

Valorisation of Corncob Residue towards the Sustainable Production of Glucuronic Acid

Abstract
:1. Introduction
2. Results and Discussion
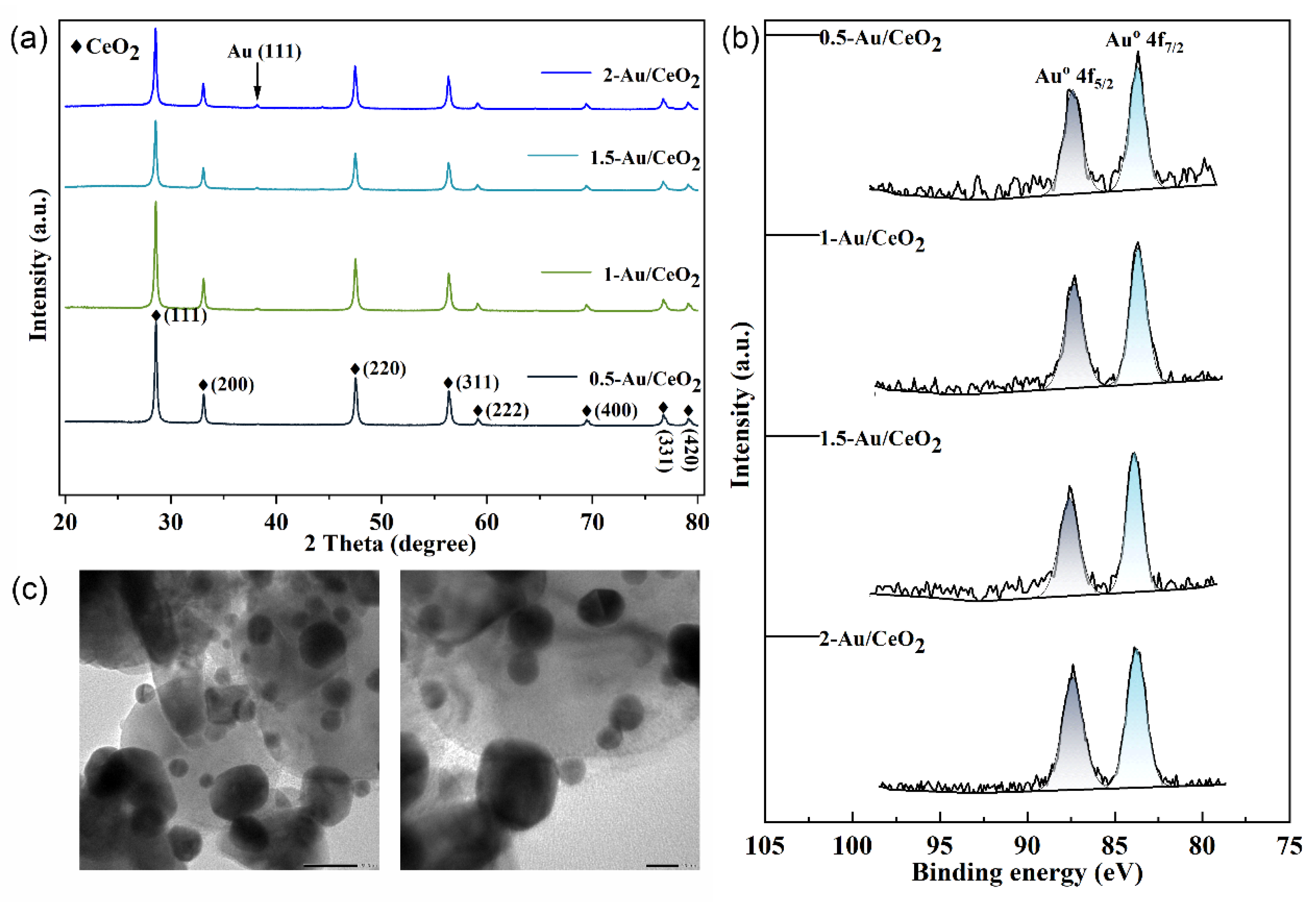
2.1. The Characterization of the Au/CeO2 Catalyst
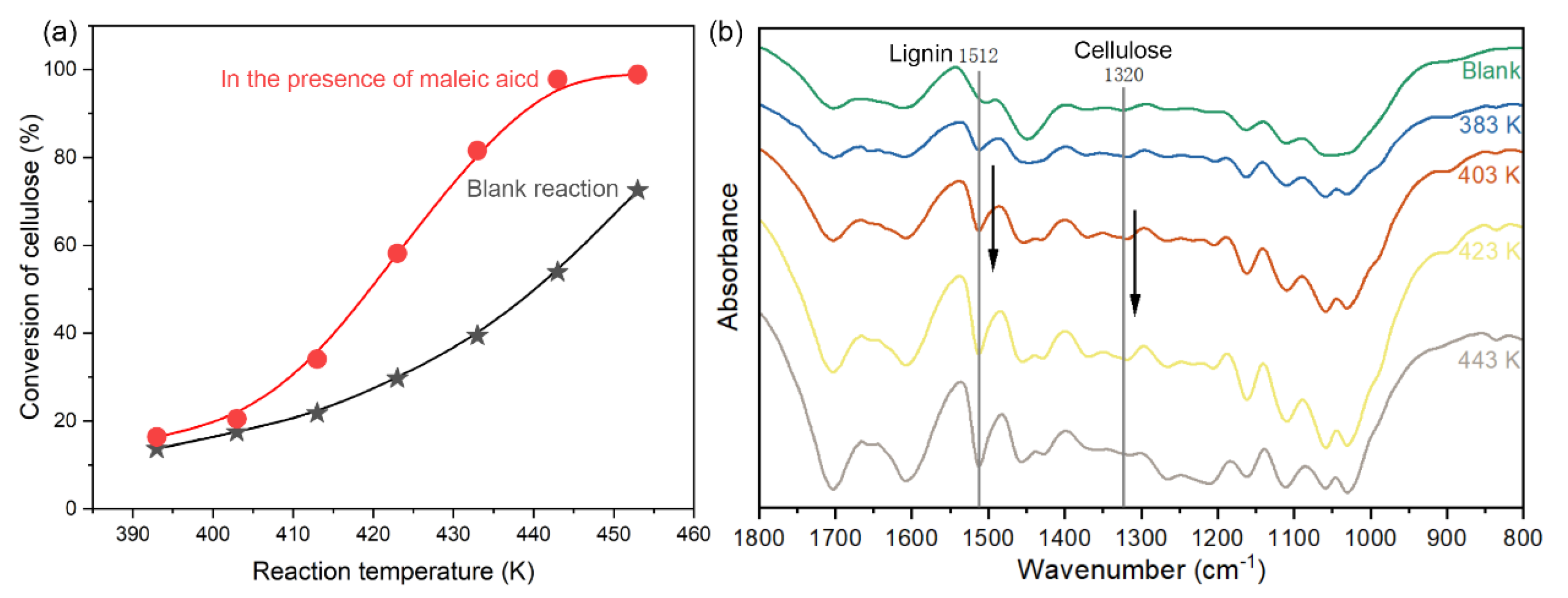
2.2. The Direct Conversion of Cellulose in Corncob Residue to GA
2.3. The Role of Maleic Acid and Au/CeO2 on the Selective Conversion of Corncob Residue
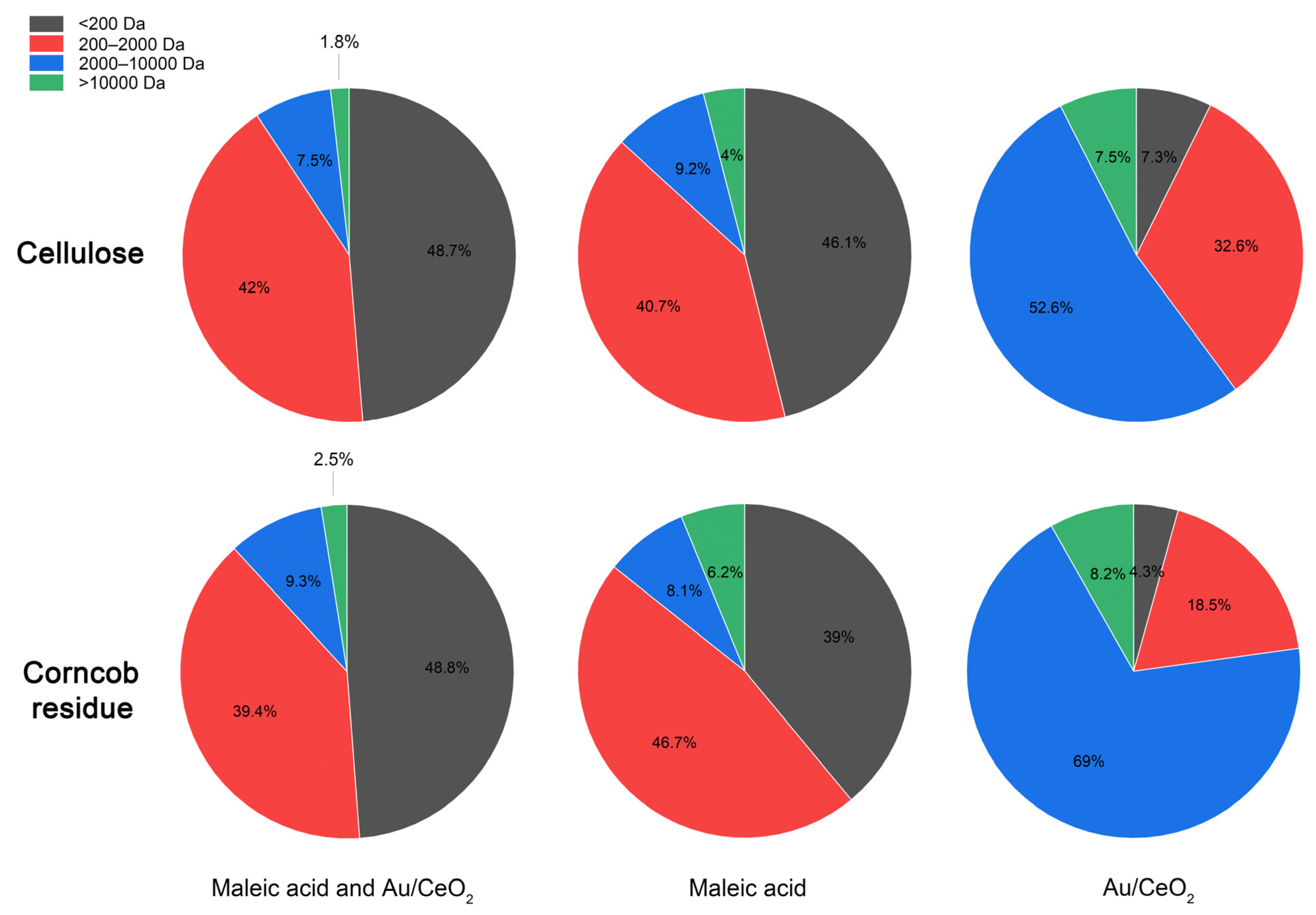
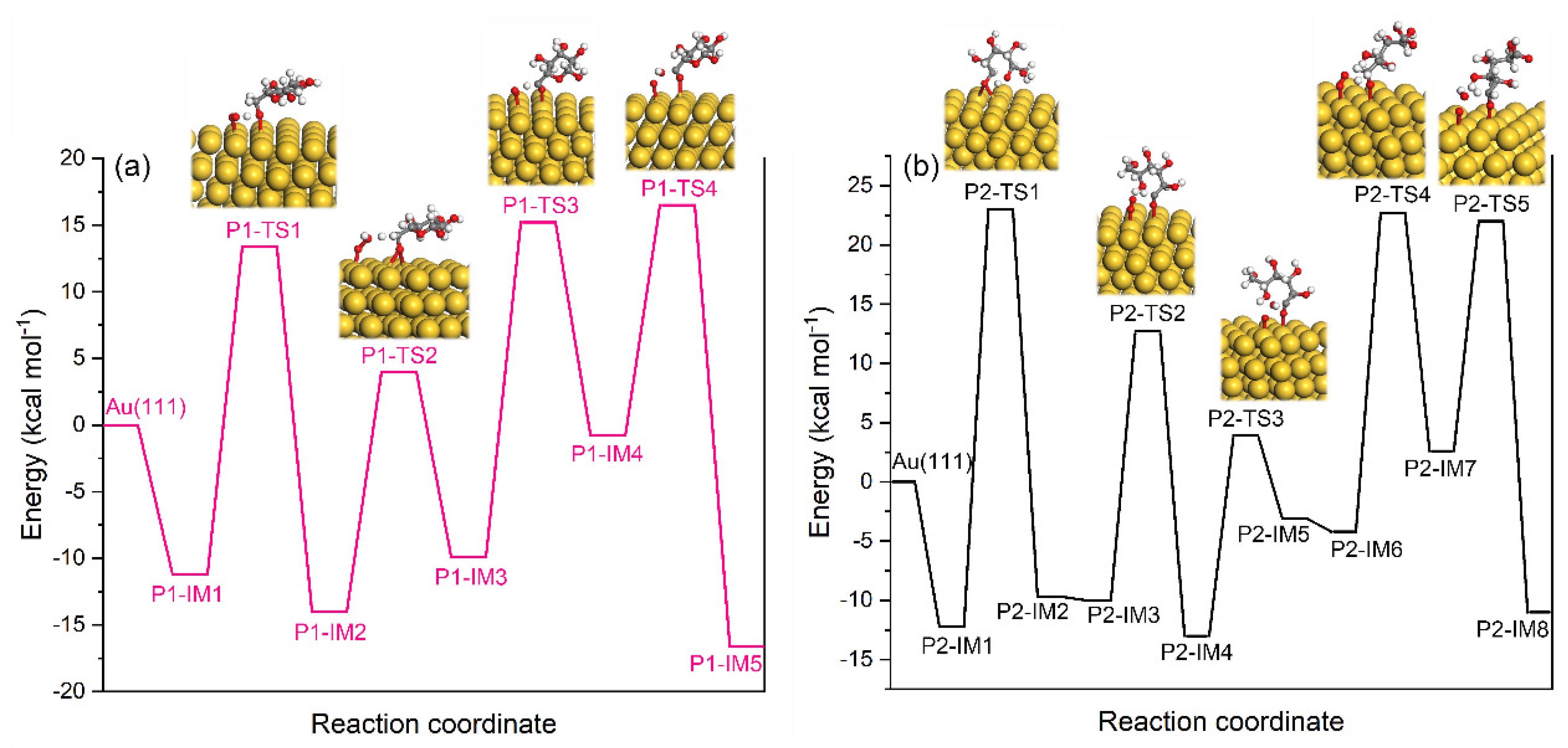
2.3.1. Fractionation of Cellulose from Corncob Residue
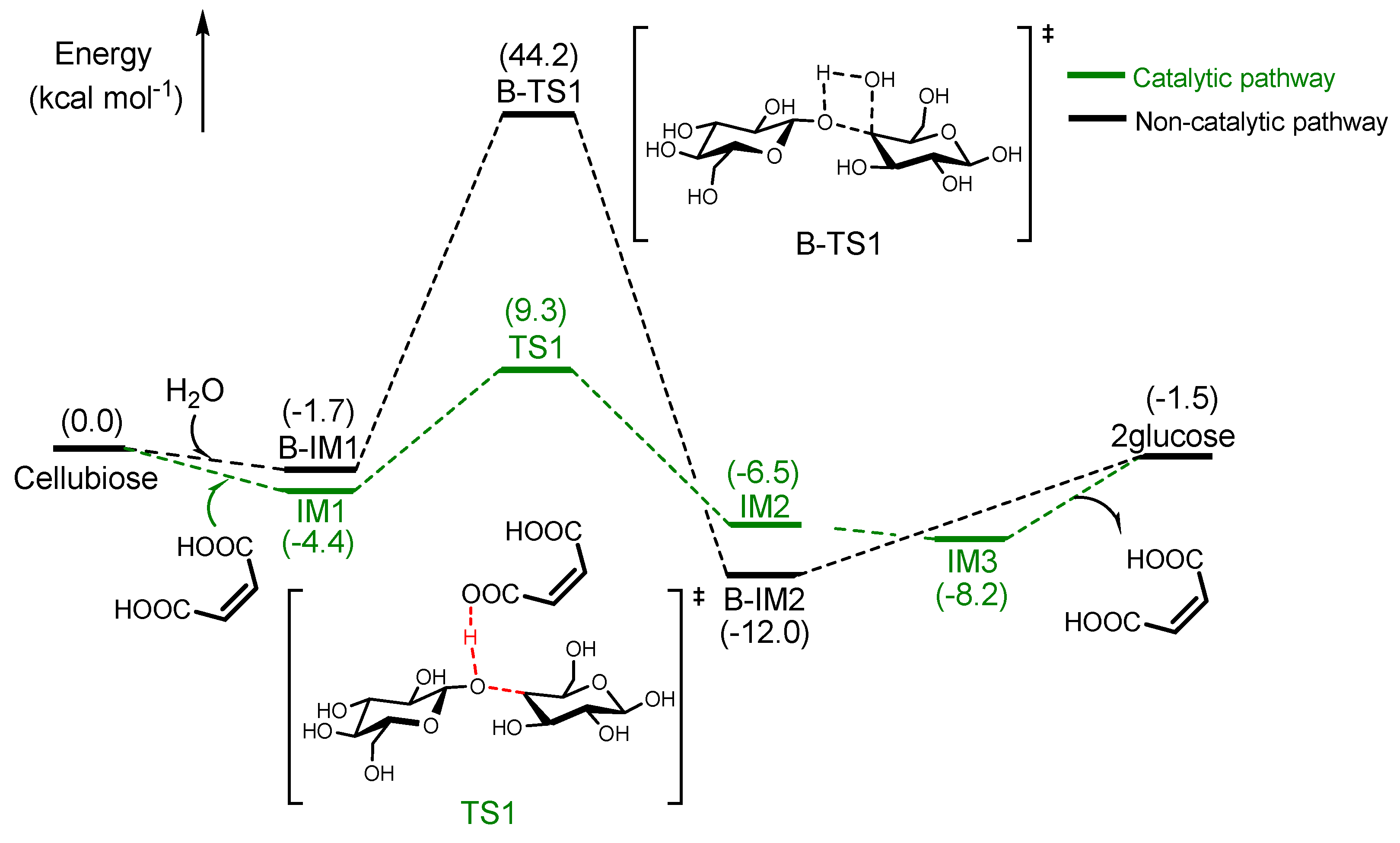
2.3.2. Depolymerisation of Polysaccharide to Glucose
2.3.3. Oxidation of Glucose to GA
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalytic Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sudarsanam, P.; Peeters, E.; Makshina, E.V.; Parvulescu, V.I.; Sels, B.F. Advances in porous and nanoscale catalysts for viable biomass conversion. Chem. Soc. Rev. 2019, 48, 2366–2421. [Google Scholar] [CrossRef]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ji, S.; Liu, Y.; Cao, X.; Tian, S.; Chen, Y.; Niu, Z.; Li, Y. Well-Defined Materials for Heterogeneous Catalysis: From Nanoparticles to Isolated Single-Atom Sites. Chem. Rev. 2020, 120, 623–682. [Google Scholar] [CrossRef] [PubMed]
- Mika, L.T.; Csefalvay, E.; Nemeth, A. Catalytic Conversion of Carbohydrates to Initial Platform Chemicals: Chemistry and Sustainability. Chem. Rev. 2018, 118, 505–613. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, J.; Han, B. Catalytic Transformation of Lignocellulose into Chemicals and Fuel Products in Ionic Liquids. Chem. Rev. 2017, 117, 6834–6880. [Google Scholar] [CrossRef]
- Zhang, Z.; Huber, G.W. Catalytic oxidation of carbohydrates into organic acids and furan chemicals. Chem. Soc. Rev. 2018, 47, 1351–1390. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Zhong, R.; Van den Bosch, S.; Coman, S.M.; Parvulescu, V.I.; Sels, B.F. Functionalised heterogeneous catalysts for sustainable biomass valorisation. Chem. Soc. Rev. 2018, 47, 8349–8402. [Google Scholar] [CrossRef] [Green Version]
- Maki-Arvela, P.; Simakova, I.L.; Salmi, T.; Murzin, D.Y. Production of lactic acid/lactates from biomass and their catalytic transformations to commodities. Chem. Rev. 2014, 114, 1909–1971. [Google Scholar] [CrossRef]
- Amaniampong, P.N.; Trinh, Q.T.; De Oliveira Vigier, K.; Dao, D.Q.; Tran, N.H.; Wang, Y.; Sherburne, M.P.; Jerome, F. Synergistic Effect of High-Frequency Ultrasound with Cupric Oxide Catalyst Resulting in a Selectivity Switch in Glucose Oxidation under Argon. J. Am. Chem. Soc. 2019, 141, 14772–14779. [Google Scholar] [CrossRef]
- Ikeda, Y.; Park, E.Y.; Okuda, N. Bioconversion of waste office paper to gluconic acid in a turbine blade reactor by the filamentous fungus Aspergillus niger. Bioresour. Technol. 2006, 97, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xue, Y.; Cao, Z.; Zhou, T.; Alnadari, F. Characterization of a uronate dehydrogenase from Thermobispora bispora for production of glucaric acid from hemicellulose substrate. World J. Microbiol. Biotechnol. 2018, 34, 102. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Luque, R.; Sepulveda-Escribano, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef]
- Omri, M.; Pourceau, G.; Becuwe, M.; Wadouachi, A. Improvement of Gold-Catalyzed Oxidation of Free Carbohydrates to Corresponding Aldonates Using Microwaves. ACS Sustain. Chem. Eng. 2016, 4, 2432–2438. [Google Scholar] [CrossRef]
- Samoilova, N.A.; Krayukhina, M.A.; Blagodatskih, I.V.; Vishivannaya, O.V.; Yamskov, I.A. Oxidation of glucose to gluconate on glucose oxidase/nanogold colloidal and heterogeneous catalysts. Catal. Lett. 2017, 147, 383–390. [Google Scholar]
- Wang, W.; Xie, Y.; Zhang, S.; Liu, X.; Zhang, L.; Zhang, B.; Haruta, M.; Huang, J. Highly efficient base-free aerobic oxidation of alcohols over gold nanoparticles supported on ZnO-CuO mixed oxides. Chin. J. Catal. 2019, 40, 1924–1933. [Google Scholar] [CrossRef]
- Khallouk, K.; Solhy, A.; Kherbeche, A.; Dubreucq, E.; Kouisni, L.; Barakat, A. Effective Catalytic Delignification and Fractionation of Lignocellulosic Biomass in Water over Zn3V2O8 Mixed Oxide. Chem. Eng. J. 2020, 385, 123914. [Google Scholar] [CrossRef]
- Amaniampong, P.N.; Trinh, Q.T.; Wang, B.; Borgna, A.; Yang, Y.; Mushrif, S.H. Biomass Oxidation: Formyl C-H Bond Activation by the Surface Lattice Oxygen of Regenerative CuO Nanoleaves. Angew. Chem. Int. Ed. 2015, 54, 8928–8933. [Google Scholar] [CrossRef]
- Jia-Xiong, W.U.; Yuan, H.; Zhang, P.; Zhang, H.L.; Yuan-Xin, W.U. Synthesis of Glucuronic Acid by Selective Oxidation of Methyl Glucoside with Active MnO2 under Microwave. Chem. Eng. J. 2012, 207, 72–75. [Google Scholar]
- Zhou, C.; Xiao, Y.; Xu, S.; Li, J.; Hu, C. γ-Valerolactone Production from Furfural Residue with Formic Acid as the Sole Hydrogen Resource via an Integrated Strategy on Au-Ni/ZrO2. Ind. Eng. Chem. Res. 2020, 59, 17228–17238. [Google Scholar] [CrossRef]
- Zhou, X.; Li, W.; Mabon, R.; Broadbelt, L.J. A mechanistic model of fast pyrolysis of hemicellulose. Energy Environ. Sci. 2018, 11, 1240–1260. [Google Scholar] [CrossRef]
- Liu, W.J.; Li, W.W.; Jiang, H.; Yu, H.Q. Fates of Chemical Elements in Biomass during Its Pyrolysis. Chem. Rev. 2017, 117, 6367–6398. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Huang, K.; Zheng, A.; Xiao, F.-S.; Dai, S. Hydrophobic Solid Acids and Their Catalytic Applications in Green and Sustainable Chemistry. ACS Catal. 2017, 8, 372–391. [Google Scholar] [CrossRef]
- Xu, S.; Wu, Y.; Li, J.; He, T.; Xiao, Y.; Zhou, C.; Hu, C. Directing the Simultaneous Conversion of Hemicellulose and Cellulose in Raw Biomass to Lactic Acid. ACS Sustain. Chem. Eng. 2020, 8, 4244–4255. [Google Scholar] [CrossRef]
- He, T.; Jiang, Z.; Wu, P.; Yi, J.; Li, J.; Hu, C. Fractionation for further conversion: From raw corn stover to lactic acid. Sci. Rep. 2016, 6, 38623. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Fan, J.; Budarin, V.L.; Macquarrie, D.J.; Gao, Y.; Li, T.; Hu, C.; Clark, J.H. Mechanistic understanding of salt-assisted autocatalytic hydrolysis of cellulose. Sustain. Energy Fuel. 2018, 2, 936–940. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Yi, J.; Li, J.; He, T.; Hu, C. Promoting Effect of Sodium Chloride on the Solubilization and Depolymerization of Cellulose from Raw Biomass Materials in Water. ChemSusChem 2015, 8, 1901–1907. [Google Scholar] [CrossRef]
- Xu, S.; Xiao, Y.; Zhang, W.; Liao, S.; Yang, R.; Li, J.; Hu, C. Relay catalysis of copper-magnesium catalyst on efficient valorization of glycerol to glycolic acid. Chem. Eng. J. 2022, 428, 132555. [Google Scholar] [CrossRef]
- Xu, S.; Li, J.; Li, J.; Wu, Y.; Xiao, Y.; Hu, C. D-Excess-LaA Production Directly from Biomass by Trivalent Yttrium Species. iScience 2019, 12, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; He, T.; Li, J.; Huang, Z.; Hu, C. Enantioselective synthesis of D-lactic acid via chemocatalysis using MgO: Experimental and molecular-based rationalization of the triose’s reactivity and preliminary insights with raw biomass. Appl. Catal. B Environ. 2021, 292, 120145. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox, Gaussian 09 (Revision D.01); Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R.; Kahn, L.R. Ab initio effective core potentials for molecular calculations. II. All-electron comparisons and modifications of the procedure. J. Chem. Phys. 1978, 68, 3059–3066. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.; Truhlar, D. Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose Conversion (%) | Yield of Products (%) | |||||
|---|---|---|---|---|---|---|
| GA | Glucaric Acid | Formic Acid | Glycolic Acid | Glyceric Acid | ||
| Maleic acid | ~100 | 56.0 | 0.3 | 3.4 | 0.6 | 1.3 |
| Malic acid | ~100 | 51.1 | 0.5 | 4.2 | 0.9 | 1.7 |
| Citric acid | 54 | 13.4 | 0.1 | 1.7 | 0.3 | 2.5 |
| Tartaric acid | 85 | 40.7 | 0.3 | 3.9 | 0.8 | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Xu, S.; Xu, X. Valorisation of Corncob Residue towards the Sustainable Production of Glucuronic Acid. Catalysts 2022, 12, 1603. https://doi.org/10.3390/catal12121603
Li W, Xu S, Xu X. Valorisation of Corncob Residue towards the Sustainable Production of Glucuronic Acid. Catalysts. 2022; 12(12):1603. https://doi.org/10.3390/catal12121603
Chicago/Turabian StyleLi, Wei, Shuguang Xu, and Xiang Xu. 2022. "Valorisation of Corncob Residue towards the Sustainable Production of Glucuronic Acid" Catalysts 12, no. 12: 1603. https://doi.org/10.3390/catal12121603
APA StyleLi, W., Xu, S., & Xu, X. (2022). Valorisation of Corncob Residue towards the Sustainable Production of Glucuronic Acid. Catalysts, 12(12), 1603. https://doi.org/10.3390/catal12121603