
Oxidative Coupling of Methane over Pt/Al2O3 at High Temperature: Multiscale Modeling of the Catalytic Monolith

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
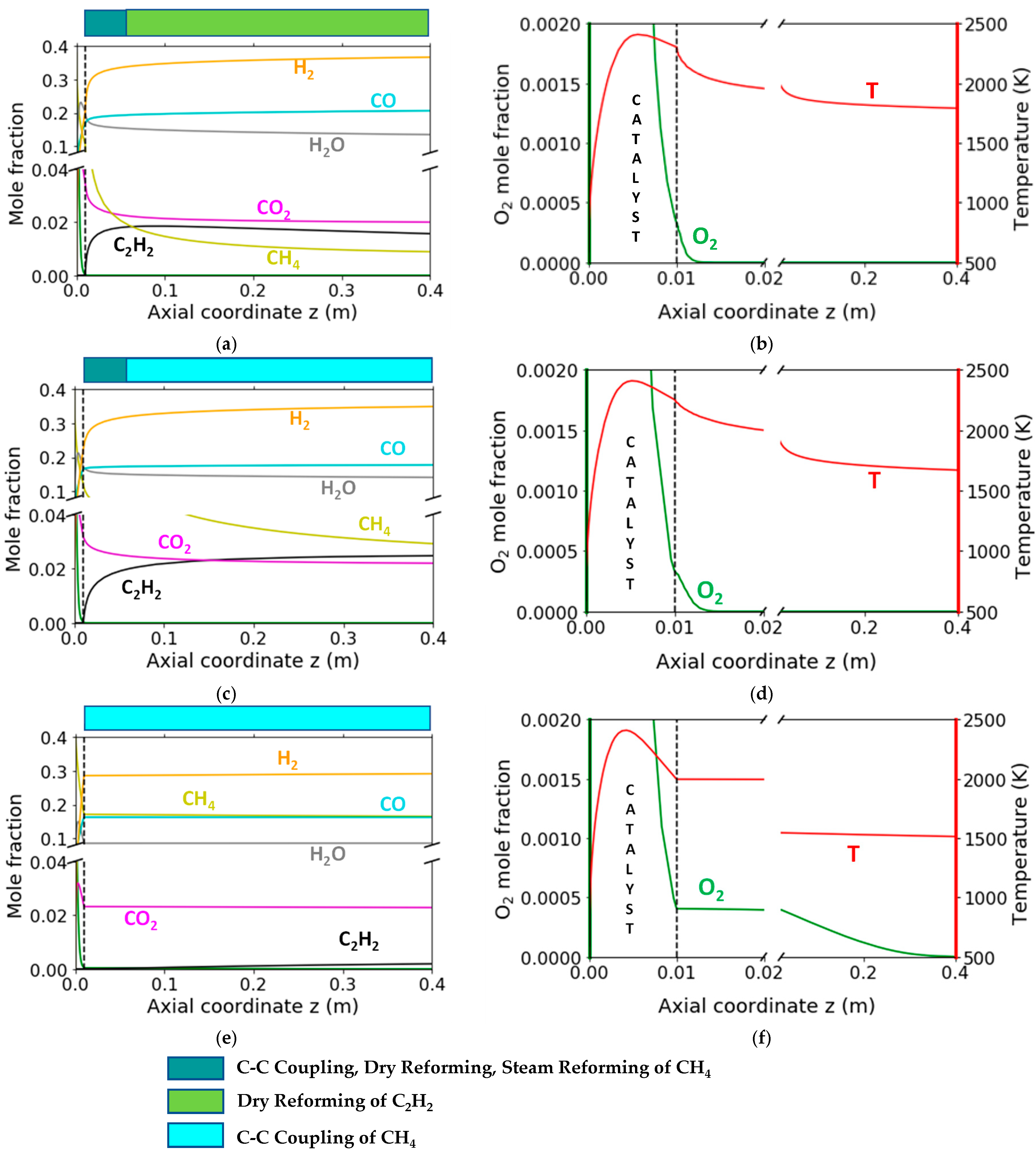
2. Results and Discussion
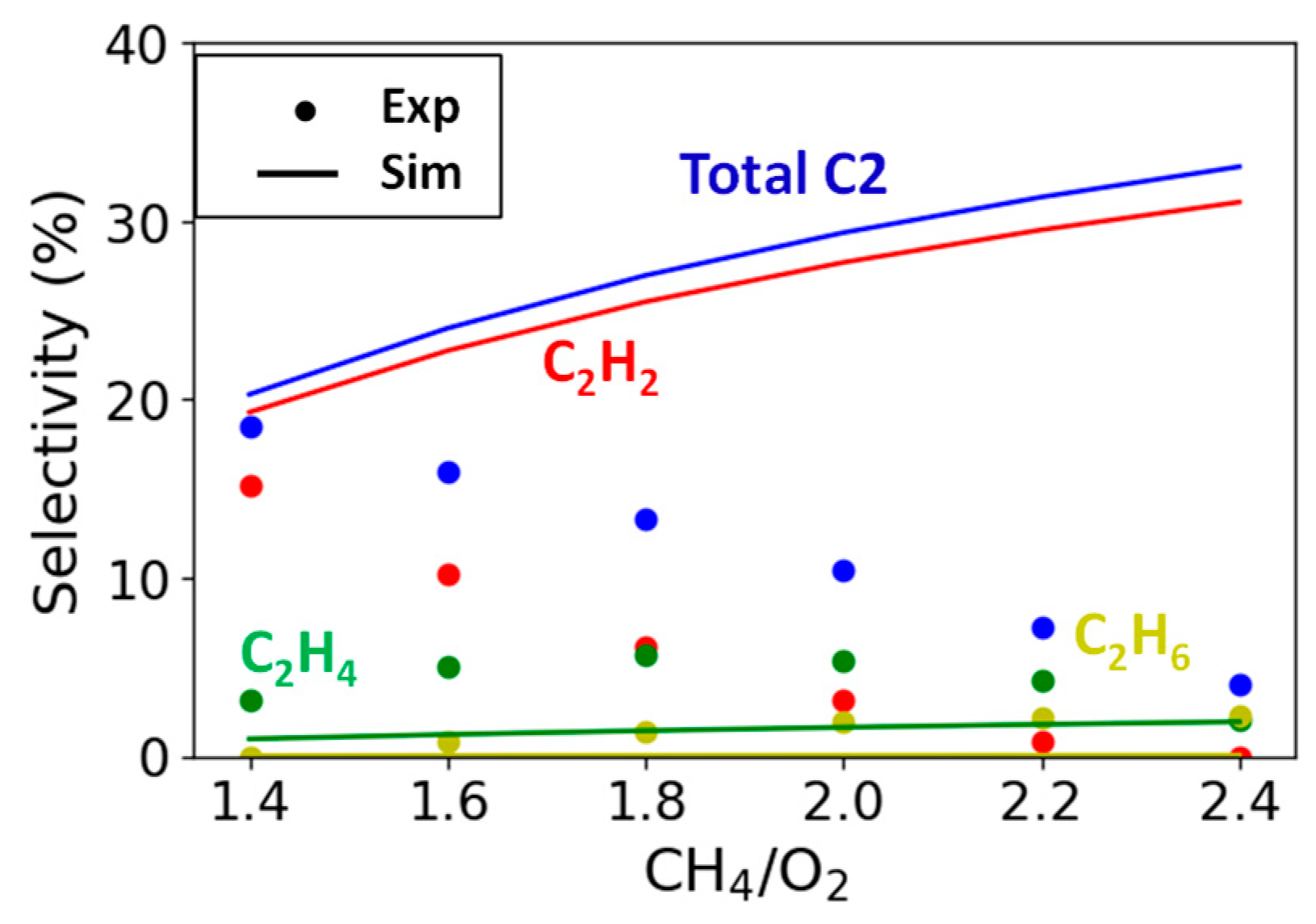
2.1. Simulation Study of Literature Experimental Results
2.2. Simulation Studies with Novel Inhouse Experimental Results
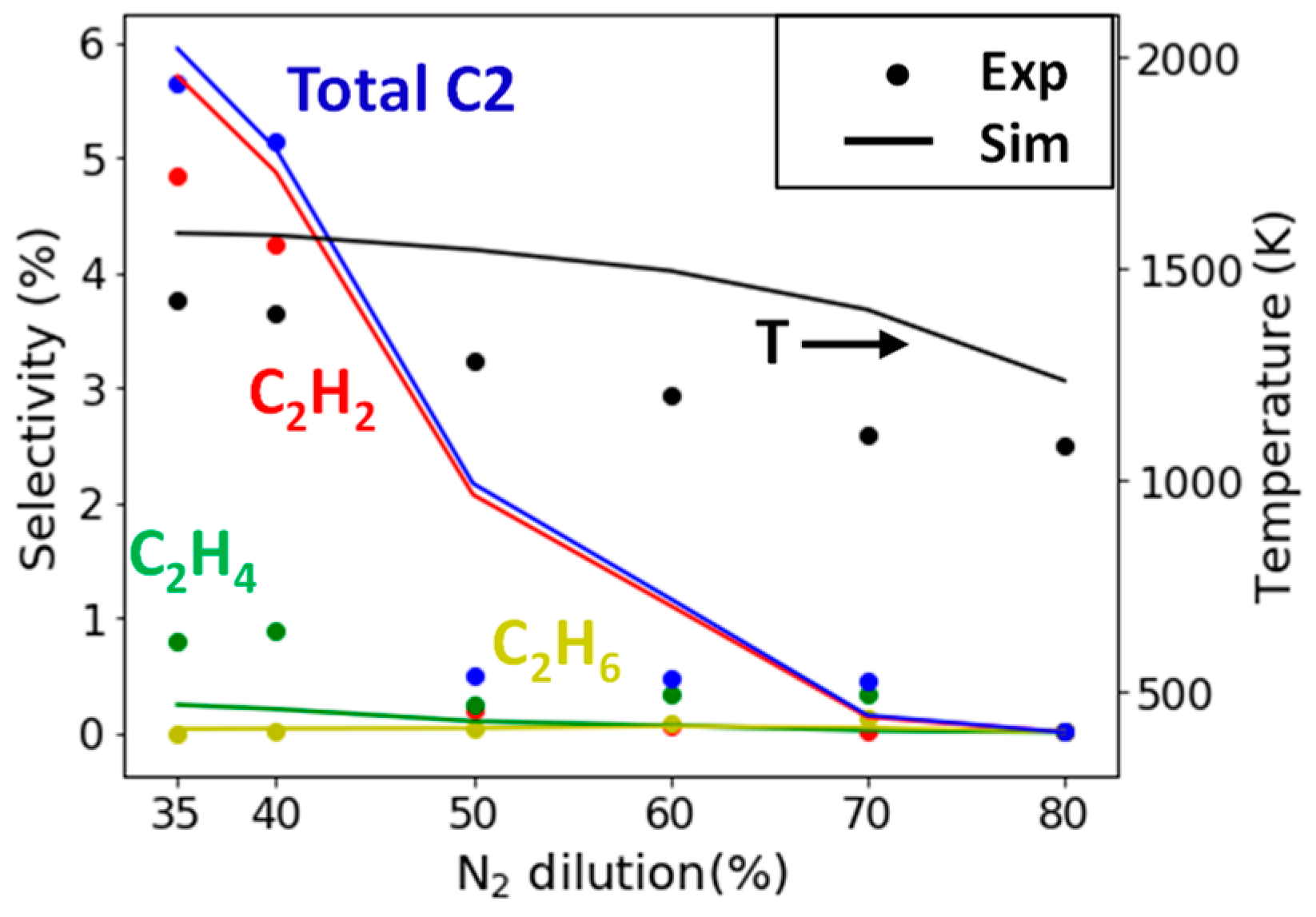
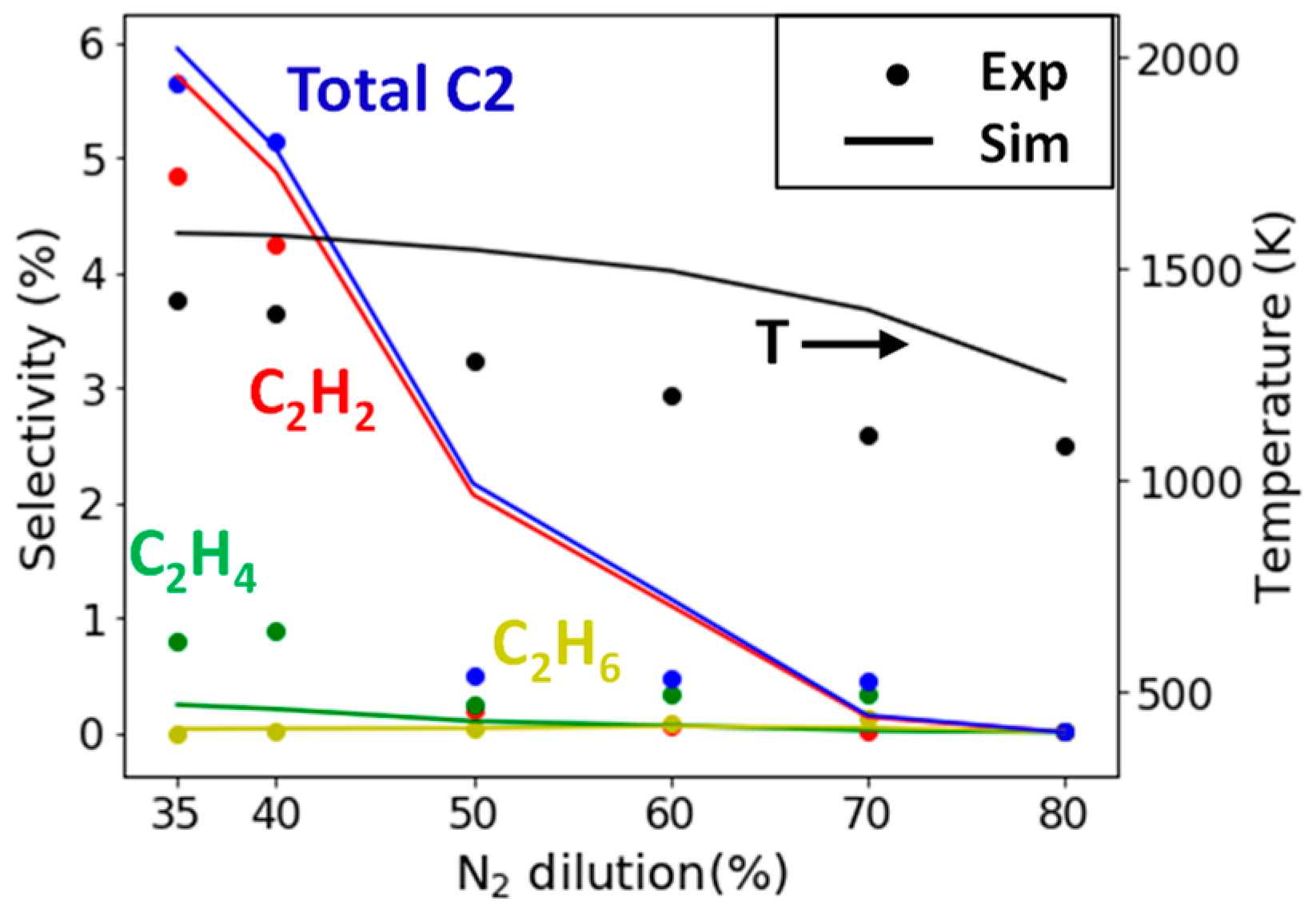
2.2.1. Study of N2 Dilution
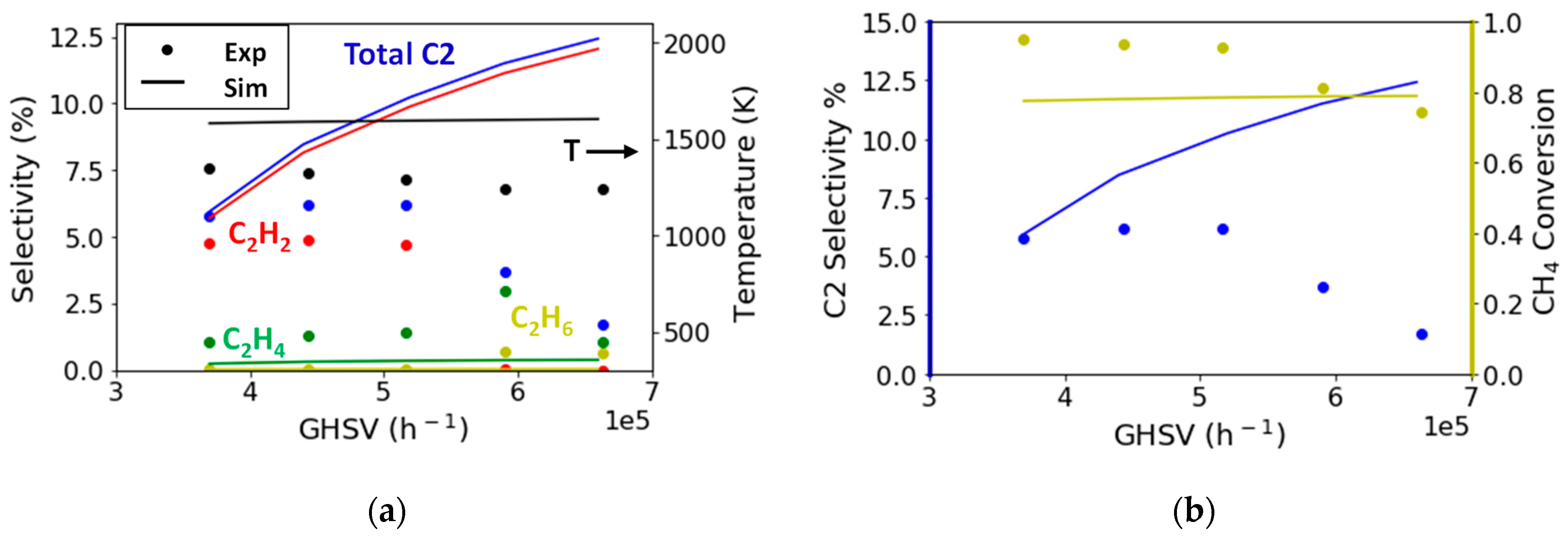
2.2.2. Variation of Flow Rate
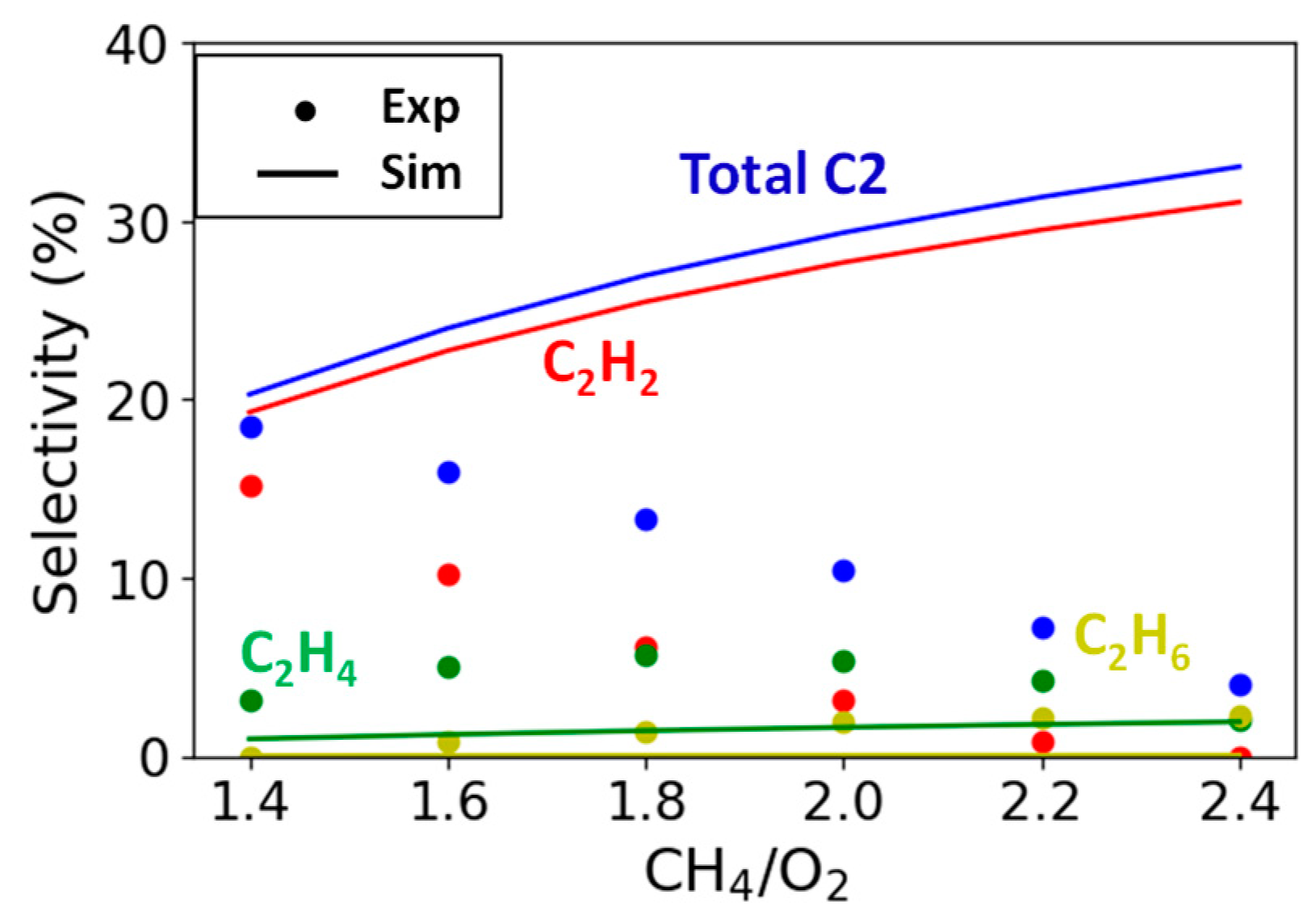
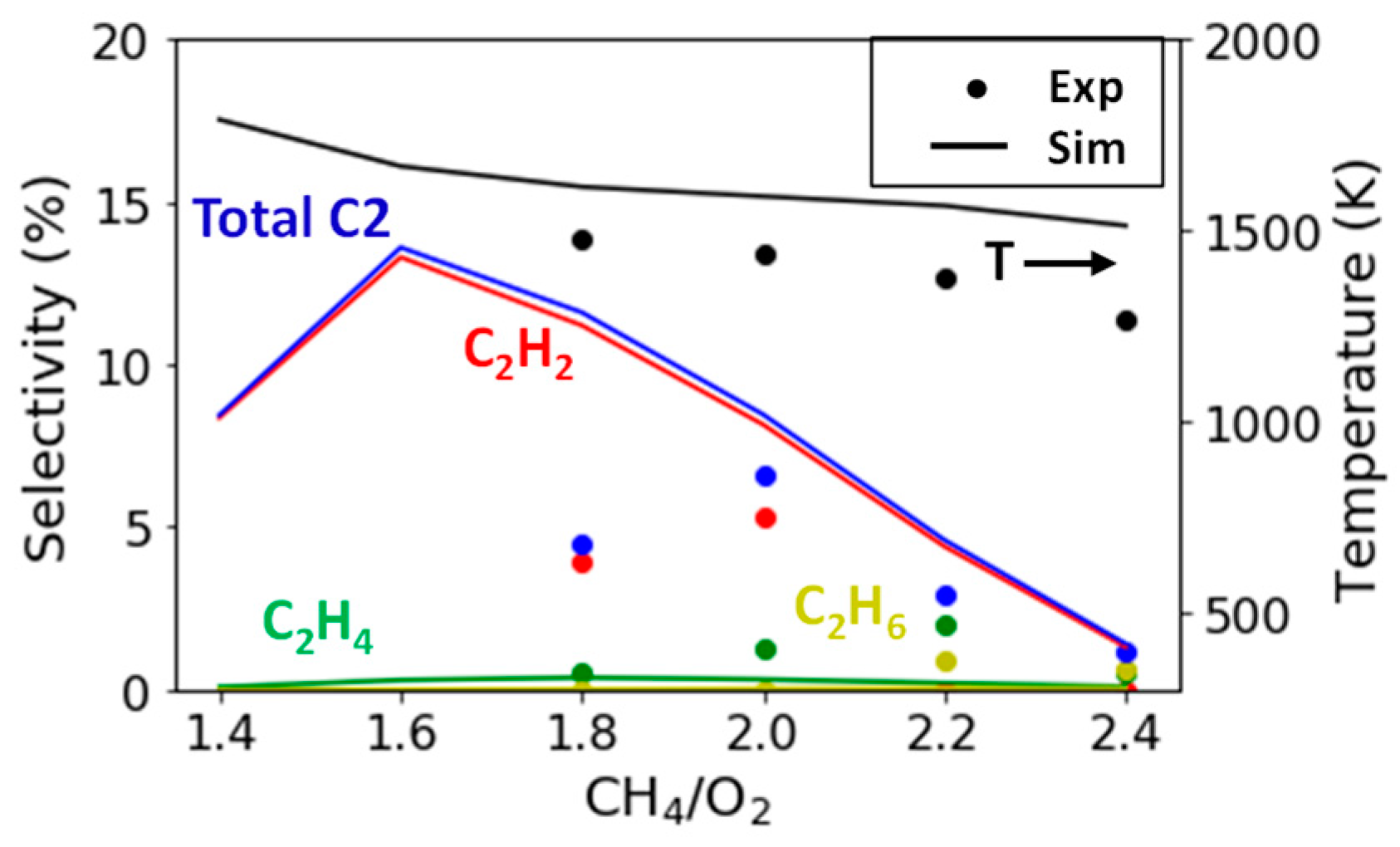
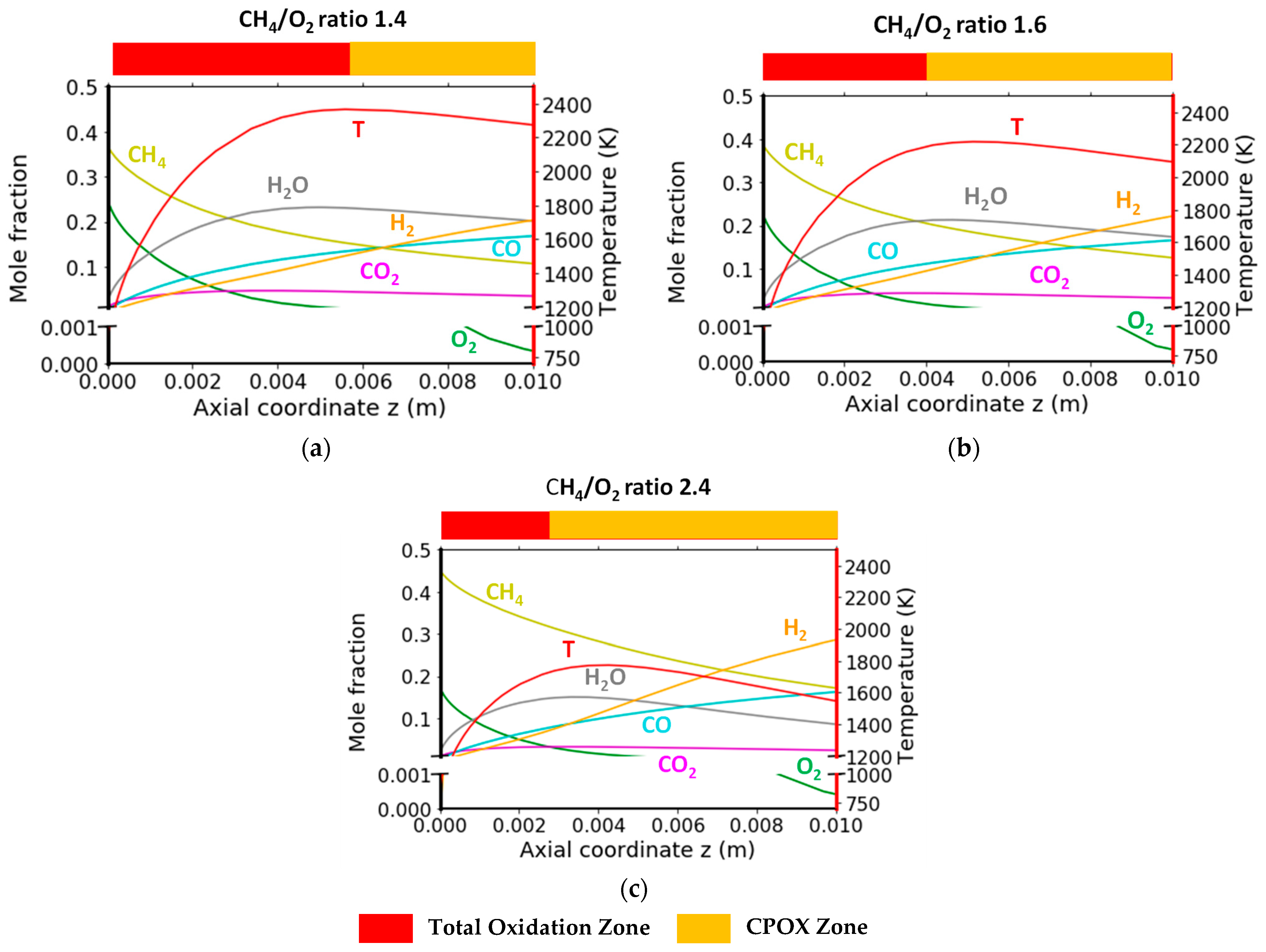
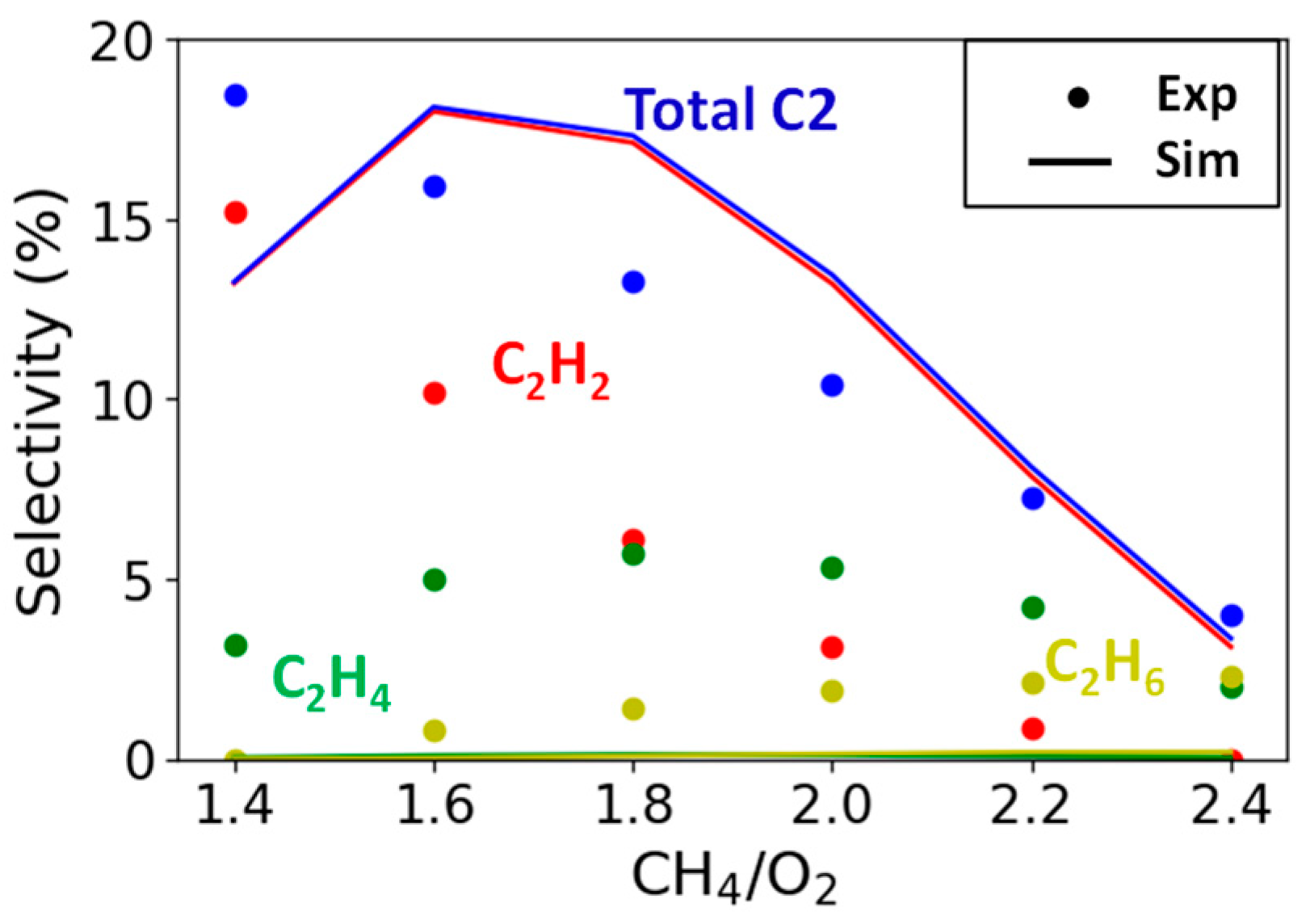
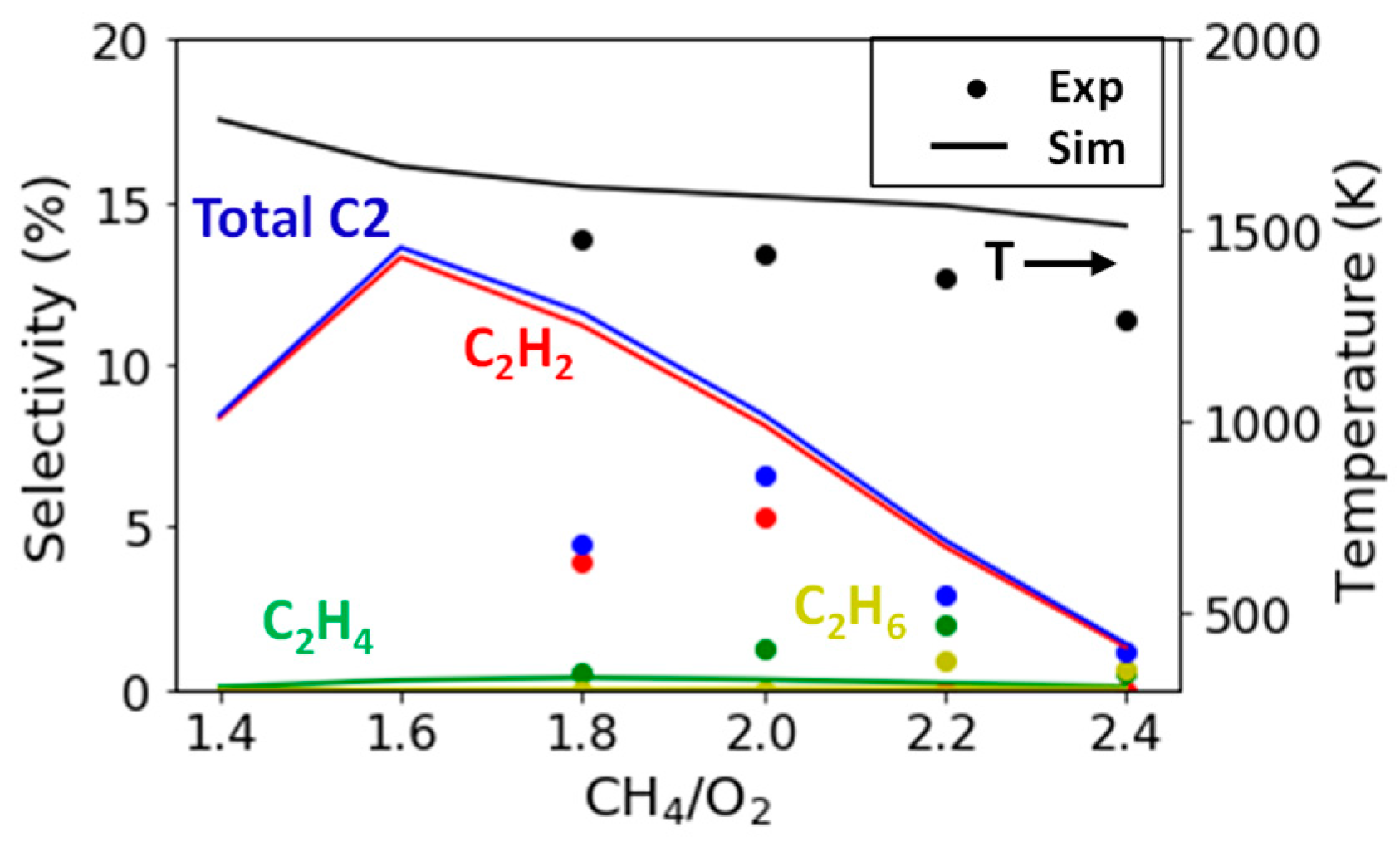
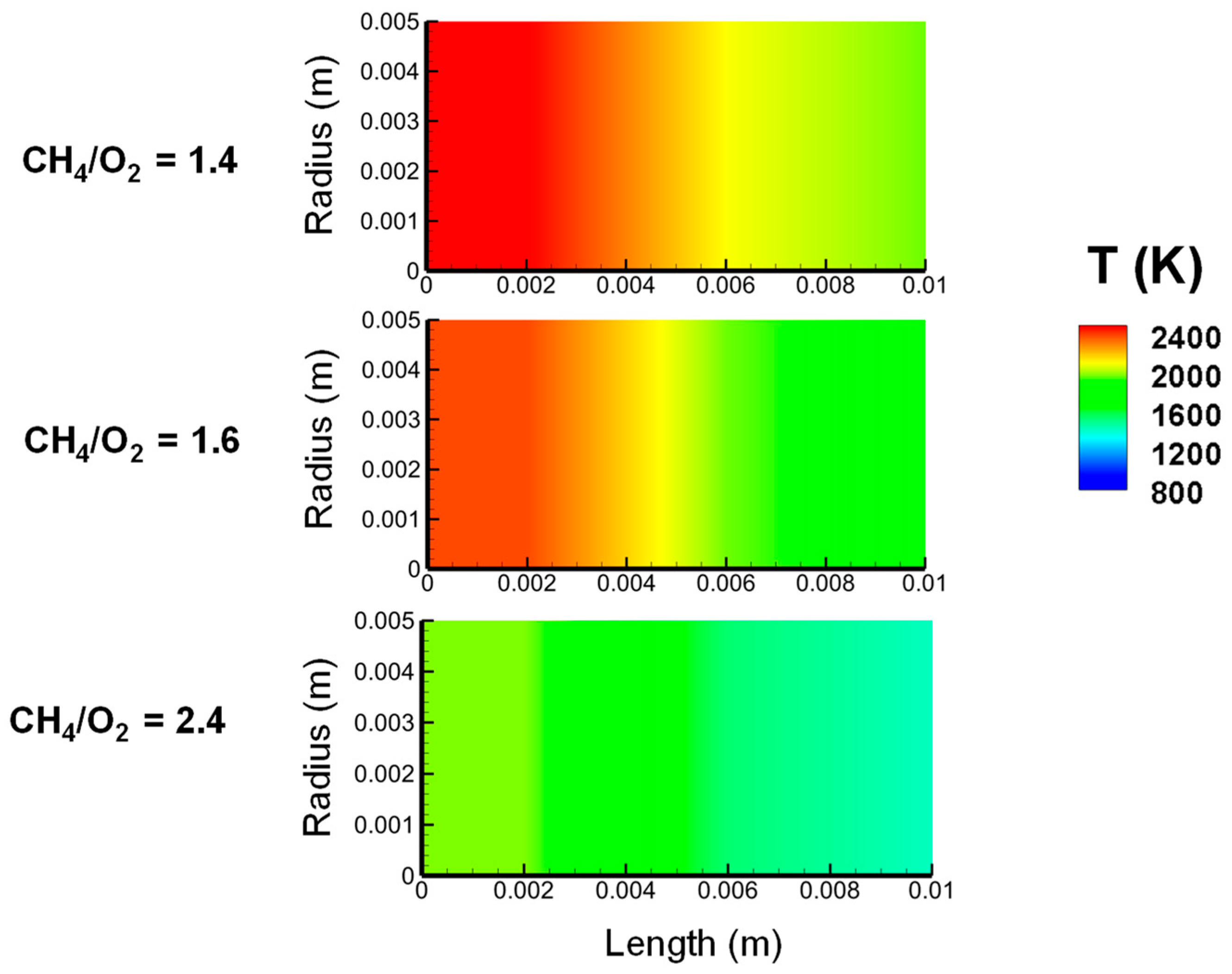
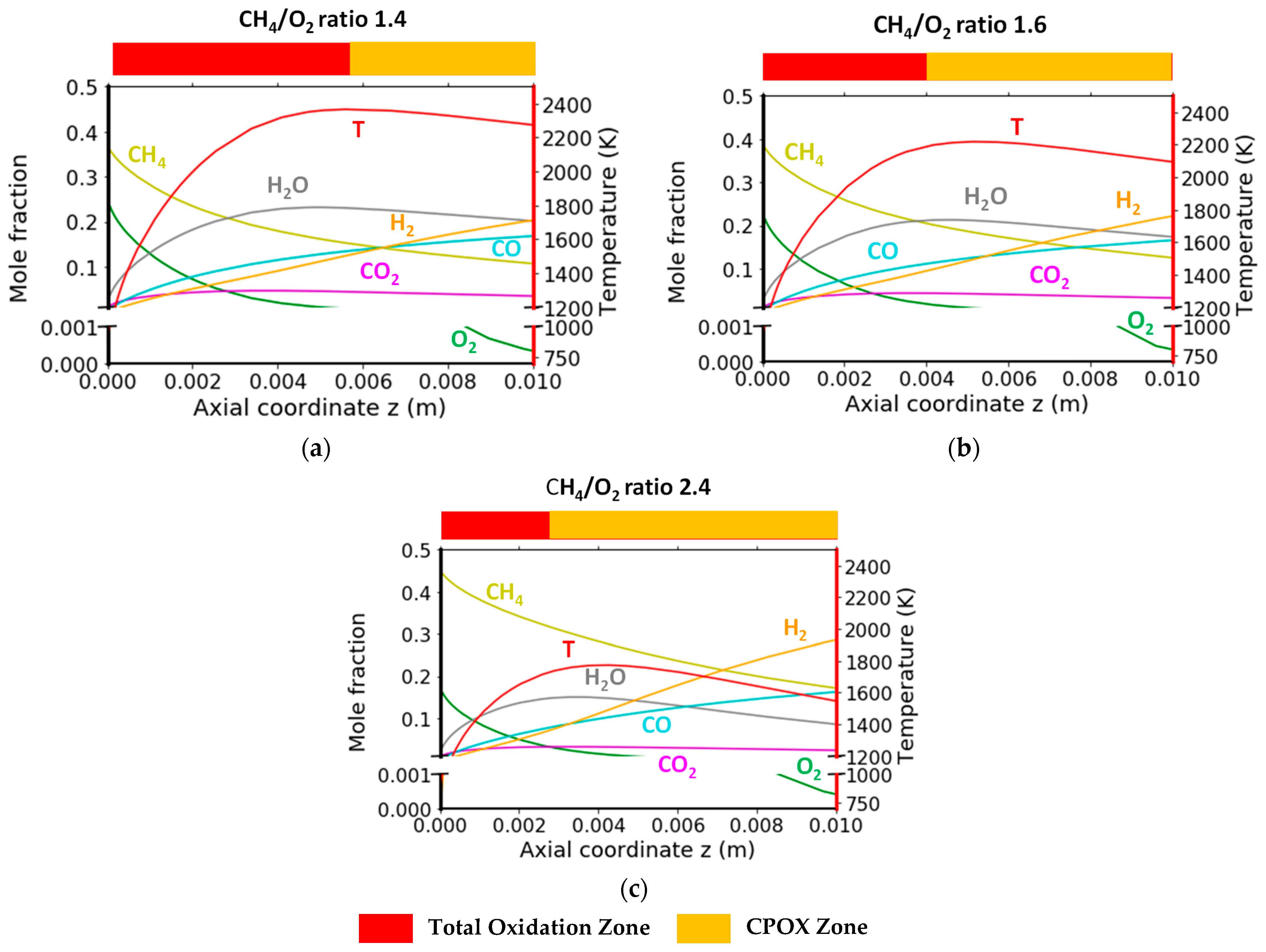
2.2.3. Variation in CH4/O2 Ratio
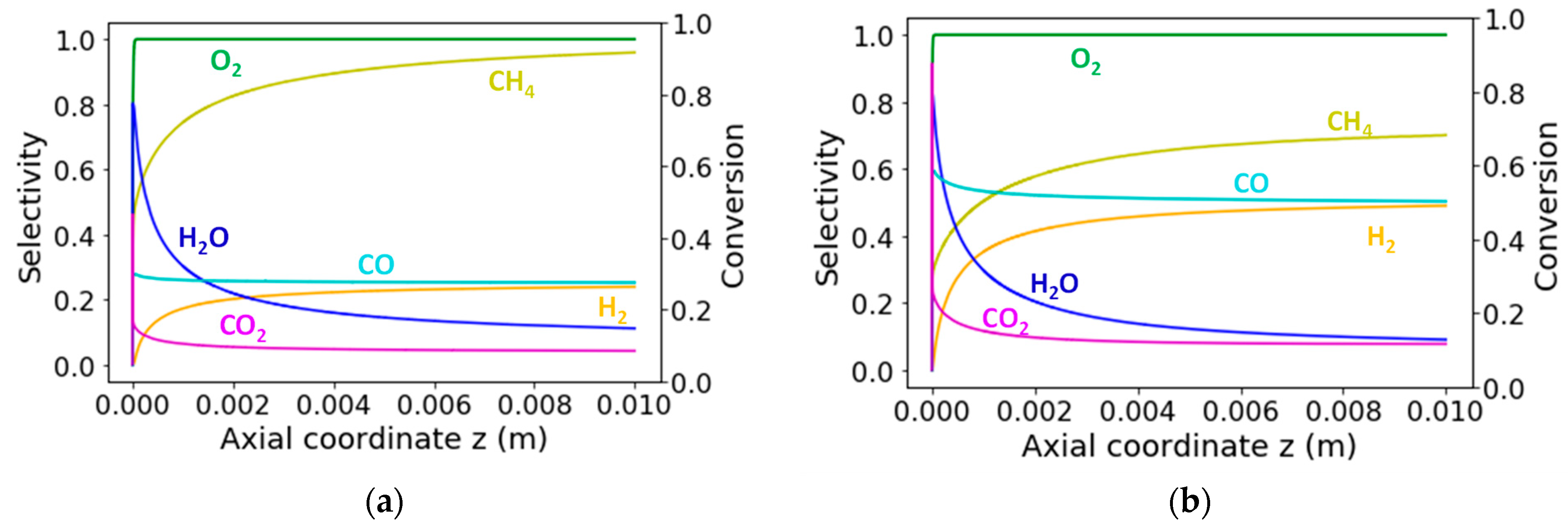
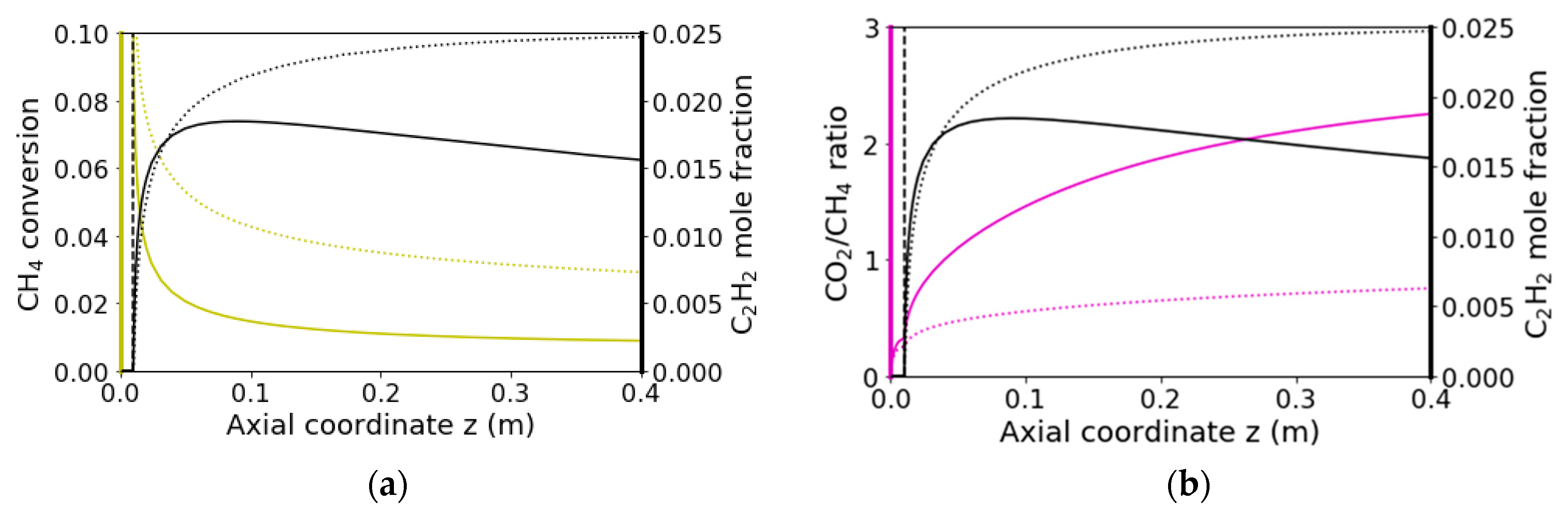
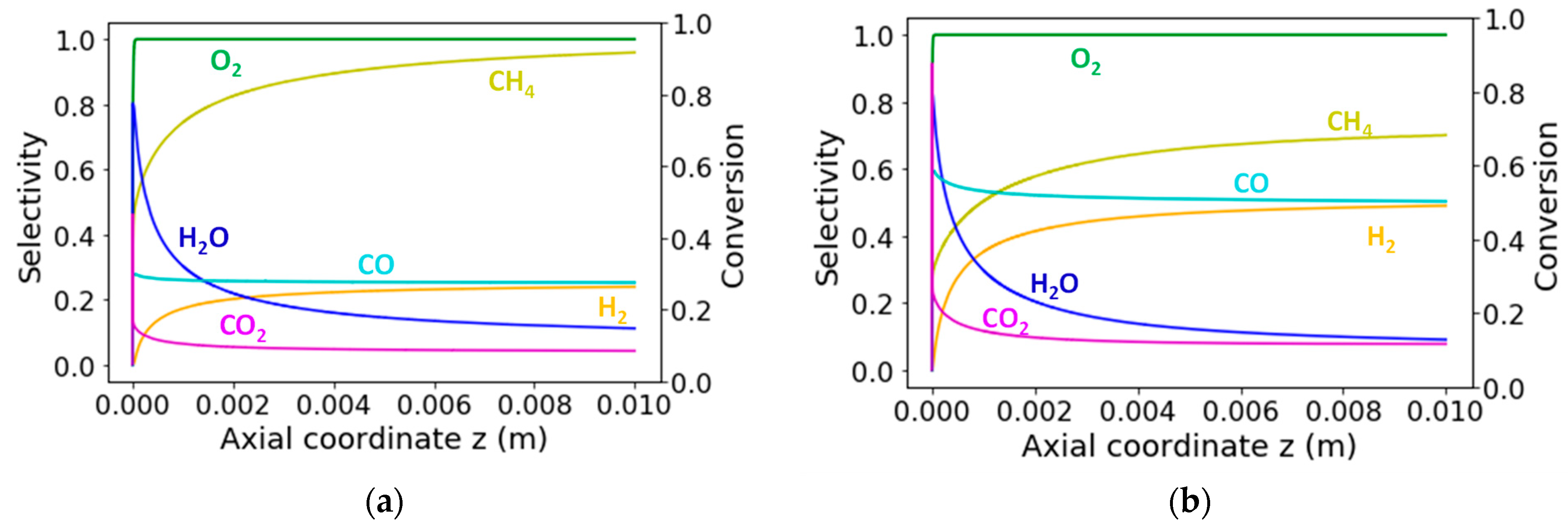
2.3. Catalytic Reaction Kinetics over the Monolith
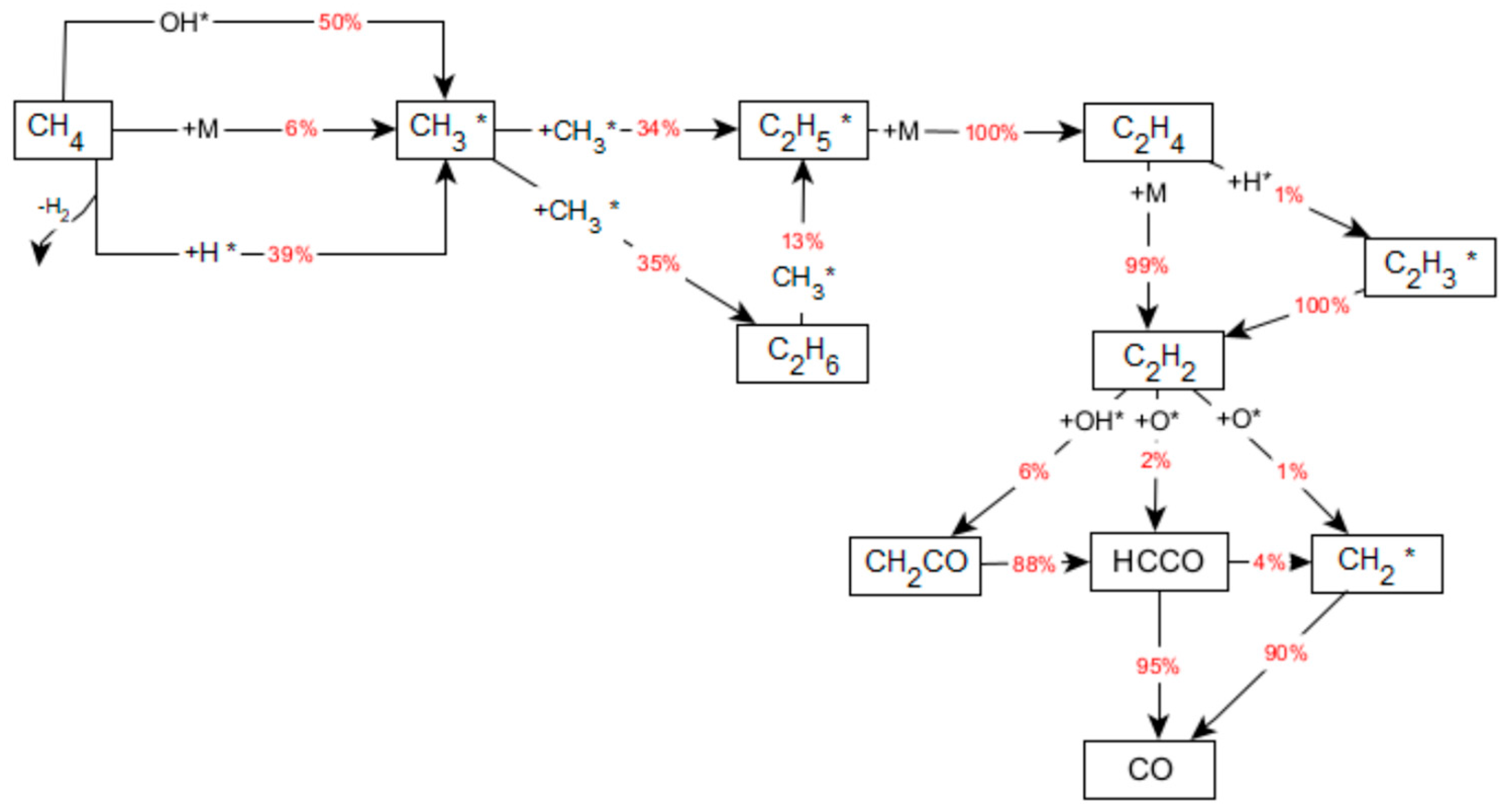
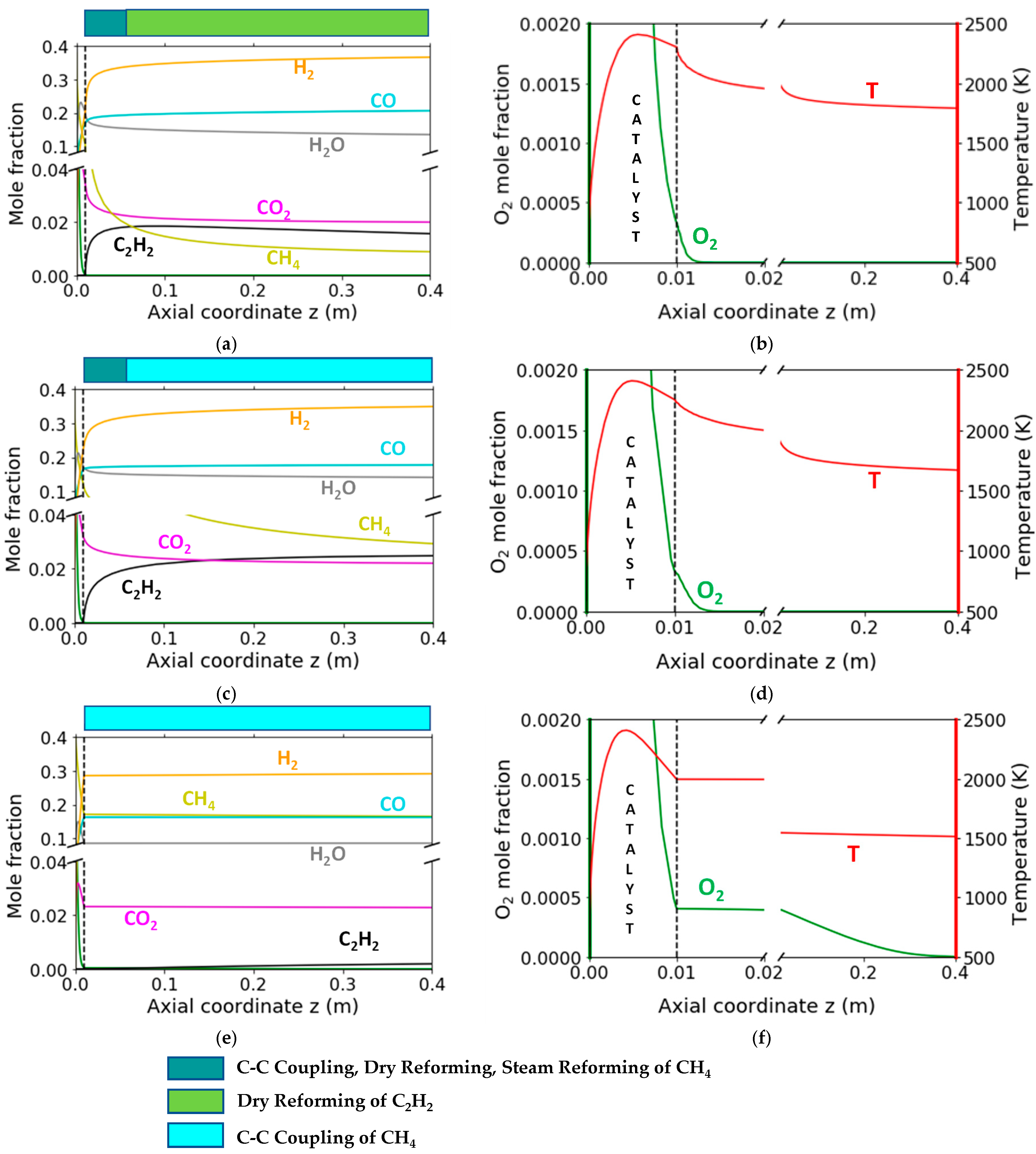
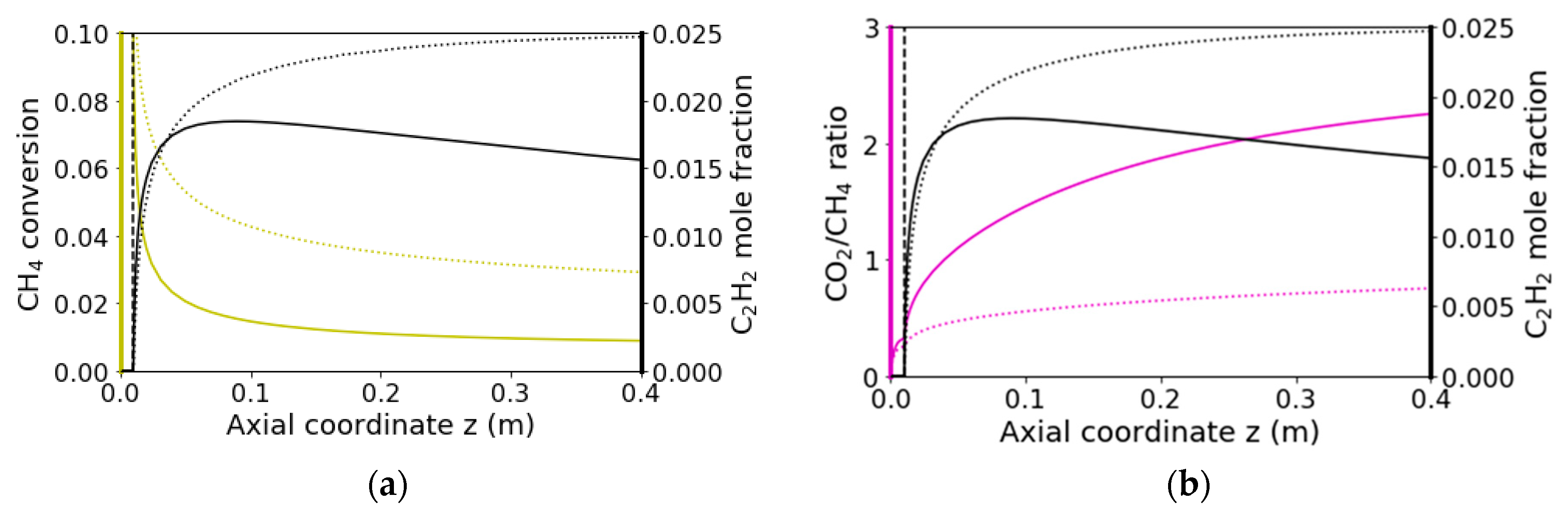
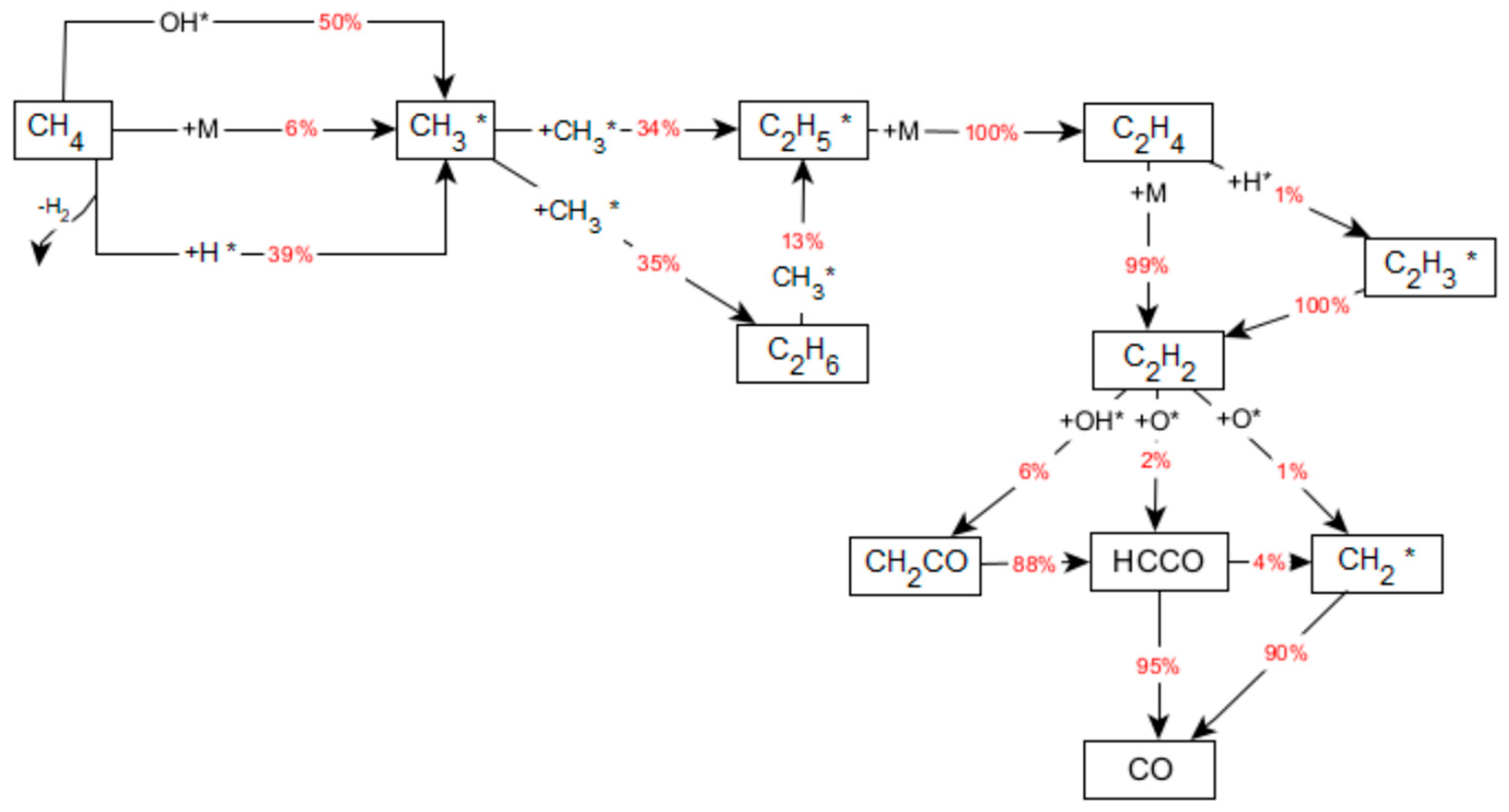
2.4. Homogeneous Reaction Kinetics after the Monolith
- The OH radicals desorbed from the catalytic surface react with methane to generate water:
- On complete consumption of available oxygen, methane releases methyl radicals by H-atom abstraction (reaction (8)) or initiation/thermal decomposition (reaction (9))
3. Materials and Methods
3.1. Experimental
3.1.1. Catalyst Preparation
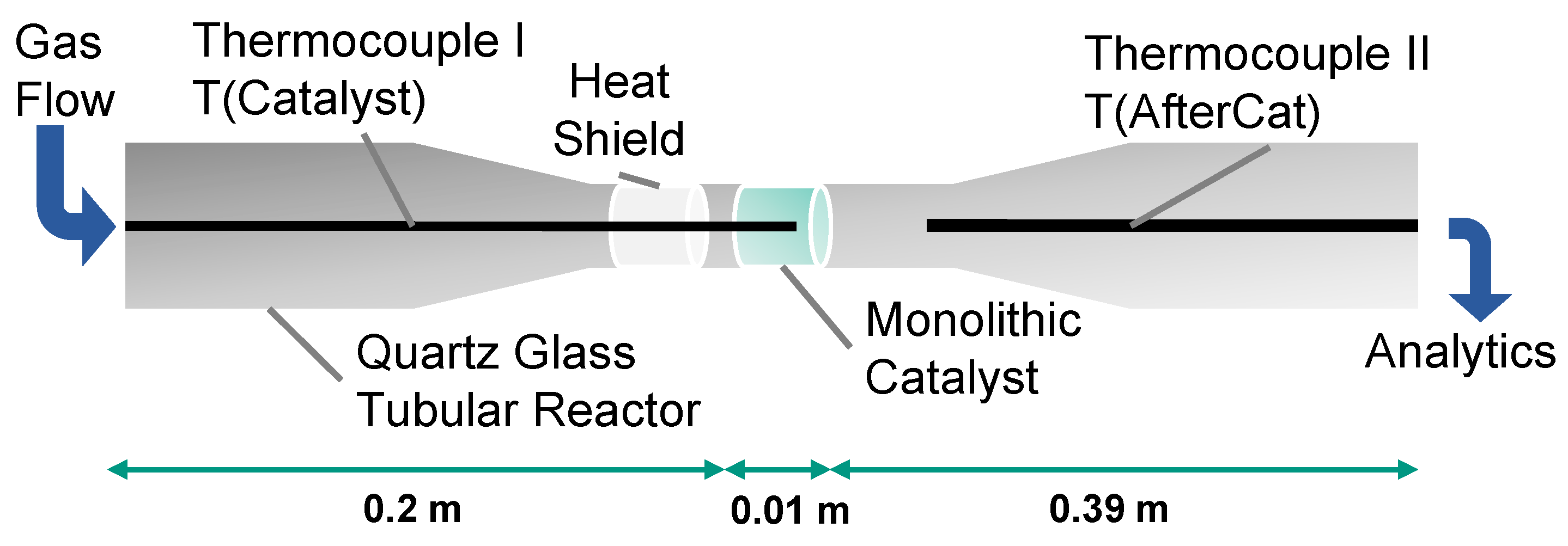
3.1.2. Reactor Setup and Experimental Procedure
3.2. Model Description and Numerical Simulation
3.2.1. Mathematical Model
3.2.2. Reaction Flow Analysis
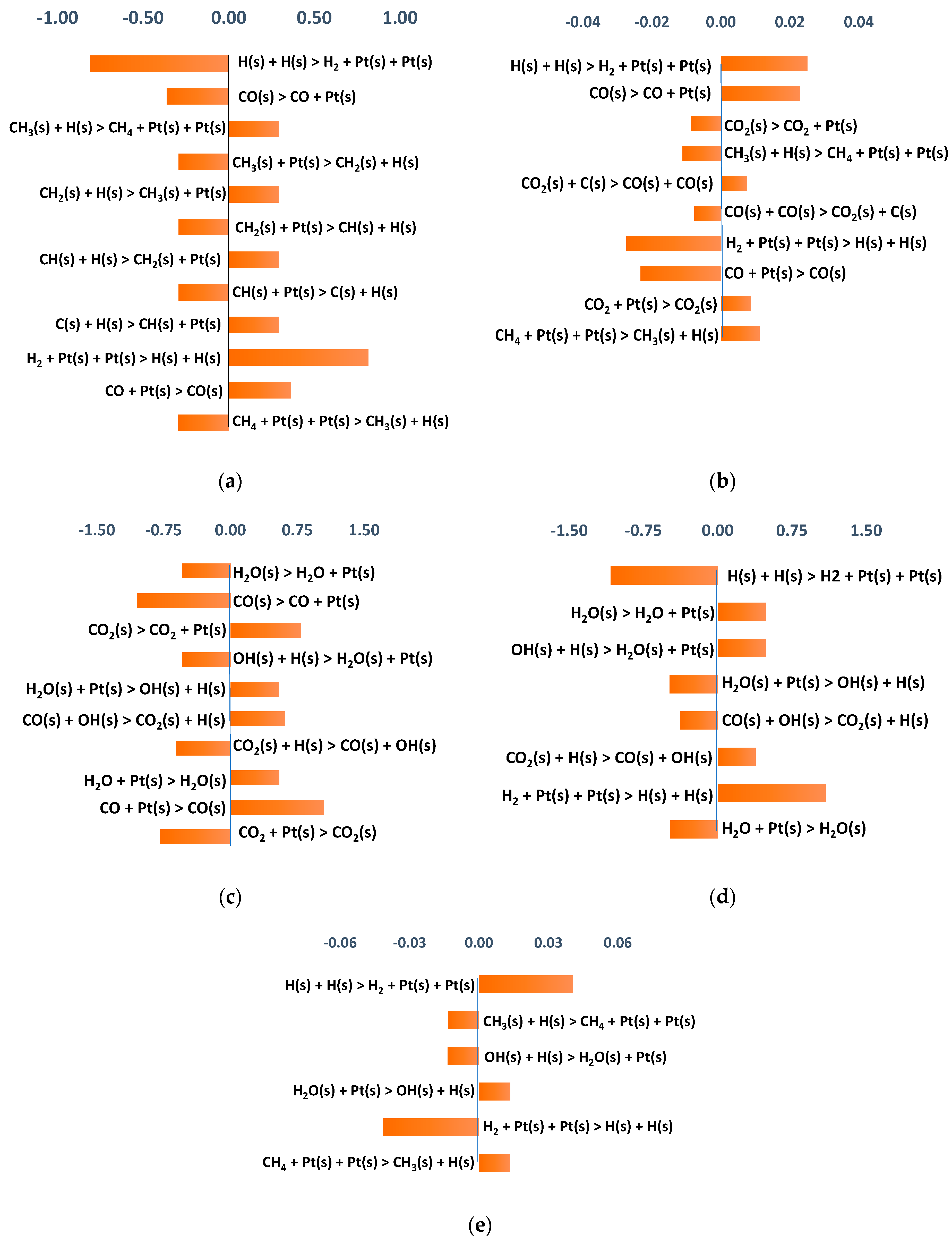
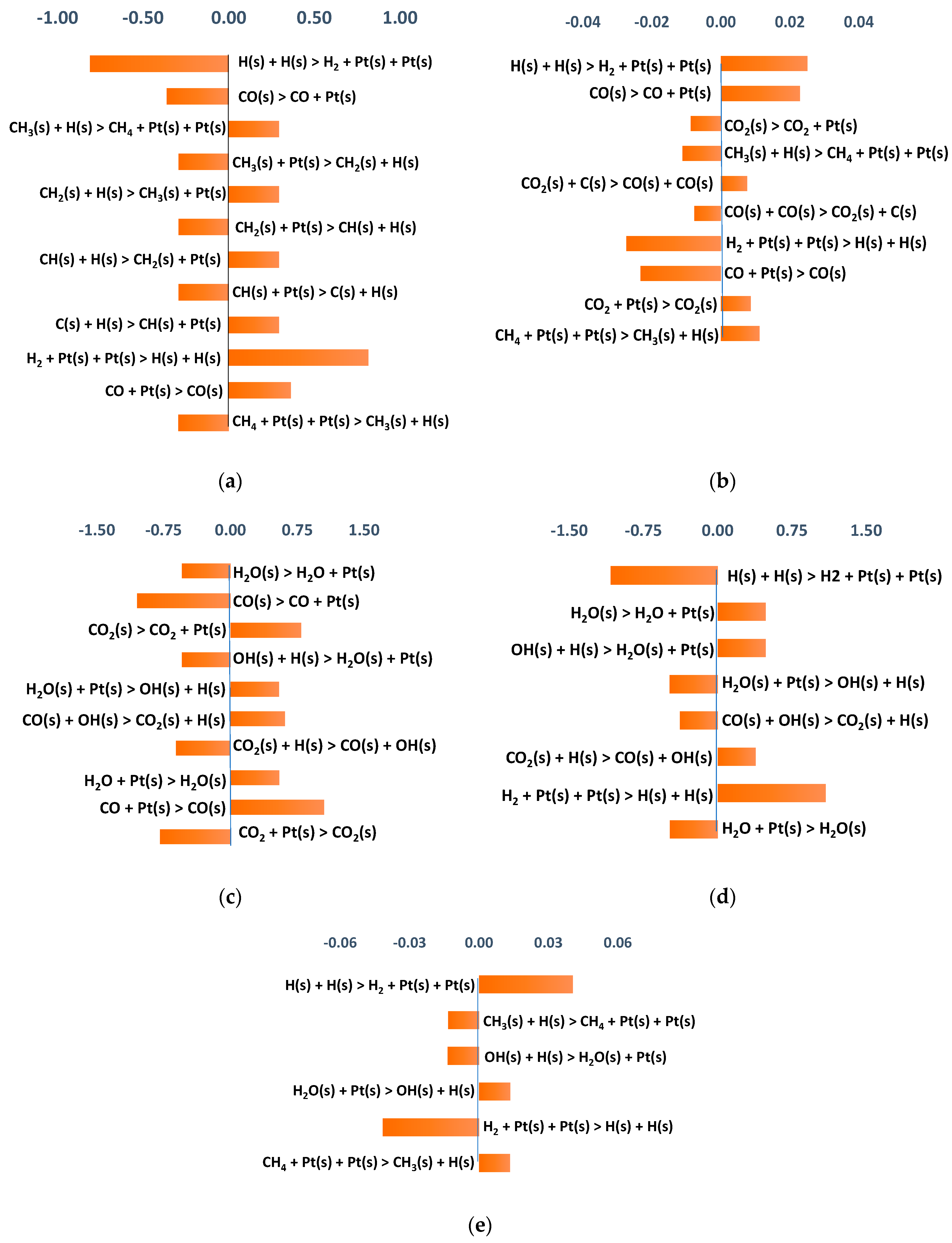
3.2.3. Sensitivity Analysis
3.2.4. Chemical Reaction System
Gas Phase Reaction Models
Surface Reaction Models
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in methane conversion processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Keller, G.E.; Bhasin, M.M. Synthesis of ethylene via oxidative coupling of methane. I. Determination of active catalysts. J. Catal. 1982, 73, 9–19. [Google Scholar] [CrossRef]
- Yunarti, R.T.; Lee, M.; Hwang, Y.J.; Choi, J.W.; Suh, D.J.; Lee, J.; Kim, I.W.; Ha, J.M. Transition metal-doped TiO2 nanowire catalysts for the oxidative coupling of methane. Catal. Commun. 2014, 50, 54–58. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Revisiting the oxidative coupling of methane to ethylene in the golden period of shale gas: A review. J. Ind. Eng. Chem. 2016, 37, 1–13. [Google Scholar] [CrossRef]
- Ghiasi, M.; Malekzadeh, A.; Hoseini, S.; Mortazavi, Y.; Khodadadi, A.; Talebizadeh, A. Kinetic study of oxidative coupling of methane over Mn and/or W promoted Na2SO4/SiO2 catalysts. J. Nat. Gas. Chem. 2011, 20, 428–434. [Google Scholar] [CrossRef]
- Conway, S.J.; Wang, D.J.; Lunsford, J.H. Selective oxidation of methane and ethane over Li+-MgO-Cl- catalysts promoted with metal oxides. Appl. Catal. A Gen. 1991, 79, L1–L5. [Google Scholar] [CrossRef]
- Spinicci, R.; Marini, P.; de Rossi, S.; Faticanti, M.; Porta, P. Oxidative coupling of methane on LaAlO3 perovskites partially substituted with alkali or alkali-earth ions. J. Mol. Catal. A Chem. 2001, 176, 253–265. [Google Scholar] [CrossRef]
- Karakaya, C.; Zhu, H.; Zohour, B.; Senkan, S.; Kee, R.J. Detailed Reaction Mechanisms for the Oxidative Coupling of Methane over La2O3/CeO2 Nanofiber Fabric Catalysts. ChemCatChem 2017, 9, 4538–4551. [Google Scholar] [CrossRef]
- Feng, Y.; Niiranen, J.; Gutman, D. Kinetic studies of the catalytic oxidation of methane. 2. Methyl radical recombination and ethane formation over 1% Sr/La2O3. J. Phys. Chem. 1991, 95, 6564–6568. [Google Scholar] [CrossRef]
- Trueba, M.; Trasatti, S.P. γ-alumina as a support for catalysts: A review of fundamental aspects. Eur. J. Inorg. Chem. 2005, 3393–3403. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, Q.; Yu, L.; Chen, L.; Crocker, M.; Shi, C. New insights into the size and support effects of γ-Al2O3 supported Au catalysts for HCHO oxidation at room temperature. Catal. Sci. Technol. 2020, 10, 4571–4579. [Google Scholar] [CrossRef]
- Korup, O.; Goldsmith, C.F.; Weinberg, G.; Geske, M.; Kandemir, T.; Schlögl, R.; Horn, R. Catalytic partial oxidation of methane on platinum investigated by spatial reactor profiles, spatially resolved spectroscopy, and microkinetic modeling. J. Catal. 2013, 297, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hohn, K.L.; Witt, P.M.; Davis, M.B.; Schmidt, L.D. Methane coupling to acetylene over Pt-coated monoliths at millisecond contact times. Catal. Lett. 1998, 54, 113–118. [Google Scholar] [CrossRef]
- Thybaut, J.W.; Sun, J.; Olivier, L.; van Veen, A.C.; Mirodatos, C.; Marin, G.B. Catalyst design based on microkinetic models: Oxidative coupling of methane. Catal. Today 2011, 159, 29–36. [Google Scholar] [CrossRef]
- Lunsford, J.H. The Catalytic Oxidative Coupling of Methane. Angew. Chem. Int. Ed. Engl. 1995, 34, 970–980. [Google Scholar] [CrossRef]
- Grace, W.R.; Angeles, L. Kinetics of the Oxidative of Methane over 1 wt-% Sr/La2O3. J. Catal. 1988, 118, 517–524. [Google Scholar]
- Labinger, J.A. Oxidative coupling of methane: An inherent limit to selectivity? Catal. Lett. 1988, 1, 371–375. [Google Scholar] [CrossRef]
- Quiceno, R.; Pérez-Ramírez, J.; Warnatz, J.; Deutschmann, O. Modeling the high-temperature catalytic partial oxidation of methane over platinum gauze: Detailed gas-phase and surface chemistries coupled with 3D flow field simulations. Appl. Catal. A Gen. 2006, 303, 166–176. [Google Scholar] [CrossRef]
- Geske, M.; Pelzer, K.; Horn, R.; Jentoft, F.C.; Schlögl, R. In-situ investigation of gas phase radical chemistry in the catalytic partial oxidation of methane on Pt. Catal. Today 2009, 142, 61–69. [Google Scholar] [CrossRef]
- Deutschmann, O.; Tischer, S.; Correa, C.; Chatterjee, D.; Kleditzsch, S.; Janardhanan, V.; Mladenov, N.; Minh, H.D.; Karadeniz, H.; Hettel, M.; et al. DETCHEM Software Package, 2.8 ed.; DETCHEM: Karlsruhe, Germany, 2020. [Google Scholar]
- Schwiedernoch, R.; Tischer, S.; Correa, C.; Deutschmann, O. Experimental and numerical study on the transient behavior of partial oxidation of methane in a catalytic monolith. Chem. Eng. Sci. 2003, 58, 633–642. [Google Scholar] [CrossRef]
- Porras, S.; Kaczmarek, D.; Herzler, J.; Drost, S.; Werler, M.; Kasper, T.; Fikri, M.; Schießl, R.; Atakan, B.; Schulz, C.; et al. An experimental and modeling study on the reactivity of extremely fuel-rich methane/dimethyl ether mixtures. Combust. Flame 2020, 212, 107–122. [Google Scholar] [CrossRef]
- Kahle, L.C.S.; Roussière, T.; Maier, L.; Herrera Delgado, K.; Wasserschaff, G.; Schunk, S.A.; Deutschmann, O. Methane dry reforming at high temperature and elevated pressure: Impact of gas-phase reactions. Ind. Eng. Chem. Res. 2013, 52, 11920–11930. [Google Scholar] [CrossRef]
- Witt, P.M.; Schmidt, L.D. Effect of flow rate on the partial oxidation of methane and ethane. J. Catal. 1996, 163, 465–475. [Google Scholar] [CrossRef]
- Sarsani, S.; West, D.; Liang, W.; Balakotaiah, V. Autothermal oxidative coupling of methane with ambient feed temperature. Chem. Eng. J. 2017, 328, 484–496. [Google Scholar] [CrossRef]
- Kooh, A.; Dubois, J.L.; Mimoun, H.; Cameron, C.J. Oxidative coupling of methane: Maximizing the yield of coupling products under cofeed operating conditions. Catal. Today 1990, 6, 453–462. [Google Scholar] [CrossRef]
- Takanabe, K.; Iglesia, E. Mechanistic aspects and reaction pathways for oxidative coupling of methane on Mn/Na2WO4/SiO2 catalysts. J. Phys. Chem. C 2009, 113, 10131–10145. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.E.; Fahr, A. High-temperature stabilities of hydrocarbons. J. Phys. Chem. 1985, 89, 3714–3725. [Google Scholar] [CrossRef]
- Savchenko, V.I.; Zimin, Y.S.; Nikitin, A.V.; Sedov, I.V.; Arutyunov, V.S. Non-Catalytic Steam Reforming of C1–C4 Hydrocarbons. Pet. Chem. 2021, 61, 762–772. [Google Scholar] [CrossRef]
- Buras, Z.J.; Safta, C.; Zádor, J.; Sheps, L. Simulated production of OH, HO2, CH2O, and CO2 during dilute fuel oxidation can predict 1st-stage ignition delays. Combust. Flame 2020, 216, 472–484. [Google Scholar] [CrossRef]
- Savchenko, V.I.; Nikitin, A.V.; Sedov, I.V.; Ozerskii, A.V.; Arutyunov, V.S. The role of homogeneous steam reforming of acetylene in the partial oxidation of methane to syngas in matrix type converters. Chem. Eng. Sci. 2019, 207, 744–751. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, D.; King, K.D. Reforming of CH4 by partial oxidation: Thermodynamic and kinetic analyses. Fuel 2001, 80, 899–905. [Google Scholar] [CrossRef]
- Hiblot, H.; Ziegler-Devin, I.; Fournet, R.; Glaude, P.A. Steam reforming of methane in a synthesis gas from biomass gasification. Int. J. Hydrogen Energy 2016, 41, 18329–18338. [Google Scholar] [CrossRef] [Green Version]
- Angeli, S.D.; Gossler, S.; Lichtenberg, S.; Kass, G.; Agrawal, A.K.; Valerius, M.; Kinzel, K.P.; Deutschmann, O. Reduction of CO2 Emission from Off-Gases of Steel Industry by Dry Reforming of Methane. Angew. Chem. Int. Ed. 2021, 60, 11852–11857. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, V.I.; Zimin, Y.S.; Nikitin, A.V.; Sedov, I.V.; Arutyunov, V.S. Utilization of CO2 in non-catalytic dry reforming of C1–C4 hydrocarbons. J. CO2 Util. 2021, 47, 101490. [Google Scholar] [CrossRef]
- Gänzler, A.M.; Casapu, M.; Doronkin, D.E.; Maurer, F.; Lott, P.; Glatzel, P.; Votsmeier, M.; Deutschmann, O.; Grunwaldt, J.-D. Unravelling the Different Reaction Pathways for Low Temperature CO Oxidation on Pt/CeO2 and Pt/Al2O3 by Spatially Resolved Structure-Activity Correlations. J. Phys. Chem. Lett. 2019, 10, 7698–7705. [Google Scholar] [CrossRef] [Green Version]
- Karinshak, K.A.; Lott, P.; Harold, M.P.; Deutschmann, O. In situ Activation of Bimetallic Pd−Pt Methane Oxidation Catalysts. ChemCatChem 2020, 12, 3712–3720. [Google Scholar] [CrossRef]
- Chan, D.; Tischer, S.; Heck, J.; Diehm, C.; Deutschmann, O. Correlation between catalytic activity and catalytic surface area of a Pt/Al2O3 DOC: An experimental and microkinetic modeling study. Appl. Catal. B Environ. 2014, 156–157, 153–165. [Google Scholar] [CrossRef]
- Bruix, A.; Margraf, J.T.; Andersen, M.; Reuter, K. First-principles-based multiscale modelling of heterogeneous catalysis. Nat. Catal. 2019, 2, 659–670. [Google Scholar] [CrossRef]
- Gossler, H.; Deutschmann, O. Numerical optimization and reaction flow analysis of syngas production via partial oxidation of natural gas in internal combustion engines. Int. J. Hydrogen Energy 2015, 40, 11046–11058. [Google Scholar] [CrossRef]
- Herrera Delgado, K.; Maier, L.; Tischer, S.; Zellner, A.; Stotz, H.; Deutschmann, O. Surface reaction kinetics of steam- and CO2-reforming as well as oxidation of methane over nickel-based catalysts. Catalysts 2015, 5, 871–904. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, W.; Lissianski, V.; Qin, Z.; Smith, G.; Golden, D.; Frenklach, M.; Eiteneer, B.; Goldenberg, M.; Moriarty, N.; Bowman, C.; et al. The GRI-Mech(TM) Model for Natural Gas Combustion and NO Formation and Removal Chemistry. In Proceedings of the Fifth International Conference on Technologies and Combustion for a Clean Environment, Lisbon, Portugal, 12–15 July 1999; pp. 153–155. [Google Scholar]
- Healy, D.; Curran, H.J.; Dooley, S.; Simmie, J.M.; Kalitan, D.M.; Petersen, D.K.; Bourque, G. Methane/propane mixture oxidation at high pressures and at high, intermediate and low temperatures. Combust. Flame 2008, 155, 451–461. [Google Scholar] [CrossRef]
- Naik, C.V.; Dean, A.M. Detailed kinetic modeling of ethane oxidation. Combust. Flame 2006, 145, 16–37. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | 2–3% Pt/Al2O3 1 | 2–3% Pt/Al2O3 1 | 1% Pt/Al2O3 2 | 1% Pt/Al2O3 2 | 1% Pt/Al2O3 2 |
|---|---|---|---|---|---|
| Inlet temperature (K) | 773 | 773 | 773 | 773 | 773 |
| Monolith length (m) | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| Monolith diameter (m) | 0.018 | 0.018 | 0.01 | 0.01 | 0.01 |
| Channel diameter (m) | 2.3 × 10−4 | 2.3 × 10−4 | 6.3 × 10−4 | 6.3 × 10−4 | 6.3 × 10−4 |
| Flow rate (slpm) | 7.5 | 7.5 | 5 | 5–9 | 6 |
| Velocity (m/s) (at Tinlet 773 K) | 2.4 | 2.4 | 3.6 | 3.6–6.5 | 4.3 |
| GHSV (×105 h−1) | 1.8 | 1.8 | 3.7 | 2.9–7.1 | 4.5 |
| Residence time (ms) | 20 | 20 | 10 | 4–10 | 8 |
| CH4/O2 ratio | 1.7 | 1.4–2.4 | 2.0 | 2.0 | 1.8–2.4 |
| N2 dilution (%) | 20 | 20 | 35–80 | 35 | 35 |
| Reference | [12] | [12] | this work | this work | this work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chawla, J.; Schardt, S.; Angeli, S.; Lott, P.; Tischer, S.; Maier, L.; Deutschmann, O. Oxidative Coupling of Methane over Pt/Al2O3 at High Temperature: Multiscale Modeling of the Catalytic Monolith. Catalysts 2022, 12, 189. https://doi.org/10.3390/catal12020189
Chawla J, Schardt S, Angeli S, Lott P, Tischer S, Maier L, Deutschmann O. Oxidative Coupling of Methane over Pt/Al2O3 at High Temperature: Multiscale Modeling of the Catalytic Monolith. Catalysts. 2022; 12(2):189. https://doi.org/10.3390/catal12020189
Chicago/Turabian StyleChawla, Jaspreet, Sven Schardt, Sofia Angeli, Patrick Lott, Steffen Tischer, Lubow Maier, and Olaf Deutschmann. 2022. "Oxidative Coupling of Methane over Pt/Al2O3 at High Temperature: Multiscale Modeling of the Catalytic Monolith" Catalysts 12, no. 2: 189. https://doi.org/10.3390/catal12020189
APA StyleChawla, J., Schardt, S., Angeli, S., Lott, P., Tischer, S., Maier, L., & Deutschmann, O. (2022). Oxidative Coupling of Methane over Pt/Al2O3 at High Temperature: Multiscale Modeling of the Catalytic Monolith. Catalysts, 12(2), 189. https://doi.org/10.3390/catal12020189