 H), 8.79* (s, 1 H, C(2)H); *lower intensity due to partial H/D exchange. 13C NMR (151 MHz, CDCl3): δ 117.5 (d, 2JC–F = 23.6 Hz), 129.3, 129.5, 129.8, 130.0, 130.1*, 130.5, 131.5, 131.9, 132.1, 132.2 (d, 4JC–F = 3.2 Hz), 146.2 (d, 1JC–F = 248.8 Hz); *broadened absorption attributed to two ortho-CH groups of the N(1)-substituent; the 3JC-F cannot be determined. 19F NMR (565 MHz, CDCl3): δ −114.0 (mc). IR (neat): ν 3071, 1506, 1485, 1398, 1364, 1295, 1223, 1162, 1110, 1005, 842, 775, 760, 693 cm–1. ESI-MS (m/z): 331 (100, [M+H]+). Elemental analysis for C21H15FN2O (330.1): calculated C 76.35, H 4.58, N 8.48; found, C 76.38, H 4.61, N 8.59.
(
R)
-1-(1-Phenyl)ethyl-4,5-dimethylimidazole 3-oxide (
1j):
Method A, 204 mg (93%), colorless crystals, m.p. 230−232 °C (Et
2O) (ref. [
43], m.p. 224 °C (decomp.)), [α]
D20−149.1 (
c = 0.16, CHCl
3) (ref. [
43], [α]
D20 = −138.5 (
c = 1.00, MeOH));
Method B, 134 mg (62%), [α]
D20 = −166.0 (
c = 0.20, CHCl
3).
1H NMR (600 MHz, CDCl
3):
δ 1.76 (d,
J = 7.1 Hz, 3 H, Me), 1.98, 2.16 (2 s, 3 H each, 2 Me), 5.20 (q,
J = 7.1 Hz, 1 H, C
HMe), 7.04–7.06, 7.27–7.34 (2 m, 2 H, 3 H), 7.89 (s, 1 H, C(2)H).
(R)-1-(1-Phenyl)ethyl-4,5-diphenylimidazole 3-oxide (
1k):
Method A, 218 mg (64%), colorless crystals, m.p. 189−191 °C (Et
2O) (ref. [
43], m.p. 217 °C (decomp.)), [α]
D20 +57.6 (
c = 0.22, CHCl
3) (ref. [
43], [α]
D20 = +30.0 (
c = 1.00, MeOH));
Method B, 150 mg (44%), [α]
D20 +63.9 (
c = 0.42, CHCl
3);
Method C, 122 mg (29%), [α]
D20 +57.7 (
c = 0.20, CHCl
3).
1H NMR (600 MHz, CDCl
3):
δ 1.77 (d,
J = 7.1 Hz, 3 H, Me), 5.24 (q,
J = 7.1 Hz, 1 H, C
HMe), 7.05–7.07, 7.13–7.15, 7.21–7.25, 7.29–7.36, 7.39–7.42, 7.53–7.55 (6 m, 2 H, 2 H, 3 H, 5 H, 1 H, 2 H), 8.12 (s, 1 H, C(2)H).
(R,R)-trans-1,1-(Cyclohexane-1,2-diyl)bis(4,5-dimethylimidazole)-3,3′-dioxide (
1l): Reactions were carried out according to general protocols using 1.1 mmol of
7a and 0.25 mmol of
11 (
Methods A and
B) or 0.5 mmol of
10 (
Method C).
Method A, 182 mg (60%), colorless crystals, m.p. 208−211 °C (Et
2O) (ref. [
44], m.p. 210 °C (decomp.)), [α]
D20 = −241.9 (
c = 0.42, CHCl
3) (ref. [
44], [α]
D20−267.6 (
c = 1.00, MeOH));
Methods B and
C: no formation of
1l was observed.
1H NMR (600 MHz, CDCl
3):
δ 1.52–1.58 (m, 2 H), 1.82–1.96 (m, 4 H), 1.94, 2.03 (2 s, 6 H each, 4 Me), 2.09–2.14 (m, 2 H), 4.31 (m
c, 2 H), 8.56 (s, 2 H, 2 C(2)H).
(R,R)-trans-1,1-(Cyclohexane-1,2-diyl)bis(4,5-diphenylimidazole)-3,3′-dioxide (
1m): Following the general protocols 1.1 mmol of
7b and 0.25 mmol of
11 (
Methods A and
B) or 0.5 mmol of diamine
10 (
Method C) were used.
Method A, 265 mg (48%), colorless crystals, m.p. 210−212 °C (Et
2O) (ref. [
44], m.p. 209 °C (decomp.)), [α]
D20 = −21.0 (
c = 0.40, CHCl
3) (ref. [
44], [α]
D20 = +6.0 (
c = 1.02, MeOH)).
Methods B and
C: no formation of
1m was observed.
1H NMR (600 MHz, CDCl
3):
δ 1.28–2.48 (m, 8 H), 4.04 (m
c, 2 H), 7.05–7.41, 7.48–7.56 (2 m, 14 H, 6 H), 8.23 (s, 2 H, 2 C(2)H).
3.2.2. Synthesis of Enolizable Imidazole 2-Thiones 13a–13d
Method D: To a stirred solution of imidazole N-oxide 1 (0.5 mmol) in CH2Cl2 (2 mL), a solution of 2,2,4,4-tetramethylcyclobutane-1,3-dithione (12, 0.26 mmol) in CH2Cl2 (2 mL) was added at room temperature, and stirring was continued until the red color of the thioketone faded (typically ca. 10 min). After the solvent was removed in vacuo, the residue was triturated with petroleum ether (10 mL), and the crystalline imidazole-2-thione 13 was filtered and washed with additional portions of petroleum ether (2 × 3 mL) to give spectroscopically pure product.
Method E: A mixture of imidazole N-oxide 1 (0.5 mmol), dithione 12 (0.26 mmol), and a zirconium ball (∅ 5.0 mm) were placed in the zirconium mechanochemical vial and the mixture was ball-milled for 3 h at 25 Hz. The resulting material was triturated with petroleum ether (10 mL), and the crystalline precipitate of product 13 was filtered and washed with two portions of petroleum ether (2 × 3 mL).
4,5-Dimethyl-1-phenyl-1H-imidazole-2(3H)-thione (13a): Method D, 170 mg (91%), colorless solid, m.p. 225−227 °C (decomp.); Method E, 179 mg (96%). 1H NMR (600 MHz, DMSO-d6): δ 1.78, 2.03 (2 s, 3 H each, 2 Me), 7.28–7.30, 7.42–7.52 (2 m, 2 H, 3 H), 12.08 (s, 1 H, NH). 13C NMR (150 MHz, DMSO-d6): δ 8.9, 9.4, 119.6, 121.3, 128.4, 128.5, 129.0, 136.6, 160.7. IR (neat): v 3164, 3053, 2922, 2706, 1662, 1593, 1495, 1435, 1383, 1353, 1215, 1032, 805, 756 cm–1. ESI-MS (m/z): 227 (100, [M+H2O+Na]+).
1,4,5-Triphenyl-1H-imidazole-2(3H)-thione (13b): Method D, 312 mg (95%), colorless solid, m.p. 240−242 °C (decomp.); Method E, 262 mg (80%). 1H NMR (600 MHz, DMSO-d6): δ 7.15–7.39 (m, 15 H, 3 Ph), 13.04 (s, 1 H, NH). 13C NMR (150 MHz, DMSO-d6): δ 125.0, 126.7*, 127.4, 127.8, 128.1, 128.2, 128.53, 128.55, 128.6, 128.7, 128.8, 129.2, 130.9, 136.3, 163.0; *higher intensity. IR (neat): v 3041, 2915, 2732, 1595, 1491, 1371, 1260, 1178, 1070, 1025, 917, 831, 764 cm–1. ESI-MS (m/z): 351 (82, [M+Na]+), 329 (100, [M+H]+).
1-(4-Methoxyphenyl)-4,5-dimethyl-1H-imidazole-2(3H)-thione (13c): Method E, 108 mg (46%), colorless solid, m.p. 227−229 °C (decomp.). 1H NMR (600 MHz, DMSO-d6): δ 1.77, 2.02 (2 s, 3 H each, 2 Me), 3.80 (s, 3 H, OMe), 7.02–7.04, 7.17–7.19 (2 m, 2 H each), 12.02 (s, 1 H, NH). 13C NMR (150 MHz, DMSO-d6): δ 8.8, 9.4, 55.4, 114.1, 119.3, 121.6, 129.2, 129.6, 158.8, 160.8. IR (neat): v 1513, 1439, 1387, 1337, 1301, 1242, 1226, 1170, 1103, 1029, 999 cm–1. ESI-MS (m/z): 257 (58, [M+Na]+), 235 (100, [M+H]+), 203 (98).
1-(4-Fluorophenyl)-4,5-dimethyl-1H-imidazole-2(3H)-thione (13d): Method E, 100 mg (45%), colorless solid, m.p. 251−254 °C (decomp.). 1H NMR (600 MHz, DMSO-d6): δ 1.79, 2.03 (2 s, 3 H each, 2 Me), 7.32–7.37 (m, 4 H), 12.11 (s, 1 H, NH). 13C NMR (150 MHz, DMSO-d6): δ 8.8, 9.3, 115.9 (d, 2JC-F = 23.0 Hz), 119.6, 121.4, 130.7 (d, 3JC-F = 8.8 Hz), 132.8 (d, 4JC-F = 3.0 Hz), 160.9, 161.5 (d, 1JC-F = 245.3 Hz). 19F NMR (565 MHz, DMSO-d6): δ −113.3 (mc). IR (neat): v 3168, 3060, 2922, 2713, 1666, 1506, 1397, 1357, 1244, 1222, 1148, 1088, 1006, 846, 782 cm–1. ESI-MS (m/z): 245 (55, [M+Na]+), 223 (100, [M+H]+).
3.2.3. Synthesis of N-benzyloxy-imidazolium Salts 14a[PF6]–14d[PF6]
Method F: A solution of imidazole N-oxide 1 (1.0 mmol) and benzyl bromide (205 mg, 1.2 mmol) in CHCl3 (1 mL) was stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was washed with few portions of Et2O (3 × 5 mL). The resulting crude imidazolium bromide was dissolved in EtOH (2 mL) and a solution of NH4PF6 (196 mg, 1.2 mmol) in distilled water (1 mL) was added dropwise under vigorous stirring. After 5 min, the crystalline product 14[PF6] was filtered and dried under reduced pressure.
Method G: Imidazole N-oxide 1 (1.0 mmol), benzyl bromide (205.2 mg, 1.2 mmol), solid NH4PF6 (196 mg, 1.2 mmol), and a zirconium ball (∅ 5.0 mm) were placed in a mechanochemical vial and the mixture was ball-milled for 0.5 h at 25 Hz. The resulting material was dissolved in CH2Cl2 (5 mL), filtered, the solvent was removed, and the product 14[PF6] was washed with Et2O (4 × 5 mL) followed by drying under reduced pressure.
3-Benzyloxy-4,5-dimethyl-1-phenylimidazolium hexafluorophosphate (14a[PF6]): Method F, 322 mg (76%), colorless crystals, m.p. 160−163 °C (EtOH/H2O); Method G, 305 mg (72%). 1H NMR (600 MHz, DMSO-d6): δ 2.14, 2.26 (2 s, 3 H each, 2 Me), 5.52 (s, 2 H, Bn), 7.48–7.52, 7.56–7.58, 7.62–7.70 (3 m, 3 H, 2 H, 5 H), 9.96 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 6.8, 8.7, 83.6, 124.5, 124.9, 126.2, 128.9, 130.1, 130.2, 130.5, 130.7, 132.1 (br)*, 132.2, 133.2; *broadened absorption of C(2). IR (neat): ν 3161, 1607, 1539, 1498, 1461, 1424, 1372, 1331, 1208, 1111, 824, 760, 697 cm–1. ESI-MS (m/z): 279 (100, [M−PF6]+). Elemental analysis for C18H19F6N2OP (424.11): calculated C 50.95, H 4.51, N 6.60; found, C 50.67, H 4.53, N 6.74.
3-Benzyloxy-1,4,5-triphenylimidazolium hexafluorophosphate (14b[PF6]). Method F, 520 mg (95%), colorless crystals, m.p. 219−221 °C (EtOH/H2O); Method G, 356 mg (65%). 1H NMR (600 MHz, DMSO-d6): δ 5.35 (s, 2 H, Bn), 7.22–7.27, 7.33–7.46, 7.49–7.59 (3 m, 4 H, 11 H, 5 H), 10.41 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 84.1, 123.1, 124.4, 128.1, 128.9, 129.0, 129.2, 129.2, 129.4, 129.7, 130.09, 130.12, 130.25, 130.29, 130.5, 131.0, 131.6, 133.8 (br), 133.9. IR (neat): ν 3153, 1539, 1510, 1450, 1372, 1238, 1159, 891, 764, 697 cm–1. ESI-MS (m/z): 421 (100, [M+H2O−PF6]+).
3-Benzyloxy-1-(4-methoxyphenyl)-4,5-dimethylimidazolium hexafluorophosphate (14c[PF6]): Method F, 286 mg (63%), colorless crystals, m.p. 172−173 °C (EtOH/H2O); Method G, 285 mg (65%). 1H NMR (600 MHz, DMSO-d6): δ 2.11, 2.25 (2 s, 3 H each, 2 Me), 3.85 (s, 3 H, OMe), 5.49 (s, 2 H, Bn), 7.18–7.21, 7.48–7.57 (2 m, 2 H, 7 H) 9.83 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 6.8, 8.6, 55.8, 83.6, 115.0, 124.2, 125.2, 125.8, 127.7, 128.9, 130.2, 130.5, 132.1 (br), 132.2, 160.6. IR (neat): ν 3165, 1539, 1510, 1454, 1379, 1305, 1249, 1208, 1174, 1036, 951, 913, 828 cm–1. ESI–MS (m/z): 309 (100, [M−PF6]+). Elemental analysis for C19H21F6N2O2P (454.1): calculated, C 50.23, H 4.66, N 6.17; found, C 50.06, H 4.66, N 6.39.
3-Benzyloxy-1-(4-methoxyphenyl)-4,5-diphenylimidazolium hexafluorophosphate (14d[PF6]): Method F, 491 mg (85%), colorless crystals, m.p. 223−225 °C (EtOH/H2O); Method G, 474 mg (82%). 1H NMR (600 MHz, DMSO-d6): δ 3.78 (s, 3 H, OMe), 5.32 (s, 2 H, Bn), 7.05–7.08, 7.21–7.26, 7.33–7.35, 7.37–7.46, 7.49–7.55 (5 m, 2 H, 4 H, 2 H, 8 H, 3 H), 10.28 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 56.7, 84.0, 114.7, 123.2, 124.6, 125.9, 127.99, 128.02, 128.8, 128.9, 129.0, 129.4, 130.06, 130.13, 130.2, 130.3, 130.5, 131.0, 131.6, 133.6 (br), 160.4. IR (neat): ν 3159, 1513, 1446, 1383, 1301, 1252, 1215, 1174, 1067, 960, 828, 764, 697 cm–1. ESI–MS (m/z): 433 (100, [M−PF6]+). Elemental analysis for C29H25F6N2O2P (578.2): calculated, C 60.21, H 4.36, N 4.84; found, C 60,21, H 4.39, N 4.99.
3-Benzyloxy-1-(4-fluorophenyl)-4,5-dimethylimidazolium hexafluorophosphate (14e[PF6]): Method F, 349 mg (79%), colorless crystals, m.p. 178−180 °C (EtOH/H2O); Method G, 367 mg (83%). 1H NMR (600 MHz, DMSO-d6): δ 2.13, 2.26 (2 s, 3 H each, 2 Me), 5.50 (s, 2 H, Bn), 7.48–7.58, 7.70–7.73 (2 m, 7 H, 2 H), 9.91 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 6.7, 8.6, 83.7, 117.0 (d, 2JC–F = 23.4 Hz), 124.4, 125.2, 128.9 (d, 3JC–F = 9.4 Hz), 129.0, 129.5 (d, 4JC–F = 3.0 Hz), 130.2, 130.5, 132.2, 132.4 (br), 162.9 (d, 1JC–F = 248.4 Hz). 19F NMR (565 MHz, CDCl3): δ−70.1 (d, 1JP–F = 711.0 Hz, PF6),−109.9 (mc, C-F). IR (neat): ν 3166, 1547, 1510, 1372, 1238, 1215, 1160, 1103, 947, 910, 828, 757, 701 cm–1. ESI–MS (m/z): 297 (100, [M−PF6]+). Elemental analysis for C18H18F7N2OP (442.1): calculated, C 48.88, H 4.10, N 6.33; found, C 48.66, H 4.11, N 6.56.
3-Benzyloxy-1-(4-fluorophenyl)-4,5-diphenylimidazolium hexafluorophosphate (14f[PF6]): Method F, 402 mg (71%), colorless crystals, m.p. 225−227 °C (EtOH/H2O); Method G, 379 mg (67%). 1H NMR (600 MHz, DMSO-d6): δ 5.33 (s, 2 H, Bn), 7.21–7.27, 7.33–7.36, 7.38–7.46, 7.49–7.57 (4 m, 4 H, 2 H, 8 H, 5 H), 10.37 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 84.1, 116.8 (d, 2JC-F = 23.6 Hz), 123.1, 124.4, 128.1, 128.86, 128.88, 129.1, 129.2 (d, 3JC–F = 9.4 Hz), 129.3, 129.6 (d, 4JC-F = 2.8 Hz), 130.0, 130.1, 130.2, 130.3, 130.5, 131.0, 131.5, 133.9 (br), 162.6 (d, 1JC–F = 248.8 Hz). 19F NMR (565 MHz, CDCl3): δ−70.1 (d, 1JP–F = 711.6 Hz, PF6), −109.8 (mc, C–F). IR (neat): ν 3153, 1543, 1510, 1449, 1238, 1159, 831, 764, 701 cm–1. ESI–MS (m/z): 421 (100, [M−PF6]+). Elemental analysis for C28H22F7N2OP (566.1): calculated, C 59.37, H 3.91, N 4.95; found, C 59.28, H 3.98, N 5.22.
3.2.4. Synthesis of Non-Enolizable Imidazole-2-thiones 15 and 18 via Sulfurization of an Intermediate Carbene
Method H: To a solution of imidazolium salt 14c[PF6] or 17b[PF6] (1.0 mmol), elemental sulfur (70.4 mg, 2.4 mmol) in anhydrous pyridine (3 mL), Et3N (0.15 mL, 1.1 mmol) was added dropwise and the mixture was stirred overnight under inert atmosphere of Ar. After solvents were removed in vacuo, the resulting material was purified by preparative thin-layer chromatography (PLC: SiO2, CH2Cl2/MeOH 98:2) to give non-enolizable imidazole-2-thione 15 or 18, respectively.
MethodI: Imidazolium salt 14c[PF6] or 17b[PF6] (1.0 mmol), elemental sulfur (70.4 mg, 2.4 mmol), solid K2CO3 (415 mg, 3.0 mmol), and a zirconium ball (∅ 5.0 mm) were placed in a mechanochemical vial and the mixture was ball-milled for 3 h at 25 Hz. The resulting crude product 15 or 18, respectively, was purified on PLC as described for Method H.
1-Benzyloxy-3-(4-methoxyphenyl)-4,5-dimethyl-1H-imidazole-2(3H)-thione (15). Method H, 228 mg (67%), colorless oil; Method I, 146 mg (43%). 1H NMR (600 MHz, CDCl3): δ 1.80, 1.87 (2 s, 3 H each, 2 Me), 3.83 (s, 3 H, OMe), 5.46 (s, 2 H, Bn), 6.98–7.01, 7.19–7.22, 7.37–7.39, 7.51–7.53 (4 m, 2 H, 2 H, 3 H, 2 H). 13C NMR (151 MHz, CDCl3): δ 7.7, 9.8, 55.5, 78.1, 114.7, 118.5, 120.0, 128.63, 128.65, 129.3, 129.6, 130.4, 134.1, 158.3, 159.8. IR (neat): ν 2933, 2836, 1673, 1606, 1550, 1510, 1442, 1410, 1297, 1245, 1170, 1110, 1021, 831 cm–1. ESI–MS (m/z): 341 (100, [M+H]+).
1-Benzyl-3-(4-methoxyphenyl)-4,5-dimethyl-1H-imidazole-2(3H)-thione (18). Method H, 91 mg (28%), colorless oil; Method I, 176 mg (54%). 1H NMR (600 MHz, CDCl3): δ 1.86, 2.01 (2 s, 3 H each, 2 Me), 3.84 (s, 3 H, OMe), 5.43 (s, 2 H, Bn), 7.00–7.03, 7.23–7.27, 7.30–7.35 (3 m, 2 H, 3 H, 4 H). 13C NMR (151 MHz, CDCl3): δ 9.6, 9.8, 48.8, 55.5, 114.7, 121.5, 122.2, 127.3, 127.6, 128.7, 129.5, 129.9, 136.6, 159.7, 163.6. IR (neat): ν 1517, 1442, 1387, 1342, 1301, 1252, 1167, 1099, 1029, 995 cm–1. ESI-MS (m/z): 347 (100, [M+Na]+), 325 (26, [M+H]+).
3.2.5. Synthesis of 1,4,5-trisubstituted Imidazoles 16
To a solution of imidazole N-oxide 1 (1.0 mmol) in MeOH (2 mL) was added excess freshly prepared Raney nickel at room temperature and the vigorous stirring was continued until the starting N-oxide was fully consumed (typically ca. 30 min; TLC monitoring: SiO2, AcOEt/MeOH 1:1). The resulting mixture was filtered through a short pad of Celite® and the solvent was removed under reduced pressure to give spectroscopically pure imidazole 16. Solid products were recrystallized from a hexane/Et2O mixture.
1,4,5-Triphenyl-1H-imidazole (16a): 129 mg (64%); colorless crystals, m.p. 175–178 °C (Et2O/hexane). 1H NMR (600 MHz, CDCl3): δ 7.10–7.21, 7.24–7.29, 7.31–7.34, 7.53–7.55 (4 m, 5 H, 5 H, 3 H, 2 H), 7.78 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 126.0, 126.8, 127.4, 128.1, 128.25, 128.33, 128.7, 128.8, 129.4, 130.3, 131.0, 134.6, 136.6, 137.5, 139.1. IR (neat): ν 3112, 3056, 1599, 1502, 1442, 1371, 1271, 1245, 1129, 1074, 1025, 951, 913 cm–1. ESI-MS (m/z): 297 (100, [M+H]+).
1-(4-Methoxyphenyl)-4,5-dimethyl-1H-imidazole (16b): 145 mg (72%); brown oil. 1H NMR (600 MHz, CDCl3): δ 2.04, 2.21 (2 s, 3 H each, 2 Me), 3.84 (s, 3 H, OMe), 6.95–6.98, 7.15–7.17 (2 m, 2 H each), 7.46 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 9.1, 12.8, 55.7, 114.6, 123.4, 127.1, 129.9, 133.8, 135.3, 159.4. IR (neat): ν 2922, 2840, 1707, 1610, 1513, 1446, 1297, 1245, 1170, 1107, 1029, 943, 835, 801, 716, 671 cm–1. ESI-MS (m/z): 203 (100, [M+H]+).
1-(4-Methoxyphenyl)-4,5-diphenyl-1H-imidazole (16c): 326 mg (58%); colorless crystals, m.p. 182–184 °C (Et2O/hexane). 1H NMR (600 MHz, CDCl3): δ 3.79 (s, 3 H, OMe), 6.81–6.84, 7.03–7.05, 7.14–7.20, 7.23–7.29, 7.52–7.54 (5 m, 2 H, 2 H, 3 H, 5 H, 2 H), 7.73 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ 55.6, 114.4, 126.7, 127.26, 127.32, 128.2, 128.3, 128.7, 129.1, 129.5, 130.3, 130.9, 134.6, 137.7, 138.7, 159.2. IR (neat): ν 3041, 2956, 2904, 2836, 1886, 1599, 1530, 1439, 1301, 1249, 1174, 1111, 1070, 1029, 925, 917, 839, 772, 719 cm–1. ESI-MS (m/z): 327 (100, [M+H]+). Elemental analysis for C22H18N2O (326.1): calculated, C 80.96, H 5.56, N 8.58; found, C 80.75, H 5.76, N 8.41.
1-(4-Fluorophenyl)-4,5-diphenyl-1H-imidazole (16d): 314 (79%); colorless crystals, m.p. 172–173 °C (Et2O/hexane). 1H NMR (600 MHz, CDCl3): δ 7.00–7.04, 7.08–7.11, 7.13–7.15, 7.19–7.21, 7.24–7.31, 7.52–7.54 (6 m, 2 H, 2 H, 2 H, 1 H, 5 H, 2 H), 7.75 (s, 1 H, C(2)H). 13C NMR (151 MHz, CDCl3): δ116.4 (d, 2JC–F = 23.0 Hz), 126.9, 127.3, 127.7 (d, 3JC–F = 8.7 Hz), 128.36, 128.40, 128.8, 128.9, 130.0, 130.9, 132.7 (d, 4JC–F = 3.3 Hz), 134.4, 137.5, 139.1, 162.0 (d, 1JC–F = 248.5 Hz). 19F NMR (565 MHz, CDCl3): δ −113.0 (mc). IR (neat): ν 3064, 1599, 1510, 1485, 1372, 1219, 1185, 1101, 1066, 1025, 980, 910, 835, 809, 764, 695 cm–1. ESI-MS (m/z): 315 (110, [M+H]+). Elemental analysis for C21H15FN2 (314.1): calculated, C 80.24, H 4.81, N 8.91; found, C 80.14, H 4.79, N 8.94.
3.2.6. Synthesis of N-benzyl-imidazolium Salts 17a[PF6]–17d[PF6]
A mixture of imidazole 16 (1.0 mmol) and benzyl bromide (0.18 mL, 1.5 mmol) in MeCN (2 mL) was placed in a closed vessel and heated in a microwave reactor at 110 °C for 3 h. The mixture was cooled to room temperature, the solvent was removed and the resulting material was washed with Et2O (4 × 5 mL). The obtained crude imidazolium bromide was dissolved in EtOH (2 mL) and a solution of NH4PF6 (196 mg, 1.2 mmol) in distilled water (1 mL) was added dropwise under vigorous stirring. After 5 min, the precipitate of 17[PF6] was filtered and dried under reduced pressure.
1-Benzyl-3,4,5-triphenylimidazolium hexafluorophosphate (17a[PF6]): 425 mg (80%); colorless crystals, m.p. 224–227 °C (EtOH/H2O). 1H NMR (600 MHz, DMSO-d6): δ 5.45 (s, 2 H, Bn), 7.13–7.20, 7.22–7.34, 7.39–7.58 (3 m, 4 H, 8 H, 8 H), 9.97 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 50.7, 124.9, 125.0, 126.5, 127.9, 128.49, 128.51, 128.7, 128.9, 129.7, 129.8, 130.35, 130.36, 130.7, 130.9, 131.4, 132.0, 133.70, 133.73, 137.1 (br). IR (neat): ν 1595, 1558, 1498, 1450, 1353, 1219, 1170, 1081, 1025, 924, 831, 764, 705 cm–1. ESI-MS (m/z): 387 (100, [M−PF6]+).
1-Benzyl-3-(4-methoxyphenyl)-4,5-dimethylimidazolium hexafluorophosphate (17b[PF6]): 399 mg (91%); colorless crystals, m.p. 170–171 °C (EtOH/H2O). 1H NMR (600 MHz, DMSO-d6): δ 2.11, 2.19 (2 s, 3 H each, 2 Me), 3.85 (s, 3 H, OMe), 5.47 (s, 2 H, Bn), 7.18–7.21, 7.39–7.46, 7.58–7.61 (3 m, 2 H, 5 H, 2 H), 9.49 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 8.2, 8.6, 49.9, 55.8, 115.0, 126.2, 126.6, 127.7, 127.9, 128.0, 128.6, 129.1, 134.1, 135.7 (br), 160.5. IR (neat): ν 3168, 1562, 1517, 1454, 1359, 1305, 1256, 1236, 1197, 1167, 1109, 1033, 831, 749, 711 cm–1. ESI-MS (m/z): 293 (100, [M−PF6]+).
1-Benzyl-3-(4-methoxyphenyl)-4,5-diphenylimidazolium hexafluorophosphate (17c[PF6]): 444 mg (79%); colorless crystals, m.p. 173–175 °C (EtOH/H2O). 1H NMR (600 MHz, DMSO-d6): δ 3.78 (s, 3 H, OMe), 5.44 (s, 2 H, Bn), 7.04–7.07, 7.14–7.17, 7.23–7.32, 7.38–7.48 (4 m, 2 H, 4 H, 8 H, 5 H), 9.90 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 50.7, 55.7, 114.8, 125.0, 125.2, 126.4, 127.9, 128.0, 128.5, 128.6, 128.8, 129.0, 129.8, 130.4, 130.8, 131.0, 131.3, 132.3, 133.9, 137.1 (br), 160.3. IR (neat): ν 316, 1554, 1513, 1454, 1223, 1182, 1161, 1077, 1018, 941, 824, 760, 697 cm–1. ESI-MS (m/z): 417 (100, [M−PF6]+).
1-Benzyl-3-(4-fluorophenyl)-4,5-diphenylimidazolium hexafluorophosphate (17d[PF6]): 500 mg (91%); colorless oil (EtOH/H2O). 1H NMR (600 MHz, DMSO-d6): δ 5.44 (s, 2 H, Bn), 7.12–7.18, 7.22–7.25, 7.28–7.32, 7.36–7.40, 7.44–7.46, 7.58–7.62 (6 m, 4 H, 2 H, 6 H, 4 H, 1 H, 2 H), 9.90 (s, 1 H, C(2)H). 13C NMR (151 MHz, DMSO-d6): δ 51.1, 116.9 (d, 2JC–F = 23.4 Hz), 125.09, 125.11, 126.8, 128.2, 128.4, 128.8, 129.0, 129.2, 129.4 (d, 3JC–F = 9.3 Hz), 130.2, 130.3 (d, 4JC–F = 3.0 Hz), 130.7, 131.1, 131.2, 131.7, 132.6, 133.9, 137.4 (br), 162.8 (d, 1JC–F = 248.5 Hz). IR (neat): ν 3023, 2255, 2125, 1715, 1551, 1513, 1446, 1223, 1159, 1051, 1025, 962, 839, 760, 701 cm–1. ESI-MS (m/z): 405 (100, [M−PF6]+).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}