
Co and Ni Incorporated γ-Al2O3 (110) Surface: A Density Functional Theory Study



Abstract
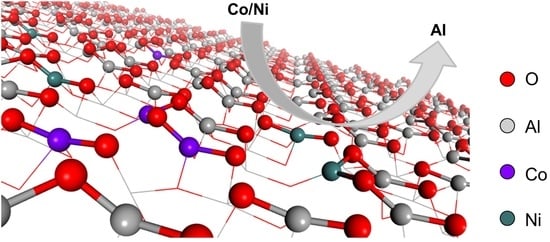
:
1. Introduction
2. Results and Discussion
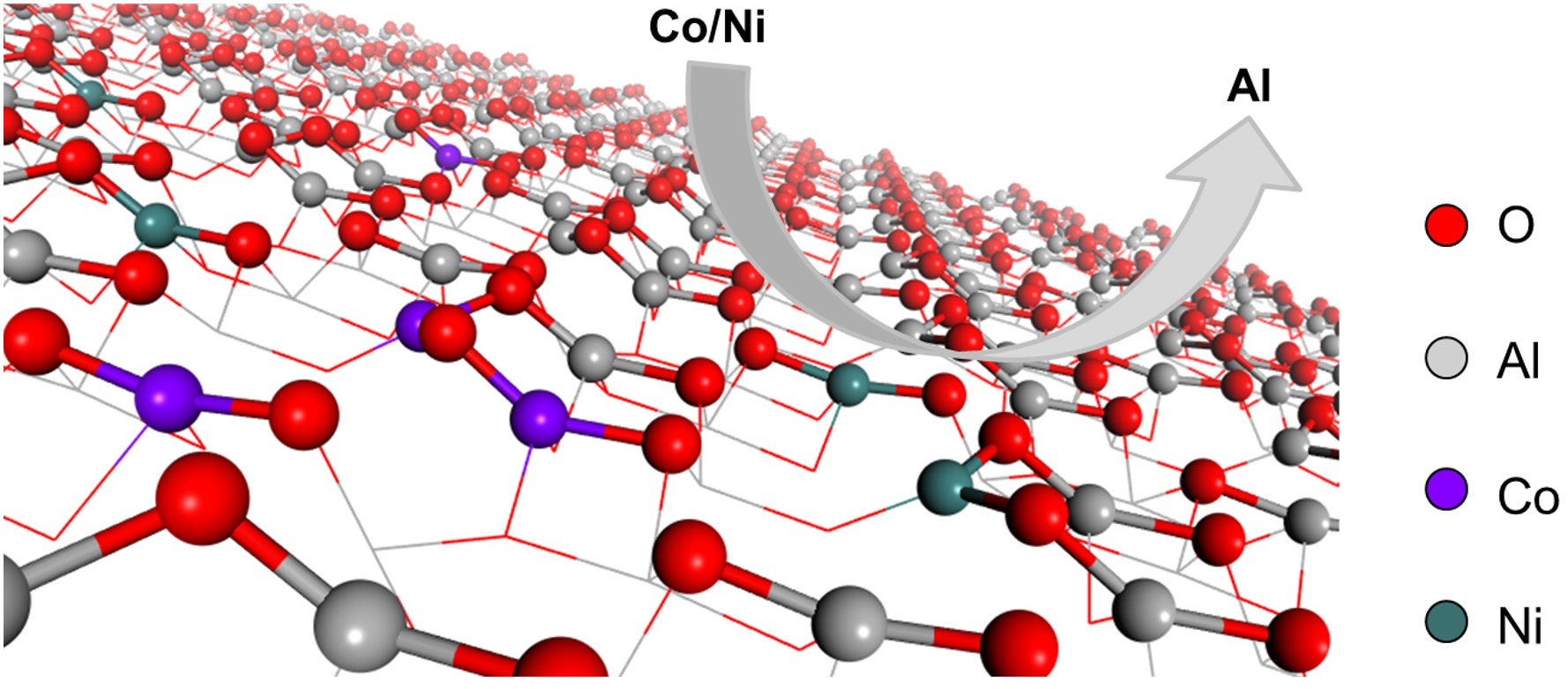
2.1. Substitution of Al3+ by Co3+ and Ni3+
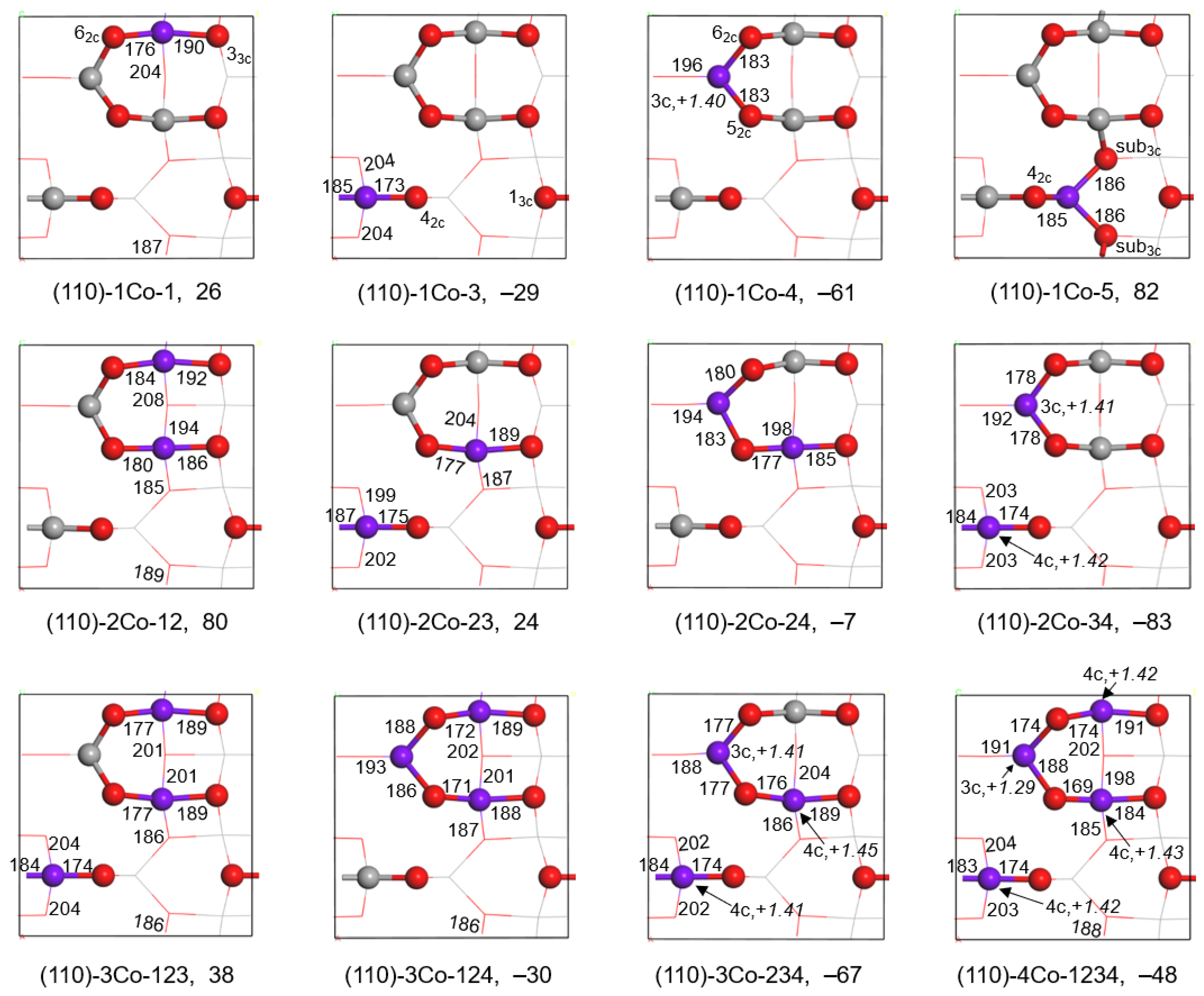
2.1.1. Substitution of Al3+ by Co3+
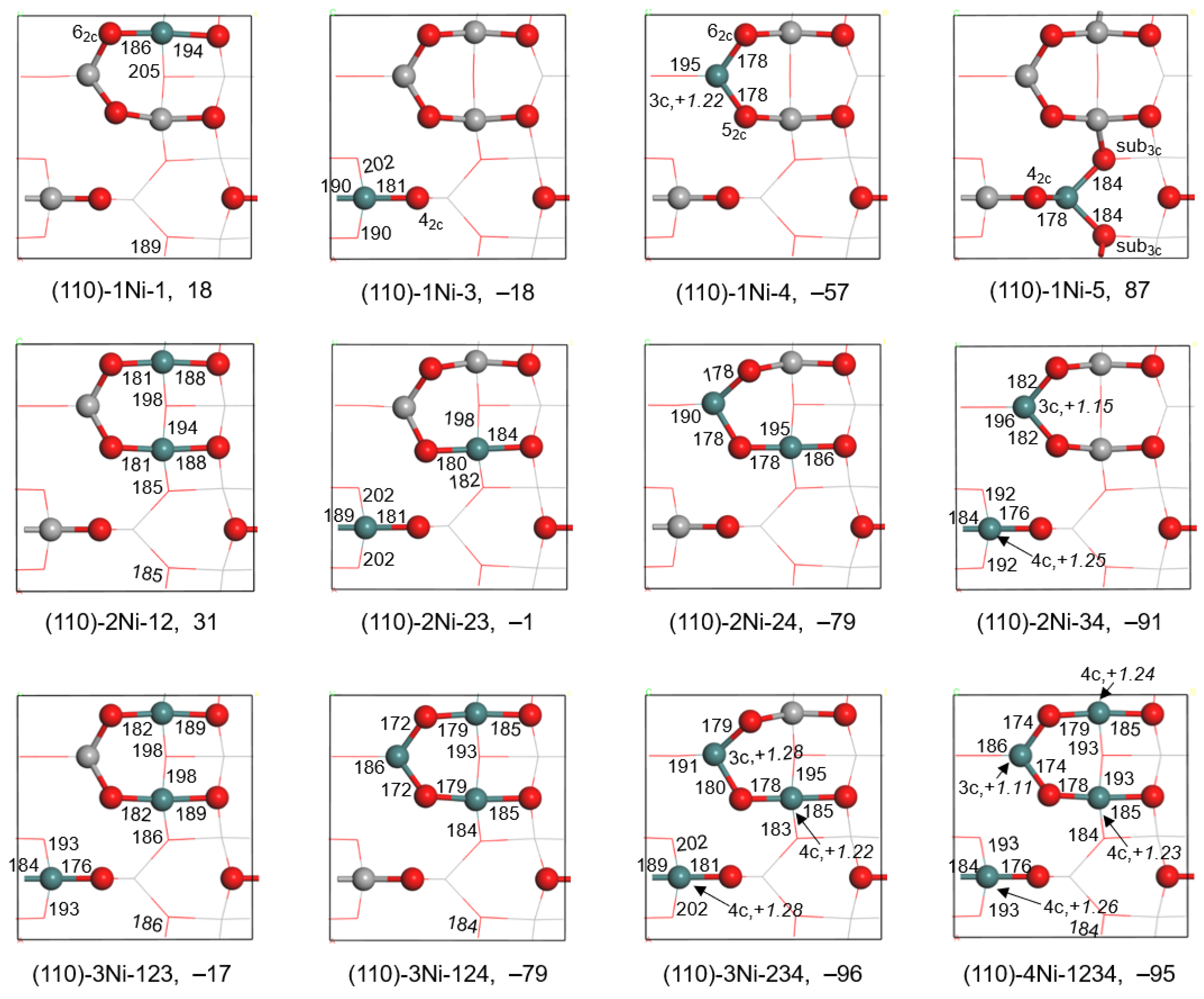
2.1.2. Substitution of Al3+ by Ni3+
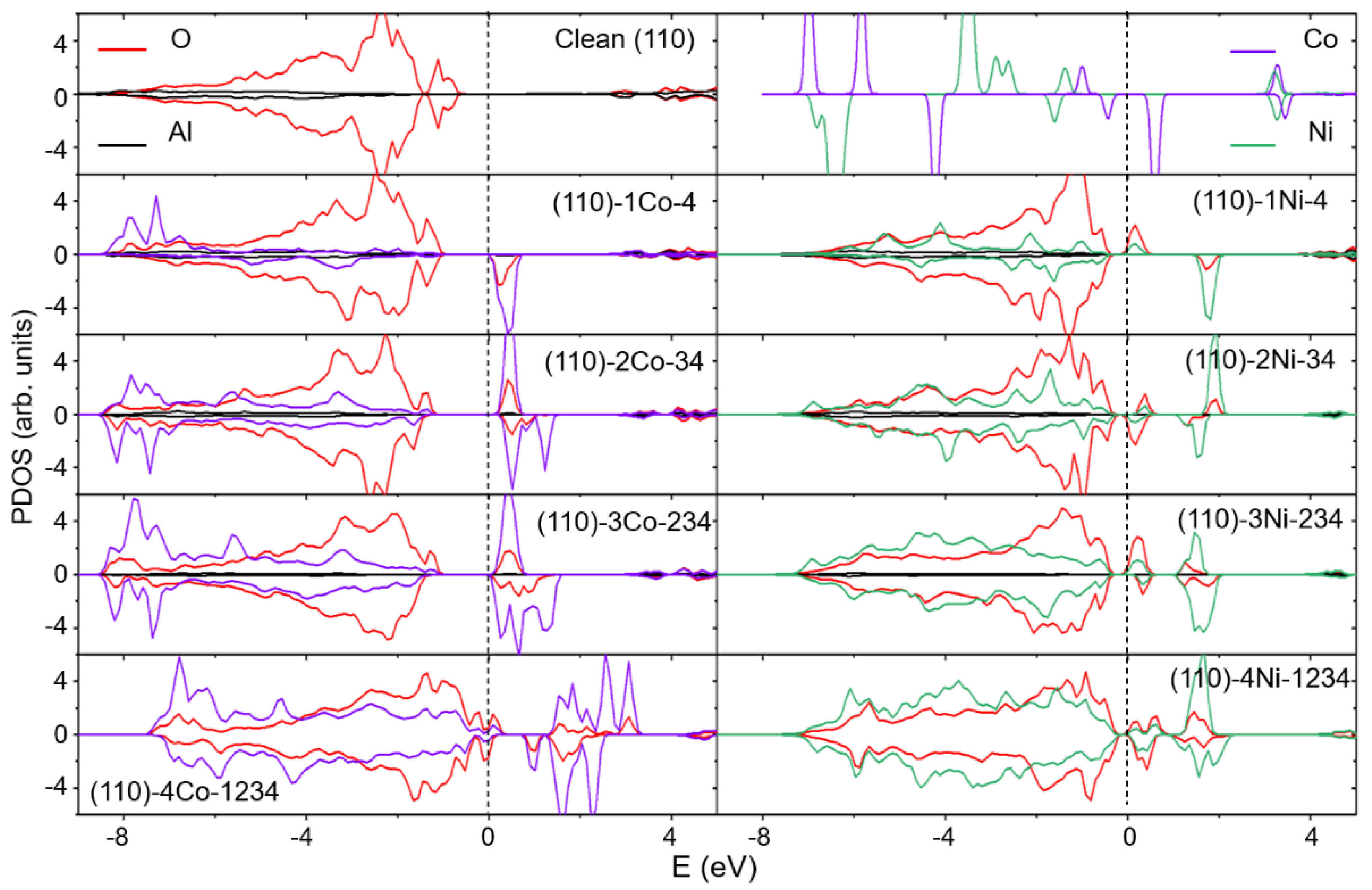
2.2. Electronic Structure Analysis
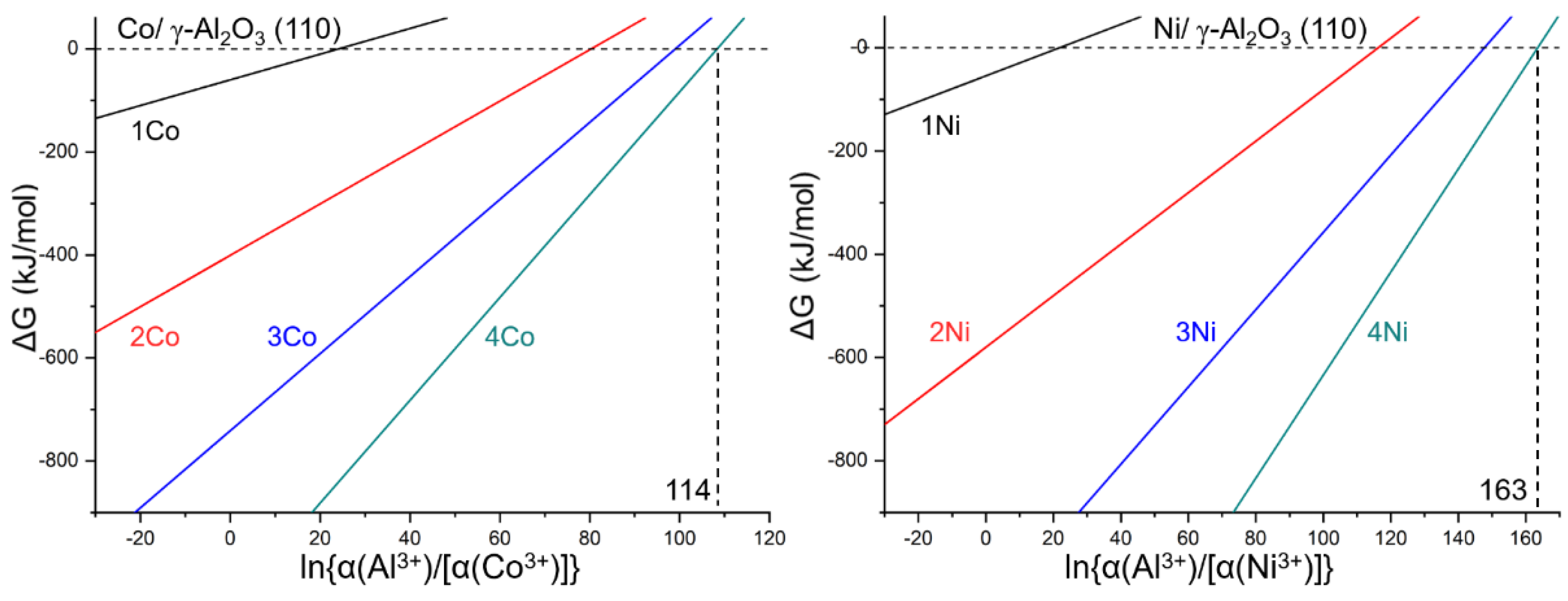
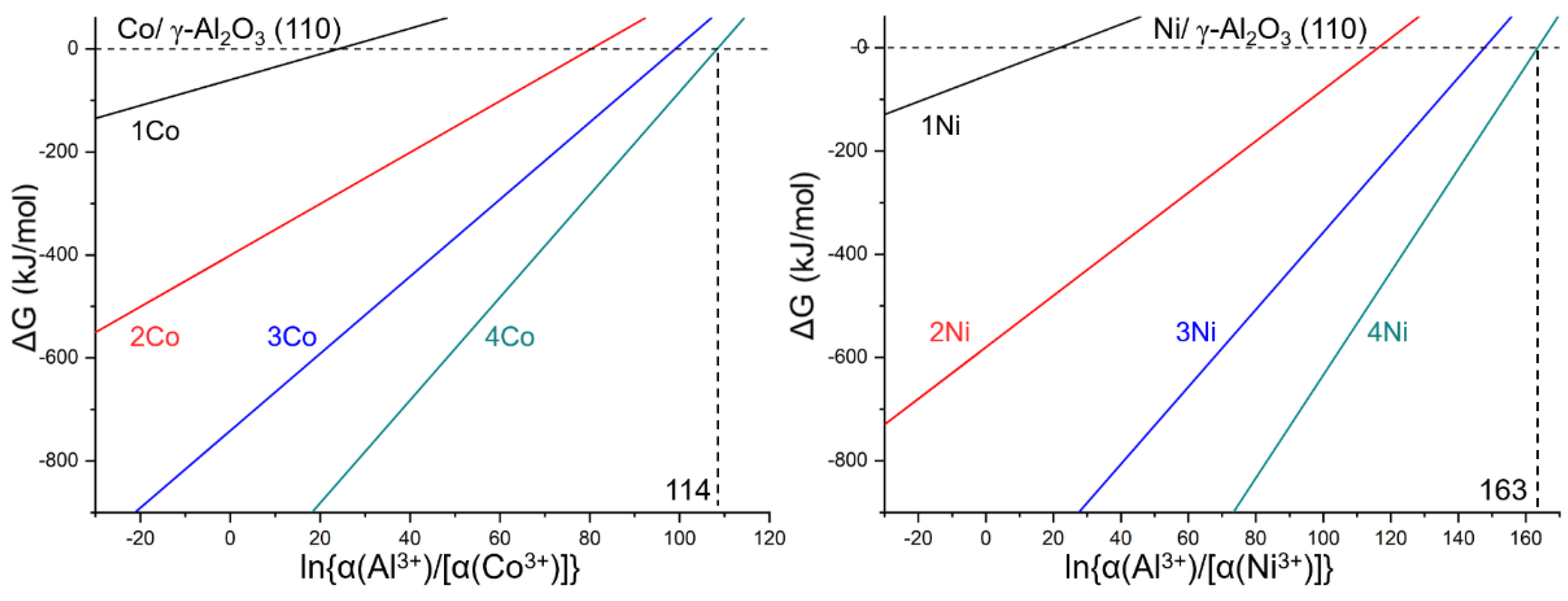
2.3. Thermodynamic Properties
2.3.1. Substitution Reaction under Given Conditions
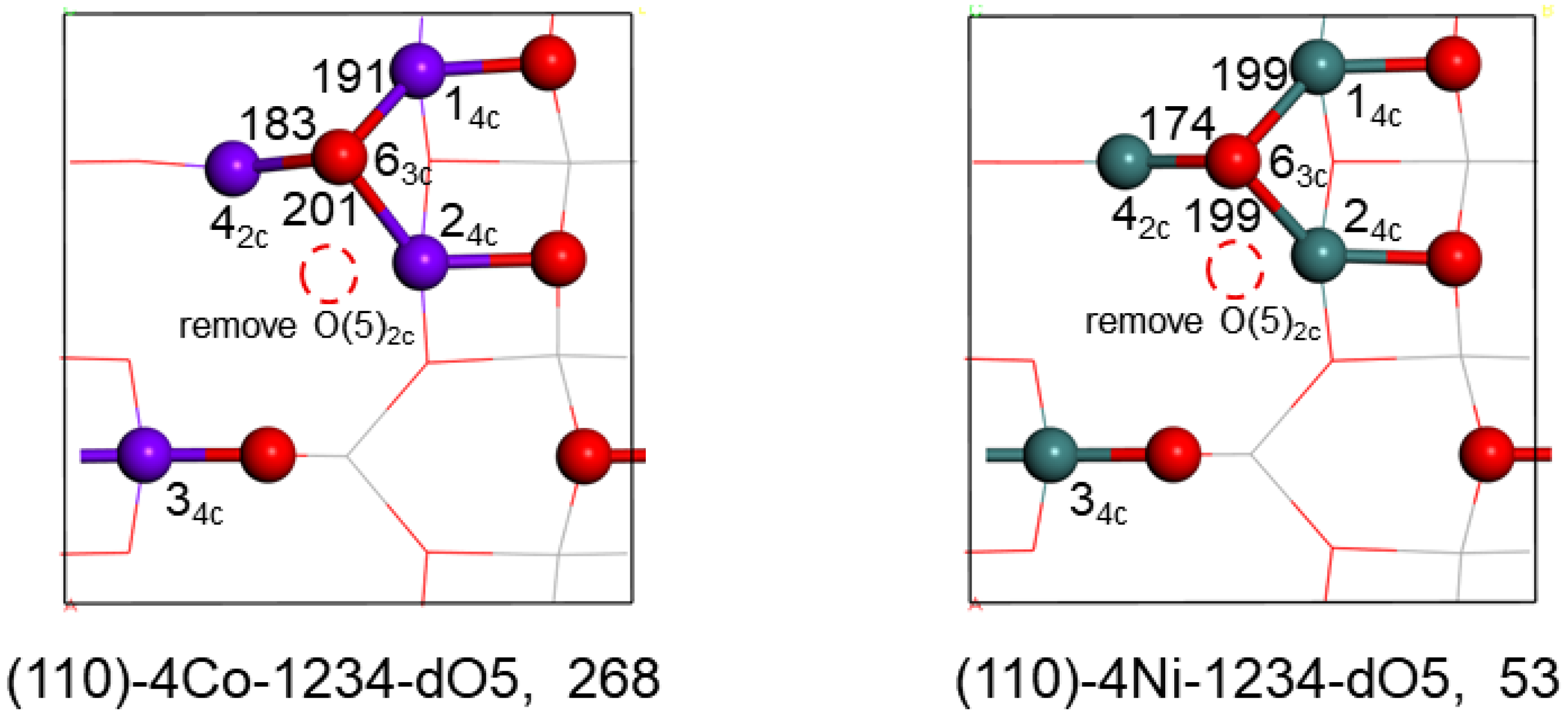
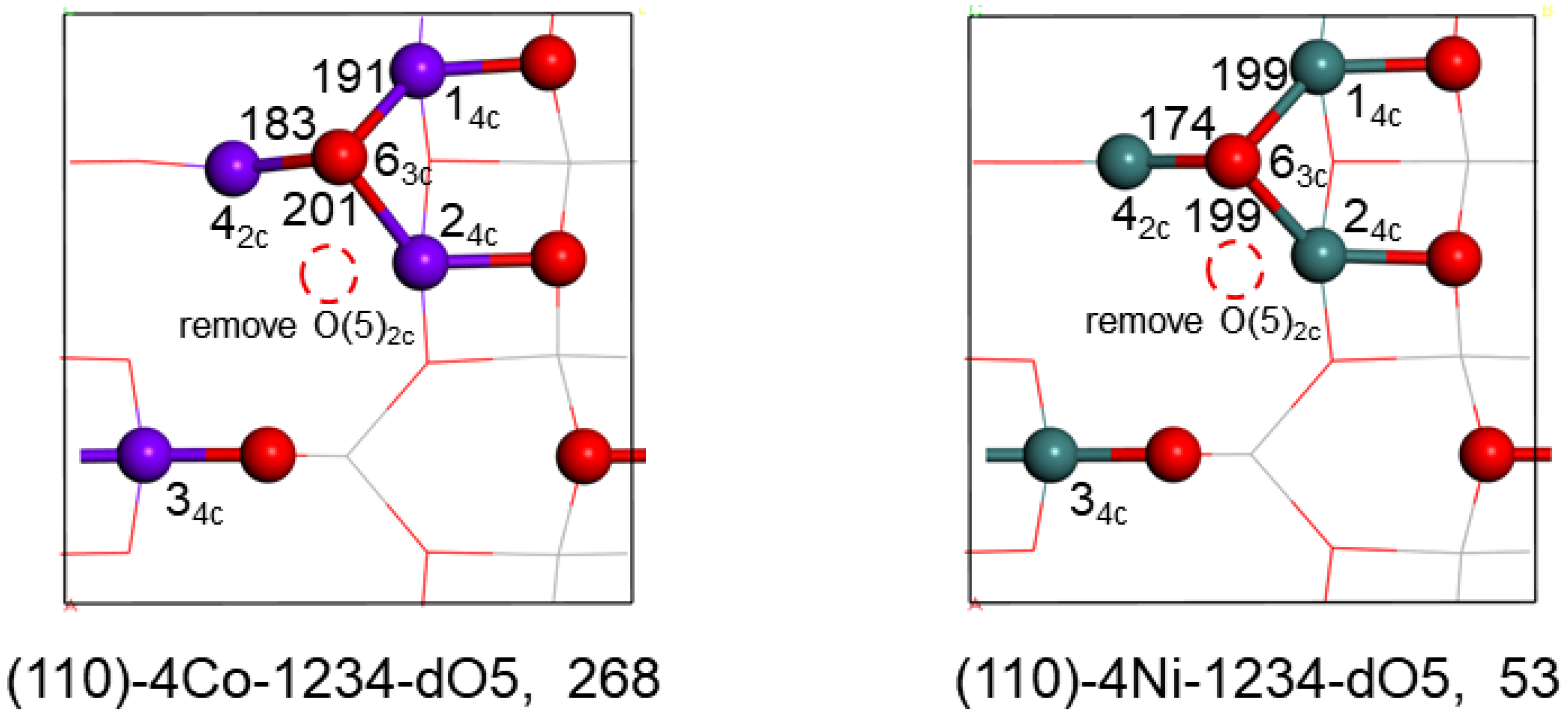
2.3.2. Surface O-Defects of the Co and Ni Substituted Surfaces
3. Computation Details
3.1. Computational Methods
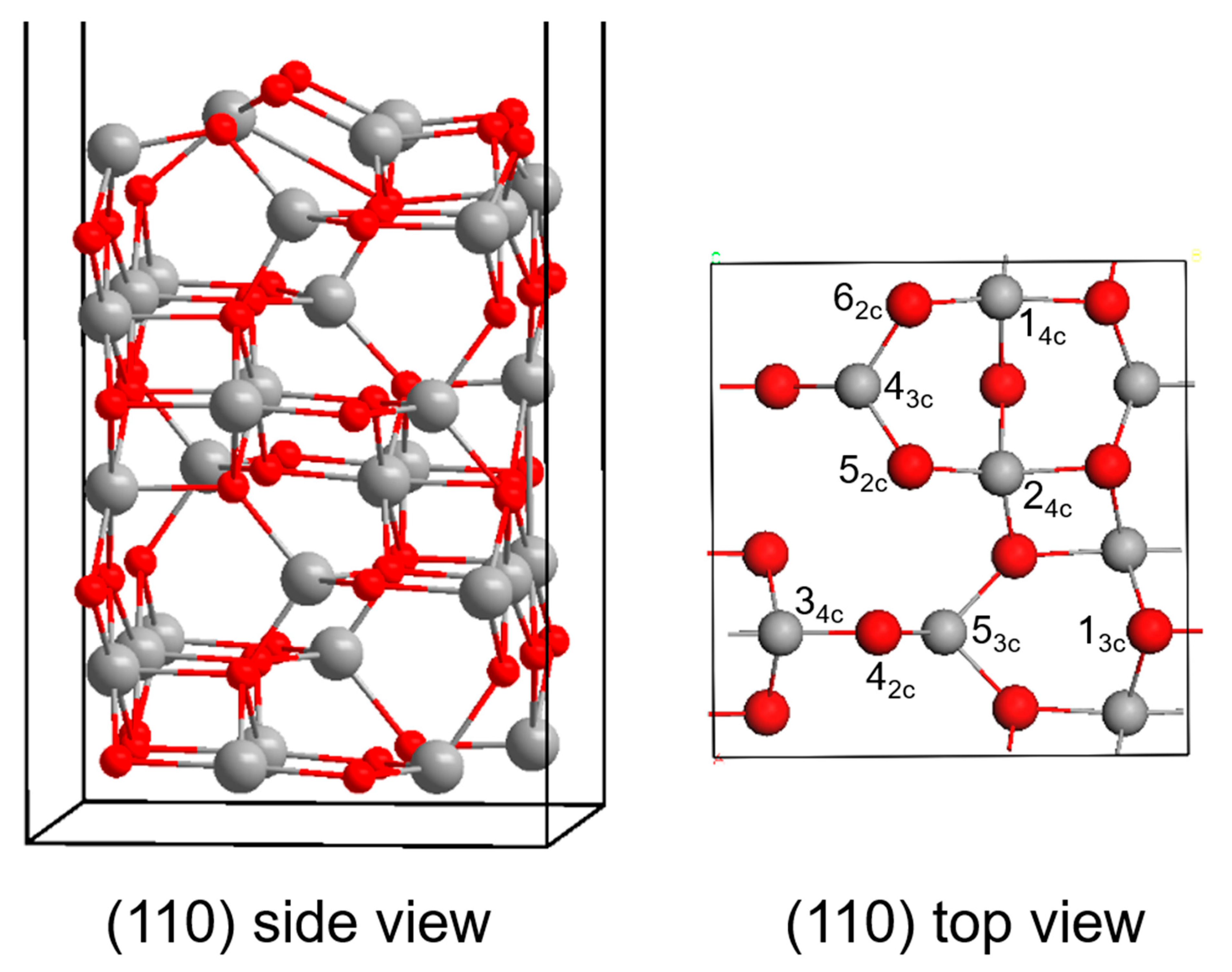
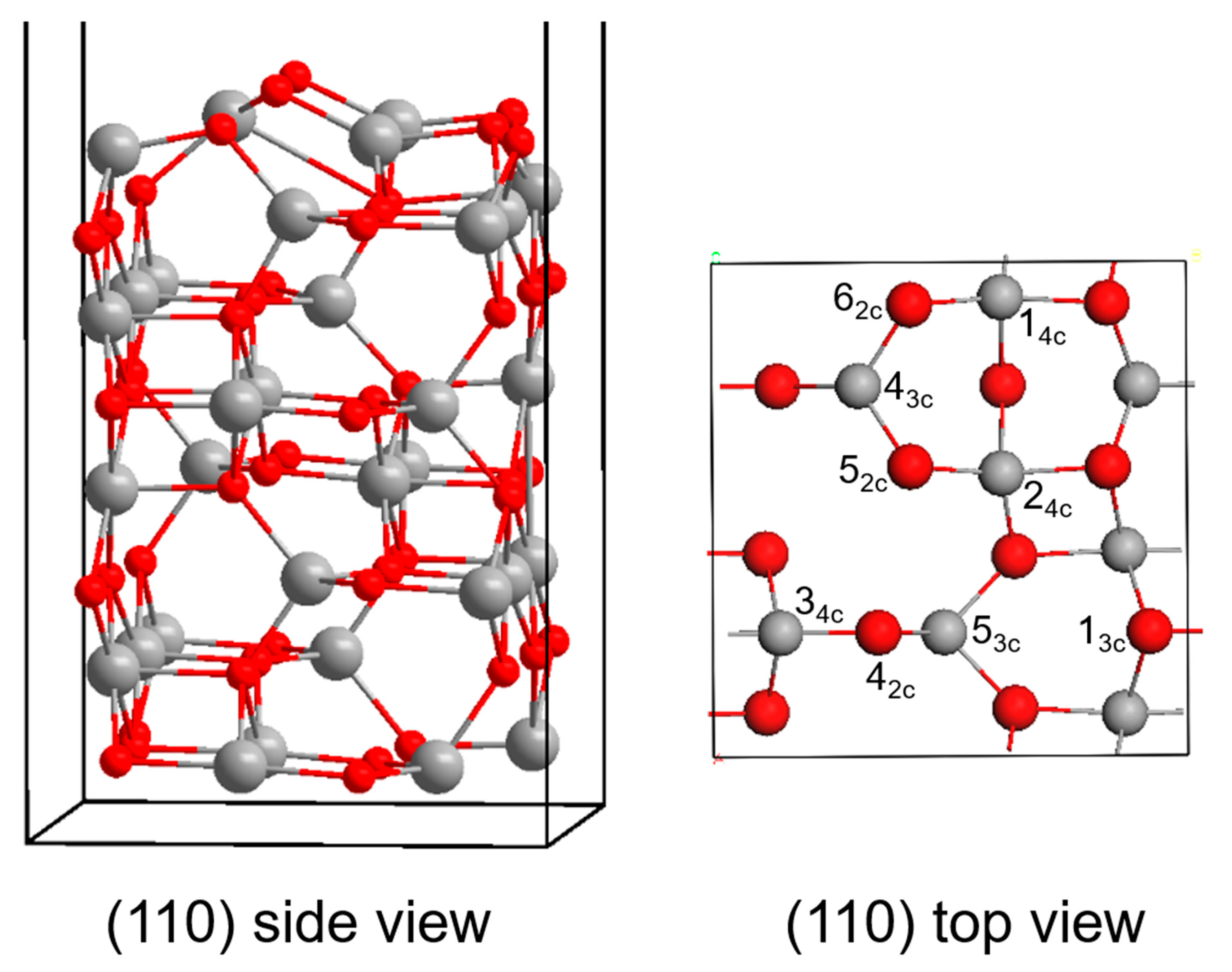
3.2. γ-Al2O3 Surface Model
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weitkamp, J.; Sing, K.S.W.; Schüth, F. Handbook of Porous Solids; Wiley-Vch: Weinheim, Germany, 2002; Volume 3, pp. 1543–1591. [Google Scholar]
- Ma, W.; Jacobs, G.; Das, T.K.; Masuku, C.M.; Kang, J.; Pendyala, V.R.R.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H. Fischer–Tropsch synthesis: Kinetics and water effect on methane formation over 25%Co/γ-Al2O3 Catalyst. Ind. Eng. Chem. Res. 2014, 53, 2157–2166. [Google Scholar] [CrossRef]
- Jacobs, G.; Patterson, P.M.; Zhang, Y.; Das, T.; Li, J.; Davis, B.H. Fischer–Tropsch synthesis: Deactivation of noble metal-promoted Co/Al2O3 catalysts. Appl. Catal. A 2002, 233, 215–226. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Davis, B.H. Fischer–Tropsch synthesis: Study of the promotion of Re on the reduction property of Co/Al2O3 catalysts by in situ EXAFS/XANES of Co K and Re LIII edges and XPS. Appl. Catal. A 2004, 264, 203–212. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, Y.; Wang, S.; Li, J. Fischer–Tropsch synthesis: The effect of Al2O3 porosity on the performance of Co/Al2O3 catalyst. Catal. Commun. 2005, 6, 512–516. [Google Scholar] [CrossRef]
- Hu, D.; Gao, J.; Ping, Y.; Jia, L.; Gunawan, P.; Zhong, Z.; Xu, G.; Gu, F.; Su, F. Enhanced investigation of CO methanation over Ni/Al2O3 catalysts for synthetic natural gas production. Ind. Eng. Chem. Res. 2012, 51, 4875–4886. [Google Scholar] [CrossRef]
- Zhu, X.; Huo, P.; Zhang, Y.-P.; Cheng, D.-G.; Liu, C.-J. Structure and reactivity of plasma treated Ni/Al2O3 catalyst for CO2 reforming of methane. Appl. Catal. B 2008, 81, 132–140. [Google Scholar] [CrossRef]
- Dissanayake, D.; Rosynek, M.P.; Kharas, K.C.; Lunsford, J.H. Partial oxidation of methane to carbon monoxide and hydrogen over a Ni/Al2O3 catalyst. J. Catal. 1991, 132, 117–127. [Google Scholar] [CrossRef]
- Alberton, A.L.; Souza, M.M.; Schmal, M. Carbon formation and its influence on ethanol steam reforming over Ni/Al2O3 catalysts. Catal. Today 2007, 123, 257–264. [Google Scholar] [CrossRef]
- Comas, J.; Mariño, F.; Laborde, M.; Amadeo, N. Bio-ethanol steam reforming on Ni/Al2O3 catalyst. Chem. Eng. J. 2004, 98, 61–68. [Google Scholar] [CrossRef]
- Niklasson, G.A.; Granqvist, C.G. Optical properties and solar selectivity of coevaporated Co-Al2O3 composite films. J. Appl. Phys. 1984, 55, 3382–3410. [Google Scholar] [CrossRef]
- Bechara, R.; Balloy, D.; Vanhove, D. Catalytic properties of Co/Al2O3 system for hydrocarbon synthesis. Appl. Catal. A 2001, 207, 343–353. [Google Scholar] [CrossRef]
- Bechara, R.; Balloy, D.; Dauphin, J.-Y.; Grimblot, J. Influence of the characteristics of γ-aluminas on the dispersion and the reducibility of supported cobalt catalysts. Chem. Mater. 1999, 11, 1703–1711. [Google Scholar] [CrossRef]
- Gonçalves, A.A.S.; Costa, M.J.F.; Zhang, L.; Ciesielczyk, F.; Jaroniec, M. One-pot synthesis of MeAl2O4 (Me = Ni, Co, or Cu) supported on γ-Al2O3 with ultralarge mesopores: Enhancing interfacial defects in γ-Al2O3 to facilitate the formation of Spinel structures at lower temperatures. Chem. Mater. 2018, 30, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Miranda, B.; Chimentao, R.J.; Santos, J.B.; Gispert-Guirado, F.; Llorca, J.; Medina, F.; Bonillo, F.L.; Sueiras, J.E. Conversion of glycerol over 10% Ni/γ-Al2O3 catalyst. Appl. Catal. B 2014, 147, 464–480. [Google Scholar] [CrossRef]
- Wang, H.; Ruckenstein, E. Conversions of methane to synthesis gas over Co/γ-Al2O3 by CO2 and/or O2. Catal. Lett. 2001, 75, 13–18. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhao, X.; Li, J.; Zhu, Q. Role of support in CO2 reforming of CH4 over a Ni/γ-Al2O3 catalyst. Appl. Catal. A 2001, 205, 31–36. [Google Scholar] [CrossRef]
- Hinklin, T.R.; Azurdia, J.; Kim, M.; Marchal, J.C.; Kumar, S.; Laine, R.M. Finding spinel in all the wrong places. Adv. Mater. 2008, 20, 1373–1375. [Google Scholar] [CrossRef] [Green Version]
- Phillips, B.; Hutta, J.; Warshaw, I. Phase equilibria in the system NiO–Al2O3–SiO2. J. Am. Ceram. Soc. 1963, 46, 579–583. [Google Scholar] [CrossRef]
- Cava, S.; Tebcherani, S.; Pianaro, S.; Paskocimas, C.; Longo, E.; Varela, J.A. Structural and spectroscopic analysis of γ-Al2O3 to α-Al2O3-CoAl2O4 phase transition. Mater. Chem. Phys. 2006, 97, 102–108. [Google Scholar] [CrossRef]
- Gassenbauer, Y.; Schafranek, R.; Klein, A.; Zafeiratos, S.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R. Surface states, surface potentials, and segregation at surfaces of tin-doped In2O3. Phys. Rev. B 2006, 73, 245312. [Google Scholar] [CrossRef]
- Rad, A.S. Adsorption of mercaptopyridine on the surface of Al-and B-doped graphenes: Theoretical study. J. Alloys Compd. 2016, 682, 345–351. [Google Scholar]
- Harris, J.; Joyce, B.; Dobson, P. Oscillations in the surface structure of Sn-doped GaAs during growth by MBE. Surf. Sci. Lett. 1981, 103, L90–L96. [Google Scholar]
- Nolan, M. Molecular adsorption on the doped (110) ceria surface. J. Phys. Chem. C 2009, 113, 2425–2432. [Google Scholar] [CrossRef]
- Li, L.; Gan, Y.-M.; Lu, Z.-H.; Yu, X.H.; Qing, S.; Gao, Z.; Zhang, R.; Feng, G. The effects of Fe, Co and Ni doping in CuAl2O4 spinel surface and bulk: A DFT study. Appl. Surf. Sci. 2020, 521, 146478. [Google Scholar] [CrossRef]
- Miran, H.A.; Jaf, Z.N.; Altarawneh, M.; Jiang, Z.T. An insight into geometries and catalytic applications of CeO2 from a DFT outlook. Molecules 2021, 26, 6485. [Google Scholar] [CrossRef]
- Zhang, S.-T.; Li, C.-M.; Yan, H.; Wei, M.; Evans, D.G.; Duan, X. Density functional theory study on the metal–support interaction between Ru cluster and anatase TiO2(101) surface. J. Phys. Chem. C 2014, 118, 3514–3522. [Google Scholar] [CrossRef]
- Gu, J.; Wang, J.; Leszczynski, J. Single site Fe on the (110) surface of gamma-Al2O3: Insights from a DFT study including the periodic boundary approach. Phys. Chem. Chem. Phys. 2021, 23, 7164–7177. [Google Scholar] [CrossRef]
- Benam, M.R.; Rahnamaye Aliabad, H.A.; Hosseini, S.M. Effect of substituted IIIB transition metals on the energy gap of α-Al2O3 by first-principle calculations. Phys. Status Solidi A 2006, 203, 2223–2228. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Hatch, C.; Orlando, R. Electronic properties and reactivity of simulated Fe3+ and Cr3+ substituted α-Al2O3 (0001) surface. J. Phys. Chem. C 2012, 116, 18847–18856. [Google Scholar] [CrossRef] [Green Version]
- Finazzi, E.; Di Valentin, C.; Pacchioni, G. Boron-doped anatase TiO2: Pure and hybrid DFT calculations. J. Phys. Chem. C 2009, 113, 220–228. [Google Scholar] [CrossRef]
- Yang, S.; Lei, G.; Xu, H.; Xu, B.; Li, H.; Lan, Z.; Wang, Z.; Gu, H. A DFT study of CO adsorption on the pristine, defective, In-doped and Sb-doped graphene and the effect of applied electric field. Appl. Surf. Sci. 2019, 480, 205–211. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, H.; Wang, B.; Ling, L. Insights into the effect of surface hydroxyls on CO2 hydrogenation over Pd/γ-Al2O3 catalyst: A computational study. Appl. Catal. B 2012, 126, 108–120. [Google Scholar] [CrossRef]
- Wang, F.; Ma, J.; Xin, S.; Wang, Q.; Xu, J.; Zhang, C.; He, H.; Cheng Zeng, X. Resolving the puzzle of single-atom silver dispersion on nanosized gamma-Al2O3 surface for high catalytic performance. Nat. Commun. 2020, 11, 529. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Huang, Y.; Lu, Z.-H.; Cen, W.; Yu, X.; Qing, S.; Gao, Z.; Zhang, R.; Feng, G. Surface property of the Cu doped γ-Al2O3: A density functional theory study. Appl. Surf. Sci. 2021, 535, 147651. [Google Scholar] [CrossRef]
- Shibiao, R.; Jinheng, Q.; Chunyan, W.; Bolian, X.; Yining, F.; Yi, C. Influence of nickel salt precursors on the hydrogenation activity of Ni/γ-Al2O3 catalyst. Chin. J. Catal. 2007, 28, 651–656. [Google Scholar]
- YU, C.-L.; ZHOU, X.-C.; WENG, W.-Z.; HU, J.-B.; Xi-rong, C.; WEI, L.-F. Effects of alkaline-earth strontium on the performance of Co/Al2O3 catalyst for methane partial oxidation. J. Fuel Chem. Technol. 2012, 40, 1222–1229. [Google Scholar] [CrossRef]
- Nagano, T.; Sato, K.; Saitoh, T.; Takahashi, S. Hydrothermal stability of mesoporous Ni-doped γ-Al2O3. J. Ceram. Soc. Jpn. 2009, 117, 832–835. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xu, Y.; Gao, C.; Zhao, Y. Structural and textural evolution of Ni/γ-Al2O3 catalyst under hydrothermal conditions. Catal. Today 2010, 158, 475–480. [Google Scholar] [CrossRef]
- Liu, J.; Peng, H.; Liu, W.; Xu, X.; Wang, X.; Li, C.; Zhou, W.; Yuan, P.; Chen, X.; Zhang, W. Tin modification on Ni/Al2O3: Designing potent coke-resistant catalysts for the dry reforming of methane. ChemCatChem 2014, 6, 2095–2104. [Google Scholar] [CrossRef]
- Digne, M. Use of DFT to achieve a rational understanding of acid-basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Krokidis, X.; Raybaud, P.; Gobichon, A.E.; Rebours, B.; Euzen, P.; Toulhoat, H. Theoretical study of the dehydration process of boehmite to γ-Alumina. J. Phys. Chem. B 2001, 105, 5121–5130. [Google Scholar] [CrossRef]
- Lee, M.H.; Cheng, C.F.; Heine, V.; Klinowski, J. Distribution of tetrahedral and octahedral Al sites in gamma alumina. Chem. Phys. Lett. 1997, 265, 673–676. [Google Scholar] [CrossRef]
- John, C.S.; Alma, N.C.M.; Hays, G.R. Characterization of transitional alumina by solid-state magic angle spinning aluminium NMR. Appl. Catal. 1983, 6, 341–346. [Google Scholar] [CrossRef]
- Zhang, R.; Duan, T.; Wang, B.; Ling, L. Unraveling the role of support surface hydroxyls and its effect on the selectivity of C2 species over Rh/γ-Al2O3 catalyst in syngas conversion: A theoretical study. Appl. Surf. Sci. 2016, 379, 384–394. [Google Scholar] [CrossRef]
- Ja Hun, K.; Jianzhi, H.; Donghai, M.; Cheol-Woo, Y.; Do Heui, K.; Peden, C.H.F.; Allard, L.F.; Janos, S. Coordinatively unsaturated Al3+ centers as binding sites for active catalyst phases of platinum on gamma-Al2O3. Science 2009, 325, 1670–1673. [Google Scholar]
- Feng, G.; Huo, C.-F.; Li, Y.-W.; Wang, J.; Jiao, H. Structures and energies of iron promoted γ-Al2O3 surface: A computational study. Chem. Phys. Lett. 2011, 510, 224–227. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton T. 2008, 21, 2832–2838. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Orgel, L.E. Electronic properties of transition-metal oxides—I: Distortions from cubic symmetry. J. Phys. Chem. Solids 1957, 3, 20–29. [Google Scholar] [CrossRef]
- Jacob, K.; Alcock, C. Thermodynamics of CuAlO2 and CuAl2O4 and phase equilibria in the system Cu2O-CuO-Al2O3. J. Am. Chem. Soc. 2010, 58, 192–195. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Pan, L.; Lu, J.; Song, J.; Li, Z.; Zhang, X.; Wang, L.; Zou, J.-J. Unraveling the facet-dependent and oxygen vacancy role for ethylene hydrogenation on Co3O4 (110) surface: A DFT+U study. Appl. Surf. Sci. 2017, 401, 241–247. [Google Scholar] [CrossRef]
- Lu, J.; Song, J.; Niu, H.; Pan, L.; Zhang, X.; Wang, L.; Zou, J.-J. Periodic density functional theory study of ethylene hydrogenation over Co3O4 (1 1 1) surface: The critical role of oxygen vacancies. Appl. Surf. Sci. 2016, 371, 61–66. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Pisani, C.; Cinquini, F.; Giordano, L.; Pacchioni, G. Cationic and anionic vacancies on the NiO(100) surface: DFT+U and hybrid functional density functional theory calculations. J. Chem. Phys. 2007, 127, 174711. [Google Scholar] [CrossRef]
- Li, J.; Shi, L.; Feng, G.; Shi, Z.; Sun, C.; Kong, D. Selective hydrogenation of naphthalene over γ-Al2O3-supported NiCu and NiZn bimetal catalysts. Catalysts 2020, 10, 1215. [Google Scholar] [CrossRef]
- Feng, G.; Huo, C.-F.; Deng, C.-M.; Huang, L.; Li, Y.-W.; Wang, J.; Jiao, H. Isopropanol adsorption on γ-Al2O3 surfaces: A computational study. J. Mol. Catal. A Chem. 2009, 304, 58–64. [Google Scholar] [CrossRef]
- Corral Valero, M.; Raybaud, P.; Sautet, P. Influence of the hydroxylation of γ-Al2O3 surfaces on the stability and diffusion of single Pd atoms: A DFT study. J. Phys. Chem. B 2006, 110, 1759–1767. [Google Scholar] [CrossRef]
- Yang, T.; Ehara, M. Probing the electronic structures of Con (n = 1–5) clusters on γ-Al2O3 surfaces using first-principles calculations. Phys. Chem. Chem. Phys. 2017, 19, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-X.; Liu, C.-J.; Ge, Q. Effect of surface hydroxyls on selective CO2 hydrogenation over Ni4/γ-Al2O3: A density functional theory study. J. Catal. 2010, 272, 227–234. [Google Scholar] [CrossRef]
- Thu Ha, N.T.; Minh Hue, V.T.; Trinh, B.C.; Ha, N.N.; Cam, L.M. Study on the adsorption and activation behaviours of carbon dioxide over copper cluster (Cu4) and alumina-supported copper catalyst (Cu4/Al2O3) by means of density functional theory. J. Chem. 2019, 2019, 4341056. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Ganduglia-Pirovano, M.V.; Huo, C.-F.; Sauer, J. Hydrogen spillover to copper clusters on hydroxylated γ-Al2O3. J. Phys. Chem. C 2018, 122, 18445–18455. [Google Scholar] [CrossRef]
- Sun, G.; Alexandrova, A.N.; Sautet, P. Pt8 cluster on alumina under a pressure of hydrogen: Support-dependent reconstruction from first-principles global optimization. J. Phys. Chem. C 2019, 151, 194703. [Google Scholar] [CrossRef] [Green Version]
- Mager-Maury, C.; Chizallet, C.; Sautet, P.; Raybaud, P. Platinum nanoclusters stabilized on γ-alumina by chlorine used as a capping surface ligand: A density functional theory study. ACS Catal. 2012, 2, 1346–1357. [Google Scholar] [CrossRef]
- Valero, M.C.; Raybaud, P.; Sautet, P. Nucleation of Pdn ( n = 1–5 ) clusters and wetting of Pd particles on γ−Al2O3 surfaces: A density functional theory study. Phys. Rev. B 2007, 75, 045427. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Först, C.J.; Schimpl, J. Projector augmented wave method: Ab initio molecular dynamics with full wave functions. Bull. Mater. Sci. 2003, 26, 33–41. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituted | Esub (kJ/mol) | Charge (e) | Co-O (pm) | Substituted | Esub (kJ/mol) | Charge (e) | Co-O (pm) |
|---|---|---|---|---|---|---|---|
| (110)-1Co-4 | −61 | +1.40 | 187 | (110)-1Ni-4 | −57 | +1.22 | 184 |
| (110)-2Co-34 | −83 | +1.42 | 183,191 | (110)-2Ni-34 | −91 | +1.10 | 187,186 |
| (110)-3Co-234 | −67 | +1.42 | 181,191,189 | (110)-3Ni-234 | −96 | +1.03 | 183,188,185 |
| (110)-4Co-1234 | −48 | +1.39 | 184,191,184,189 | (110)-4Ni-1234 | −95 | +1.08 | 178,187,185,185 |
| Substituted | Esub (kJ/mol) | Method | Ref. |
|---|---|---|---|
| Co and Ni doped γ-Al2O3 (110) | −61/−57 | GGA-PBE | This work |
| Fe doped γ-Al2O3 (111) | −22 | MN12-L | [28] |
| Cu doped γ-Al2O3 (110) | −261 | GGA-PBE | [35] |
| Fe doped γ-Al2O3 (110) | −53/−52 | GGA-PW91/PBE | [47] |
| Co and Ni doped Al of CuAl2O4 (110) | −30/−61 | GGA-PBE | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Shi, L.; Jin, C.; Ye, R.; Zhang, R. Co and Ni Incorporated γ-Al2O3 (110) Surface: A Density Functional Theory Study. Catalysts 2022, 12, 111. https://doi.org/10.3390/catal12020111
Li H, Shi L, Jin C, Ye R, Zhang R. Co and Ni Incorporated γ-Al2O3 (110) Surface: A Density Functional Theory Study. Catalysts. 2022; 12(2):111. https://doi.org/10.3390/catal12020111
Chicago/Turabian StyleLi, Huaxi, Liu Shi, Chengkai Jin, Runping Ye, and Rongbin Zhang. 2022. "Co and Ni Incorporated γ-Al2O3 (110) Surface: A Density Functional Theory Study" Catalysts 12, no. 2: 111. https://doi.org/10.3390/catal12020111
APA StyleLi, H., Shi, L., Jin, C., Ye, R., & Zhang, R. (2022). Co and Ni Incorporated γ-Al2O3 (110) Surface: A Density Functional Theory Study. Catalysts, 12(2), 111. https://doi.org/10.3390/catal12020111