Abstract
Imidazole N-oxides are attractive starting materials for the preparation of complex molecules containing an imidazole ring. Dipolar cycloaddition between 1,1-difluoroalkenes and imidazole N-oxides bearing a chiral auxiliary performed in the presence of oxygen or nitrogen nucleophiles was found to provide access to esters and amides bearing a 2-azaheteroaryl substituent at the α position of the imidazole ring as mixtures of diastereomers that, in most cases, are readily separable. This three-component reaction introduces a new tertiary or quaternary all-carbon stereocenter into the heterocyclic ring at a position originally occupied by hydrogen. Importantly, products containing a trifluoromethyl group attached to this stereocenter are readily available as well.
1. Introduction
An imidazole ring is present in the structure of a large number of natural products and biologically active substances [1,2,3,4]. Imidazole derivatives are also among the most popular ionic liquids and catalysts based upon them [5,6]. In consequence, several classical methods of preparation of imidazole derivatives based upon five-membered ring construction have been and are still being actively developed [7,8,9,10,11,12,13]. Functionalization of a carbon–hydrogen bond in a preformed imidazole ring is a much less common approach, but it is an emerging synthetic strategy that relies upon either transition-metal-catalyzed processes or metal-free reactions [3,14,15,16].
In general, stereoselective formation of all-carbon quaternary stereocenters remains an important challenge in synthetic organic chemistry [17,18,19,20,21]. In particular, stereocenters of this kind bearing a trifluoromethyl substituent are interesting from the point of view of medicinal chemistry and potential applications in drug development programs [22,23,24,25,26,27,28]. An attractive and conceptually simple approach to the problem of the synthesis of such stereodefined quaternary centers is the asymmetric dipolar cycloaddition reaction [29,30,31], in particular, the 1,3-dipolar cycloaddition of fluorinated tetrasubstituted dipolarophiles [32,33].
In our laboratory, we have developed a method for the functionalization of azines and azoles via cycloaddition of the respective N-oxides to 1,1-difluoroalkenes in the presence of nucleophiles, which allows for the preparation of structurally diverse esters and amides of acetic acids bearing a 2-azaheteroaryl substituent at the α position, that is, derivatives of bis(aryl)methanes [34,35]. For medicinal chemistry projects, it is crucial to control the stereochemistry of the target products, and thus, it appeared worthwhile and important to us to explore the possibility of performing this reaction in a stereoselective manner and controlling the configuration of the newly formed stereocenter. Unlike azine N-oxides, N-oxides of imidazoles contain an additional functionalizable heteroatom in the ring. The problem of the stereocontrol of cycloaddition can therefore be approached by using imidazole N-oxides bearing chiral substituents on nitrogen [36,37,38]. Based upon our earlier studies, the planned reaction sequence is outlined in Scheme 1: dipolar cycloaddition of 1,1-difluorostyrenes to chiral N-oxides produces bicyclic isoxazolidine adducts I, the rearomatization of which provides acyl fluorides II by N–O bond cleavage. Their reaction with nucleophiles leads to diastereoisomers of the target esters and amides.
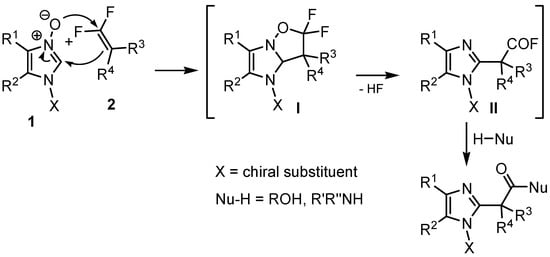
Scheme 1.
Cycloaddition of chiral imidazole N-oxides and 1,1-difluoroalkenes.
2. Results
Considering that the bond between the imidazole ring and the newly established stereocenter is formed in the dipolar cycloaddition step (see Scheme 1), control of stereoselectivity can be achieved, in principle, by employing either a chiral N-oxide or chiral difluoroalkene. Modification difluorostyrene substrates to introduce chirality would be a complicated task and would reduce the generality of the method. Therefore, the use of chiral N-oxides seemed to be the strategy of choice.
In our initial experiments, however, we explored an even simpler possibility, that is, reactions between achiral N-oxides and difluorostyrenes performed in the presence of a chiral, readily available nucleophile—(S)-1-phenylethanol—with the aim of using the chiral “handle” introduced by the nucleophilic partner to separate diastereoisomers of the products (Scheme 2).
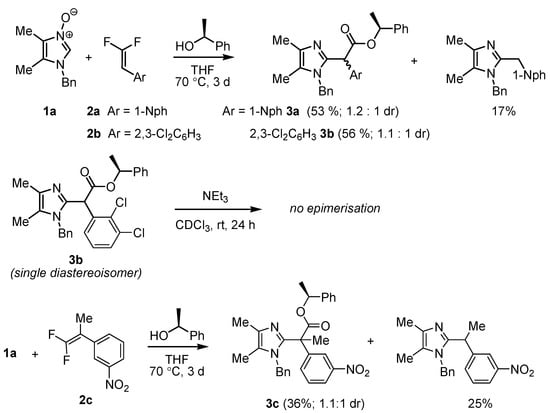
Scheme 2.
Cycloaddition of achiral imidazole N-oxide 1a with 1,1-difluorostyrenes in the presence of (S)-1-phenylethanol.
Based upon our previous studies [34,35], cycloaddition reactions of this kind are best performed in moderately polar solvents (AcOEt or THF) at slightly elevated temperatures (70–100 °C) or in DMF at RT, but the last solvent has a high boiling point, and its removal after the reaction is more tedious. After some experimentation, the reactions of 1a with difluorostyrenes 2a–c were optimally performed by heating all reactants to 70 °C in dry THF under an inert atmosphere to avoid unwanted hydrolysis of acyl fluoride intermediates II. The target esters 3a–c were obtained in moderate yields and, as expected, with very low diastereoselection. Considerable amounts of arylmethylene derivatives of imidazole as side products were still formed in these reactions, probably due to the low nucleophilicity of the secondary alcohol—(S)-1-phenylethanol—and its lower ability to scavenge the intermediate acyl fluoride. In these and all subsequent cycloaddition experiments, due to the high sterical demand of the substrates, long reaction times (3 days) were necessary to achieve reasonable yields. The reactions were run until the consumption of the N-oxide partner (used in 1.5-fold excess), which decomposed slowly under these conditions (TLC monitoring).
Unfortunately, the diastereoisomers of 3a–c could only be resolved by chromatography to a low extent. The stereogenic centers in these compounds are tertiary and can possibly undergo epimerization. To verify if the observed ratios reflect the relative stability of two diastereoisomeric esters, a single diastereoisomer of ester 3b was treated with catalytic amounts (0.2 equiv.) of bases of varying strength (NEt3 or DBU) for several hours in a THF-d8 solution, but no epimerization was observed in NMR measurements.
As mentioned in the introduction, imidazole N-oxides can be readily functionalized with chiral auxiliaries attached to the ring nitrogen. A series of chiral imidazoles were prepared from chiral primary amines and oximes of 1,2-diketones using the method developed by Mlostoń and co-workers [38,39] (Scheme 3).
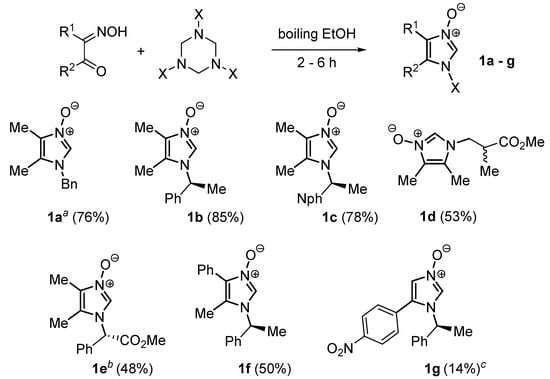
Scheme 3.
Preparation of chiral N-oxides. a: Reference [39]. b: Reference [38]. c: Reaction in HCO2H at RT. Nph = 1-naphthyl.
Imidazoles 1b–e were then tested in cycloaddition reactions with tetrasubstituted difluoroalkenes and MeOH as a nucleophile to verify the efficiency of various chiral auxiliary groups in inducing diastereoselectivity. As shown in Scheme 4, imidazoles 1b and 1c substituted with 1-phenylethyl and 1-naphthylethyl, respectively, resulted in higher selectivity, but the latter substrate resulted in a lower yield of the target methyl ester, even at higher temperature (100 °C). Oxides 1d and 1e produced inferior results, probably due to the planar nature of the ester substituent and further separation of the chirality center from the reactive N-oxide ring in the case of 1e. Therefore, 1b was employed in most of the further studies.
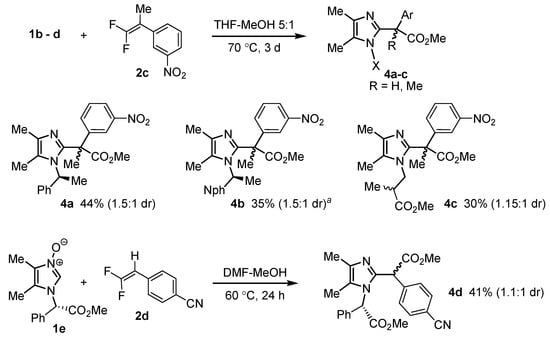
Scheme 4.
Cycloaddition of imidazole N-oxides with different chiral groups and 1,1-difluoroalkenes. a: Reaction at 100 °C.
The scope of the formation of methyl esters containing a quaternary center at the α position from various N-oxides and 1,1-difluorostyrenes was subsequently investigated (Scheme 5). Despite rather long reaction times and elevated temperatures (100 °C in most cases), the yields of products 4 (Scheme 4 and Scheme 5) were moderate at best, which is undoubtedly associated with high sterical congestion around the newly formed quaternary stereocenter. A clear influence of electronic effects can be observed: difluorostyrenes bearing electron-poor aryl rings resulted in considerably higher yields than those substituted with alkyl or Cl. A less marked positive effect on the reactivity of the dipolarophile was exerted by the fourth substituent at the double bond (Me vs. CF3 in 4g and 4e).
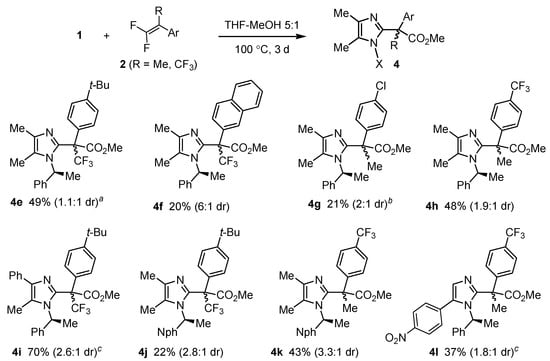
Scheme 5.
Cycloaddition of chiral imidazole N-oxides and 1,1-difluoroalkenes in the presence of MeOH. a: 29% (1.4:1) at 70 °C; b: 13% (2.5:1) at 70 °C; c: reaction at 70 °C.
Unlike the model reactions of Scheme 4, some examples of Scheme 5 demonstrate that a 1-naphthylethyl substituent at the ring nitrogen can be more useful in inducing diastereoselectivity than 1-phenylethyl. Lower temperature (70 °C) generally improved diastereoselectivity, but at the expense of yield. The highest diastereoselectivity (6:1) was observed in the formation of the highly sterically crowded β-naphthyl-substituted ester 4f. More importantly, diastereoisomers of most of esters 4 (particularly 4e, 4g, 4h, 4i and 4k) could be readily separated.
Products of even higher complexity could be obtained when performing the cycloaddition reaction in the presence of nitrogen nucleophiles. Both secondary and primary amines, including an amino acid methyl ester, reacted with 1 and tri- or tetrasubstituted difluoroalkenes to produce amides 5a–f (Scheme 6). Unlike esterification (see Scheme 5), such cycloadditions with amines as nucleophiles led to better results when performed in the presence of triethylamine (2 equivalents), probably because it binds HF and ensures the appropriate concentration of free primary or secondary amine [40].
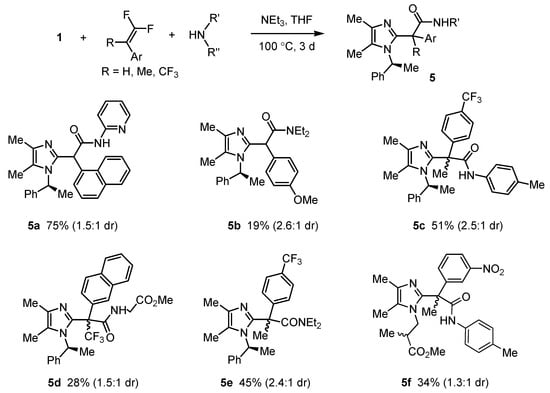
Scheme 6.
Cycloaddition of chiral imidazole N-oxides and 1,1-difluoroalkenes in the presence of amines.
As follows from the general reaction pathway shown in Scheme 1, the reaction performed in the presence of water, or in the absence of a suitable nucleophile and followed by aqueous work-up, is expected to produce free carboxylic acids. Such acids are very labile and readily undergo decarboxylation, so in consequence, a reaction of N-oxide with water and 1,1-difluoroalkene containing a tetrasubstituted double bond will eventually lead to a product containing only a tertiary stereocenter. The results of such reactions with chiral N-oxides 1b,c are presented in Scheme 7. In the reaction of N-oxide 1b with alkene 2c, two separable diastereoisomers were obtained in 24% total yield and 1.1:1 ratio. On the other hand, the more sterically congested 1c resulted almost exclusively in one diastereoisomer of imidazole 6a (>10:1).
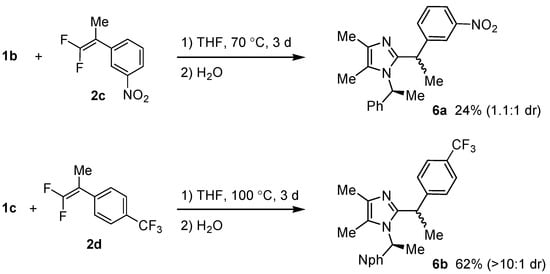
Scheme 7.
Cycloaddition of chiral imidazole N-oxides and 1,1-difluoroalkenes followed by aqueous work-up.
Ideally, a chiral auxiliary group should be removable from the final product after the stereoinduction step. Unfortunately, attempts to remove phenylethyl or naphthylethyl substituents from imidazoles 4 or 5 proved unsuccessful both under reductive (hydrogen and Pd) or oxidative (cerium(IV) ammonium nitrate) conditions.
3. Discussion
The results presented above demonstrate that imidazole N-oxides substituted with chiral phenylethyl or naphthylethyl substituents indeed undergo dipolar cycloaddition with difluorostyrenes in a moderately diastereoselective manner. Exclusive formation of products of types 3, 4, 5 and 6 demonstrates that the cycloaddition step is completely regioselective.
Concerning the general trends of stereoinduction and the actual configuration of the newly formed stereocenter, it is interesting to examine some properties of the NMR spectra of the series of esters 4. In each case, the singlet of the ester methyl group appears in the range of 3.80–3.97 ppm for the major diastereoisomer and in the 3.26–3.39 ppm (for N-phenylethyl derivatives) or even 2.36–2.56 ppm range (for N-naphthylethyl derivatives) for the minor one. In each case, the doublet of the CHCH3 methyl occurs at higher chemical shift values for the major isomer. These trends strongly suggest that, in each case, the same configuration of the product predominates.
Theoretical calculations were performed to shed some light on the course of the cycloaddition step and to predict the preferred configuration of the forming stereocenter. First, cycloaddition between simple models, 1,4,5-trimethylimidazole-3-oxide and 1,1-difluoro-2-phenylethene, was examined using DFT calculations at the B3LYP/6-31G(d,p) level of theory. The concerted cycloaddition was found to prefer exo mode (by 2.2 kcal/mol), with the formation of the CF2–O bond being more advanced than C–C in the transition state (for details, please see the Supplementary Materials). On the other hand, a putative intermediate of the stepwise reaction, resulting from nucleophilic addition of the N-oxide oxygen on the CF2 terminus of styrene, could not be optimized to a minimum, even after applying solvation with polar solvents or more advanced basis sets that included polarization functions.
Next, transition states for the exo cycloaddition of N-oxide 1b and 1,1-difluoro-2-phenylethene were compared. Two lowest-energy TSs were identified as resulting from the attack of the alkene opposite to the side of the imidazole ring shielded by the phenyl ring of the N-phenylethyl group (Figure 1). They differ in orientation of the methyl of the N-phenylethyl, which is directed either towards the incoming alkene (Figure 1b) or away from it (Figure 1a). The latter TS was found to be slightly preferred (by 0.1 kcal/mol). Such a small difference in energy explains the relatively low levels of diastereoselection in the studied reactions and does not allow for conclusive determination of the absolute configuration of major diastereoisomers. Such a determination would be possible only by means of X-ray crystallographic analysis, but unfortunately, products 3, 4, 5 and 6 are oils.
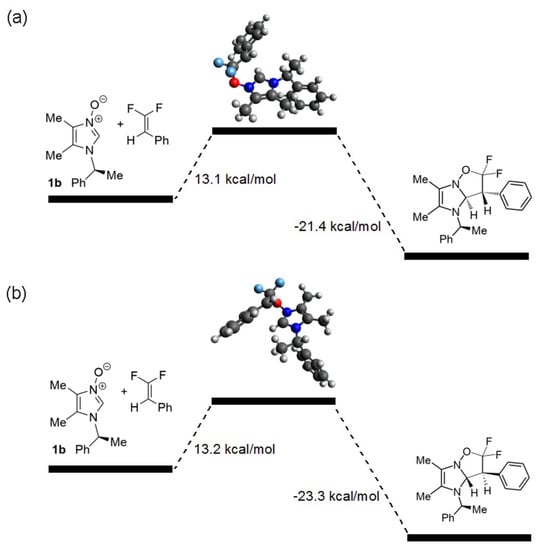
Figure 1.
Transition states in cycloaddition of N-oxide 1b to 1,1-difluorostyrene, with the methyl group of the chiral auxiliary pointing away from incoming alkene (a) or towards it (b).
Failure to remove the chiral auxiliary from the imidazole rings of the products is a drawback of the synthetic method reported herein, but our work provides insight into structural features required for reasonable diastereoselection and the separation of diastereoisomers of the products. Further studies are needed to develop chiral groups that would also be sufficiently labile. This goal could perhaps be achieved by designing imidazole N-oxides derived from chiral amines that have been reported to be useful and readily removable chiral auxiliaries in other types of chemical transformations, for example, α-phenylglycinol [41], 7-oxanorbornene-derived amino acids [42] or ephedrine derivatives [43]. More sophisticated and sterically crowded auxiliaries could also help improve the diastereoselectivity of the studied cycloadditions.
4. Materials and Methods
Analytical grade solvents were used as received. Hexanes and dichloromethane (DCM) used for extraction and chromatography were distilled before use. Commercially available dry DMF and THF were used for cycloaddition reactions. All commercially available reagents were used as received. All 1H and 13C NMR spectra were recorded at 298 K in CDCl3 solutions using a 200 MHz, 400 MHz or 500 MHz spectrometer (as indicated for each compound). 1H and 13C NMR chemical shifts are given relative to the TMS signal at 0.0 ppm and relative to CFCl3 for 19F spectra. Mass spectra and HR MS measurements were obtained using a mass spectrometer equipped with an electrospray ion source and q-TOF-type mass analyzer (ES) or a magnetic sector mass spectrometer equipped with an electron impact (EI) ion source and the EBE double-focusing geometry mass analyzer. IR spectra were obtained using an FT-IR spectrometer. Column chromatography was performed using silica gel 60 (0.040−0.063 mm). Analytical thin-layer chromatography (TLC) was performed using precoated silica gel plates (0.20 mm thickness) and visualized under a UV lamp. Flash chromatography was performed using silica gel 60 (0.040–0.063 mm).
All 1,1-difluorostyrenes used in cycloaddition reactions, as well as arylmethyleneimidazole side products of Scheme 2, are known compounds and were described by us previously [34,35].
Calculations were performed using Gaussian 16 [44].
General procedure for cycloaddition reactions—synthesis of methyl esters 4. N-Oxide 1 (0.75 mmol) and difluorostyrene 2 (0.5 mmol) were added under an argon atmosphere to a mixture of dry THF (1 mL; or DMF, mL in the case of 4d) and MeOH (0.2 mL) in a glass pressure tube (ca. 3.5 mL volume) equipped with a Teflon valve and a magnetic stirring element. The reaction mixture was then heated to 70 or 100 °C for 72 h with vigorous stirring (for exact conditions for each example, see Scheme 4 and Scheme 5). The reaction was then cooled down to room temperature, saturated aqueous NaHCO3 (1 mL) and brine (5 mL) were added, and the product was extracted with AcOEt (3 × 1.5 mL). The organic phase was washed with brine (5 mL), dried over anhydrous Na2SO4 and evaporated. The products were isolated by column chromatography on silica gel with hexane–AcOEt 5:1, hexane–AcOEt 2:1 or pure AcOEt as eluent.
General procedure for cycloaddition reactions—synthesis of amides 5. N-Oxide 1 (0.75 mmol), difluorostyrene 2 (0.5 mmol), amine (0.6 mmol) and NEt3 (1.0 mmol, 139 μL) were added under an argon atmosphere to dry THF (1 mL) in a glass pressure tube (ca. 3.5 mL volume) equipped with a Teflon valve and a magnetic stirring element. The reaction mixture was heated to 100 °C for 72 h with vigorous stirring. The reaction was then cooled down to room temperature, saturated aqueous NaHCO3 (1 mL) and brine (5 mL) were added, and the product was extracted with AcOEt (3 × 1.5 mL). The organic phase was washed with brine (5 mL), dried with anhydrous Na2SO4 and evaporated. The products were isolated using column chromatography on silica gel with hexane–AcOEt 2:1 or pure AcOEt as eluent.
The procedure for esters 5 were the same as the general procedure for cycloaddition reactions—synthesis of bis(aryl)ethanes 6 but without the addition of MeOH.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12020177/s1, Characterization data and copies of NMR spectra for new compounds and details of the theoretical calculations.
Author Contributions
Conceptualization, R.L.; methodology, R.L.; investigation and synthesis, P.B. and R.L.; writing, R.L.; funding acquisition, R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre, Poland, grant number UMO-2011/03/D/ST5/06066. The APC was funded by the Institute of Organic Chemistry, Polish Academy of Sciences, Warsaw.
Data Availability Statement
Data available in a publicly accessible repository: https://doi.org/10.6084/m9.figshare.17708753.v1 (accessed date 28 December 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Luca, L. Naturally Occurring and Synthetic Imidazoles: Their Chemistry and Their Biological Activities. Curr. Med. Chem. 2006, 13, 1–23. [Google Scholar] [PubMed]
- Narasimhan, B.; Sharma, D.; Kumar, P. Biological importance of imidazole nucleus in the new millennium. Med. Chem. Res. 2011, 20, 1119–1140. [Google Scholar] [CrossRef]
- Du, H.; He, Y.; Sivappa, R.; Lovely, C.J. New Methods of Imidazole Functionalization–From Imidazole to Marine Alkaloids. Synlett 2006, 2006, 965–992. [Google Scholar] [CrossRef]
- Mumtaz, A.; Saeed, A.; Fatima, N.; Dawood, M.; Rafique, H.; Iqbal, J. Imidazole and its derivatives as potential candidates for drug development. Bangladesh J. Pharmacol. 2016, 11, 756–764. [Google Scholar] [CrossRef][Green Version]
- Dupont, J.; de Souza, R.F.; Suarez, P.A.Z. Ionic Liquid (Molten Salt) Phase Organometallic Catalysis. Chem. Rev. 2002, 102, 3667–3692. [Google Scholar] [CrossRef]
- Maiuolo, L.; Algieri, V.; Olivito, F.; De Nino, A. Recent Developments on 1,3-Dipolar Cycloaddition Reactions by Catalysis in Green Solvents. Catalysts 2020, 10, 65. [Google Scholar] [CrossRef]
- Dai, L.; Yu, S.; Lv, N.; Ye, X.; Shao, Y.; Chen, Z.; Chen, J. Synthesis of Imidazoles and Oxazoles via a Palladium-Catalyzed Decarboxylative Addition/Cyclization Reaction Sequence of Aromatic Carboxylic Acids with Functionalized Aliphatic Nitriles. Org. Lett. 2021, 23, 5664–5668. [Google Scholar] [CrossRef]
- Hu, B.; Wang, Z.; Ai, N.; Zheng, J.; Liu, X.-H.; Shan, S.; Wang, Z. Catalyst-Free Preparation of 1,2,4,5-Tetrasubstituted Imidazoles from a Novel Unexpected Domino Reaction of 2-Azido Acrylates and Nitrones. Org. Lett. 2011, 13, 6362–6365. [Google Scholar] [CrossRef]
- Adiyala, P.R.; Borra, S.; Kamal, A.; Maurya, R.A. Access to Imidazole Derivatives by Silver(I) Carbonate Mediated Coupling of Vinyl Azides with Secondary Amines. Eur. J. Org. Chem. 2016, 2016, 1269–1273. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, H.; Chen, W.; Dong, J.; Chen, Z.; Cao, H. Silver-catalyzed [3+2] domino reaction: An efficient strategy to synthesize imidazole-5-carbaldehydes. Org. Biomol. Chem. 2017, 15, 6463–6466. [Google Scholar] [CrossRef]
- Das, U.K.; Shimon, L.J.W.; Milstein, D. Imidazole synthesis by transition metal free, base-mediated deaminative coupling of benzylamines and nitriles. Chem. Commun. 2017, 53, 13133–13136. [Google Scholar] [CrossRef] [PubMed]
- Raghu, M.S.; Kumar, C.B.P.; Prasad, K.N.N.; Prashanth, M.K.; Kumarswamy, Y.K.; Chandrasekhar, S.; Veeresh, B. MoS2-Calix [4] arene Catalyzed Synthesis and Molecular Docking Study of 2,4,5-Trisubstituted Imidazoles As Potent Inhibitors of Mycobacterium tuberculosis. ACS Comb. Sci. 2020, 22, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Beuvin, M.; Manneveau, M.; Diab, S.; Picard, B.; Sanselme, M.; Piettre, S.R.; Legros, J.; Chataigner, I. New synthesis of imidazole derivatives from cyanobenzenes. Tetrahedron Lett. 2018, 59, 4487–4491. [Google Scholar] [CrossRef]
- Loska, R. Ring C–H Functionalization of Aromatic N-Oxides. In Advances in Organic Synthesis; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Al Sharjah, United Arab Emirates, 2018; Volume 11, p. 233. [Google Scholar]
- Akulov, A.A.; Varaksin, M.V.; Charushin, V.N.; Chupakhin, O.N. Direct Functionalization of C(sp2)−H Bond in Nonaromatic Azaheterocycles: Palladium-Catalyzed Cross-Dehydrogenative Coupling (CDC) of 2H-Imidazole 1-Oxides with Pyrroles and Thiophenes. ACS Omega 2019, 4, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Varaksin, M.; Moseev, T.; Chupakhin, O.; Charushin, V.; Trofimov, B. Metal-free C–H functionalization of 2H-imidazole 1-oxides with pyrrolyl fragments in the design of novel azaheterocyclic ensembles. Org. Biomol. Chem. 2017, 15, 8280–8284. [Google Scholar] [CrossRef] [PubMed]
- Fuji, K. Asymmetric creation of quaternary carbon centers. Chem. Rev. 1993, 93, 2037–2066. [Google Scholar] [CrossRef]
- Quasdorf, K.W.; Overman, L.E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, S.-J.; Liu, W.-B.; Stoltz, B.M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. [Google Scholar] [CrossRef]
- Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Catalytic Enantioselective Desymmetrization Reactions to All-Carbon Quaternary Stereocenters. Chem. Rev. 2016, 116, 7330–7396. [Google Scholar] [CrossRef]
- Ling, T.; Rivas, F. All-carbon quaternary centers in natural products and medicinal chemistry: Recent advances. Tetrahedron 2016, 72, 6729–6777. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef] [PubMed]
- Jafari, E.; Chauhan, P.; Kumar, M.; Chen, X.-Y.; Li, S.; von Essen, C.; Rissanen, K.; Enders, D. Organocatalytic Asymmetric Synthesis of Trifluoromethylated Tetrahydrocarbazoles by a Vinylogous Michael/Aldol Formal [4 + 2] Annulation. Eur. J. Org. Chem. 2018, 2462–2465. [Google Scholar] [CrossRef]
- Enders, D.; Gottfried, K.; Raabe, G. Organocatalytic Enantioselective Strecker Synthesis of α-Quaternary α-Trifluoromethyl Amino Acids. Adv. Synth. Catal. 2010, 352, 3147–3152. [Google Scholar] [CrossRef]
- Chen, P.; Yue, Z.; Zhang, J.; Lv, X.; Wang, L.; Zhang, J. Phosphine-Catalyzed Asymmetric Umpolung Addition of Trifluoromethyl Ketimines to Morita–Baylis–Hillman Carbonates. Angew. Chem. Int. Ed. 2016, 55, 13316–13320. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, H.-H.; Zhang, J. Cu (II)-Catalyzed Enantioselective β-Boration of β-Trifluoromethyl, β,β-Disubstituted Enones and Esters: Construction of a CF3- and Boron-Containing Quaternary Stereocenter. ACS Catal. 2018, 8, 8318–8323. [Google Scholar] [CrossRef]
- Crotti, S.; Di Iorio, N.; Mazzanti, A.; Righi, P.; Bencivenni, G. Enantioselective Synthesis of Trifluoromethyl α,β-Unsaturated δ-Lactones via Vinylogous Aldol-Lactonization Cascade. J. Org. Chem. 2018, 83, 12440–12448. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, A.; Nazish, M.; Gupta, N.; Khan, N.H.; Kureshy, R.I. Enantioselective Addition of Cyanide to CF3-Substituted Alkylidenemalonates: Construction of Trifluoromethylated All-Carbon Quaternary Stereocenters. ChemistrySelect 2018, 3, 4838–4843. [Google Scholar] [CrossRef]
- Gothelf, K.V.; Jørgensen, K.A. Asymmetric 1,3-Dipolar Cycloaddition Reactions. Chem. Rev. 1998, 98, 863–909. [Google Scholar] [CrossRef]
- Revuelta, J.; Cicchi, S.; Goti, A.; Brandi, A. Enantiopure Cyclic Nitrones: A Useful Class of Building Blocks for Asymmetric Syntheses. Synthesis 2007, 485–504. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions. Chem. Rev. 2015, 115, 5366–5412. [Google Scholar] [CrossRef]
- Xu, S.; Liu, B.; Zhang, Z.-M.; Xu, B.; Zhang, J. Copper (I)-Catalyzed Asymmetric [3 + 2]-Cycloaddition of α-Substituted Iminoesters with α-Trifluoromethyl α,β-Unsaturated Esters. Chin. J. Chem. 2018, 36, 421–429. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Z.-M.; Xu, B.; Xu, S.; Wu, H.-H.; Zhang, J. Cu (I)-Ming-phos Catalyzed Enantioselective [3 + 2] Cycloadditions of Glycine ketimines to β-Trifluoromethyl Enones. Adv. Synth. Catal. 2018, 360, 2144–2150. [Google Scholar] [CrossRef]
- Loska, R.; Szachowicz, K.; Szydlik, D. Synthesis of Alkyl Aryl (heteroaryl)acetates from N-Oxides, 1,1-Difluorostyrenes, and Alcohols. Org. Lett. 2013, 15, 5706–5709. [Google Scholar] [CrossRef] [PubMed]
- Loska, R.; Bukowska, P. A three-component synthesis of aryl (heteroaryl) acylamides. Org. Biomol. Chem. 2015, 13, 9872–9882. [Google Scholar] [CrossRef]
- Mlostoń, G.; Wróblewska, A.; Obijalska, E.; Heimgartner, H. Optically active imidazole N-oxides derived from L-prolinamine. Tetrahedron Asymmetry 2013, 24, 958–965. [Google Scholar] [CrossRef][Green Version]
- Wróblewska, A.; Mlostoń, G.; Heimgartner, H. Synthesis of optically active polycyclic N-heterocycles derived from L-prolinamine. Tetrahedron Asymmetry 2015, 26, 505–509. [Google Scholar] [CrossRef][Green Version]
- Jasiński, M.; Mlostoń, G.; Linden, A.; Heimgartner, H. Synthesis and Selected Transformations of 1H-Imidazole 3-Oxides Derived from Amino Acid Esters. Helv. Chim. Acta 2008, 91, 1916–1933. [Google Scholar] [CrossRef]
- Mlostoń, G.; Gendek, T.; Heimgartner, H. First Examples of Reactions of Azole N-Oxides with Thioketones: A Novel Type of Sulfur-Transfer Reaction. Helv. Chim. Acta 1998, 81, 1585–1595. [Google Scholar] [CrossRef]
- Loska, R.; Mąkosza, M. New Synthesis of 2-Heteroarylperfluoropropionic Acids Derivatives by Reaction of Azine N-Oxides with Hexafluoropropene. Chem. Eur. J. 2008, 14, 2577–2589. [Google Scholar] [CrossRef]
- Chakraborty, T.K.; Hussain, K.A.; Reddy, G.V. α-Phenylglycinol as chiral auxiliary in diastereoselective Strecker synthesis of α-amino acids. Tetrahedron 1995, 51, 9179–9190. [Google Scholar] [CrossRef]
- Basso, A.; Banfi, L.; Riva, R.; Guanti, G. A Novel Highly Selective Chiral Auxiliary for the Asymmetric Synthesis of L- and D-α-Amino Acid Derivatives via a Multicomponent Ugi Reaction. J. Org. Chem. 2005, 70, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Alonso, B.; Ocejo, M.; Carrillo, L.; Vicario, J.L.; Reyes, E.; Uria, U. Using Heteroaryl-lithium Reagents as Hydroxycarbonyl Anion Equivalents in Conjugate Addition Reactions with (S,S)-(+)-Pseudoephedrine as Chiral Auxiliary; Enantioselective Synthesis of 3-Substituted Pyrrolidines. J. Org. Chem. 2013, 78, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).