

Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
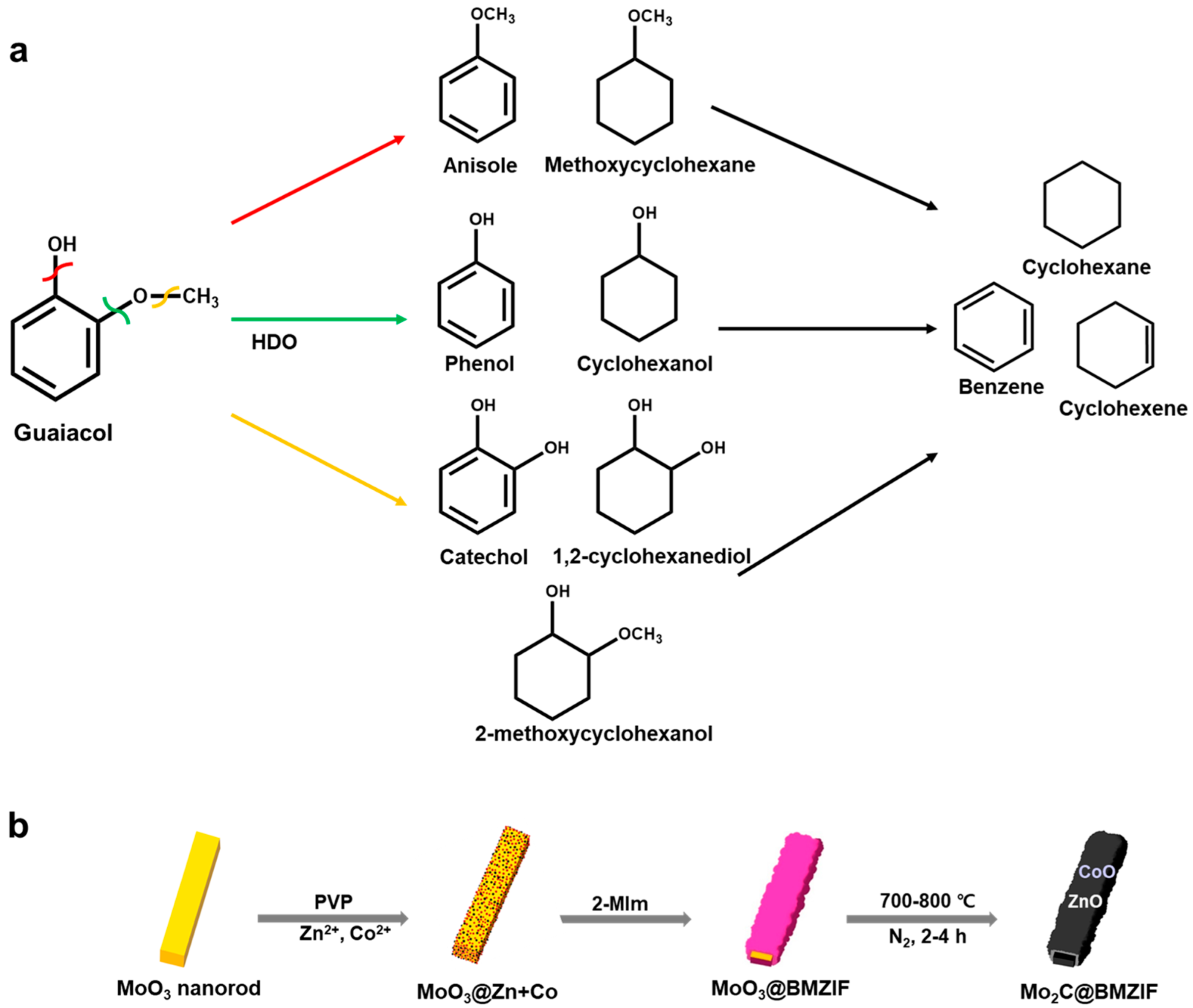
2. Results and Discussion
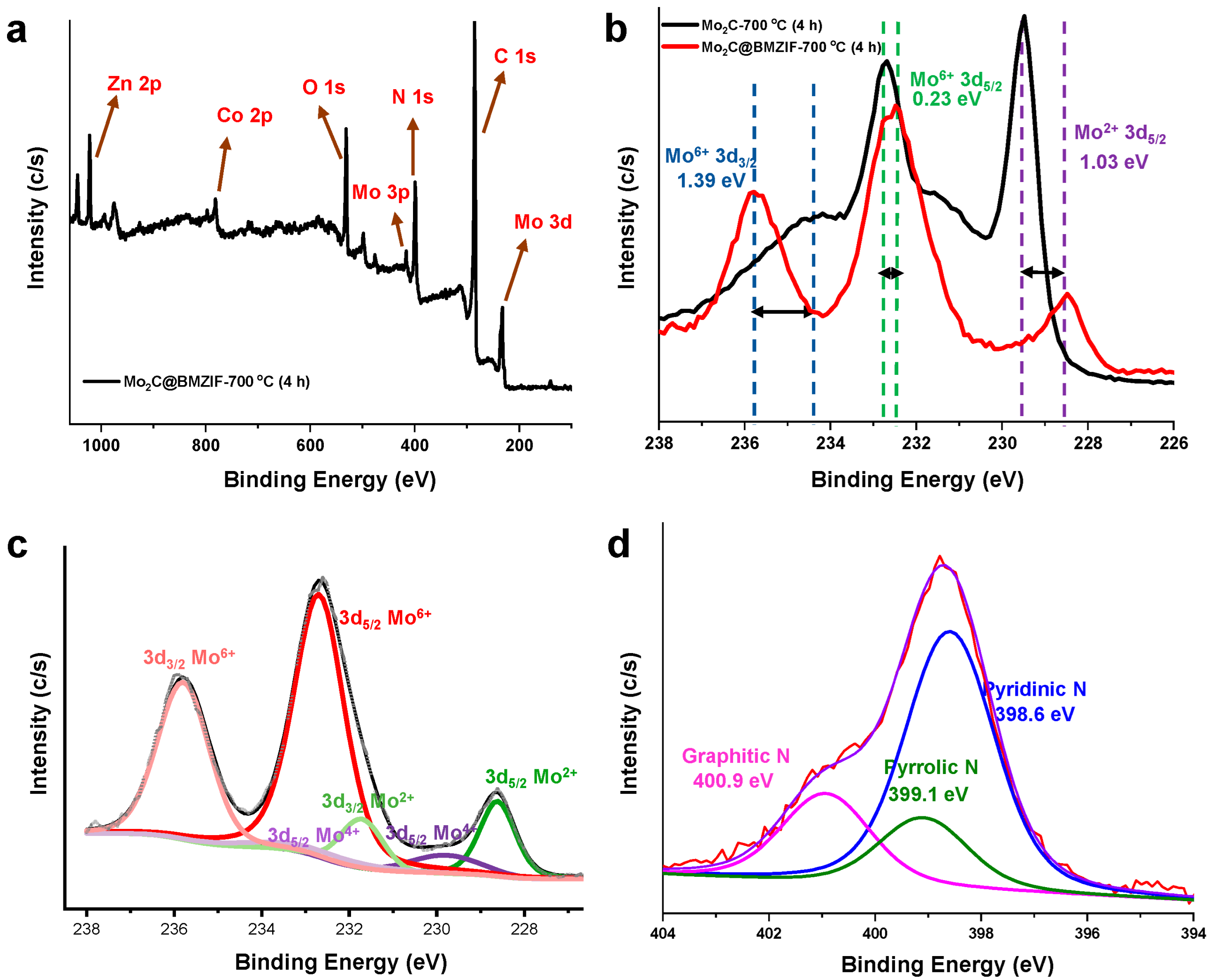
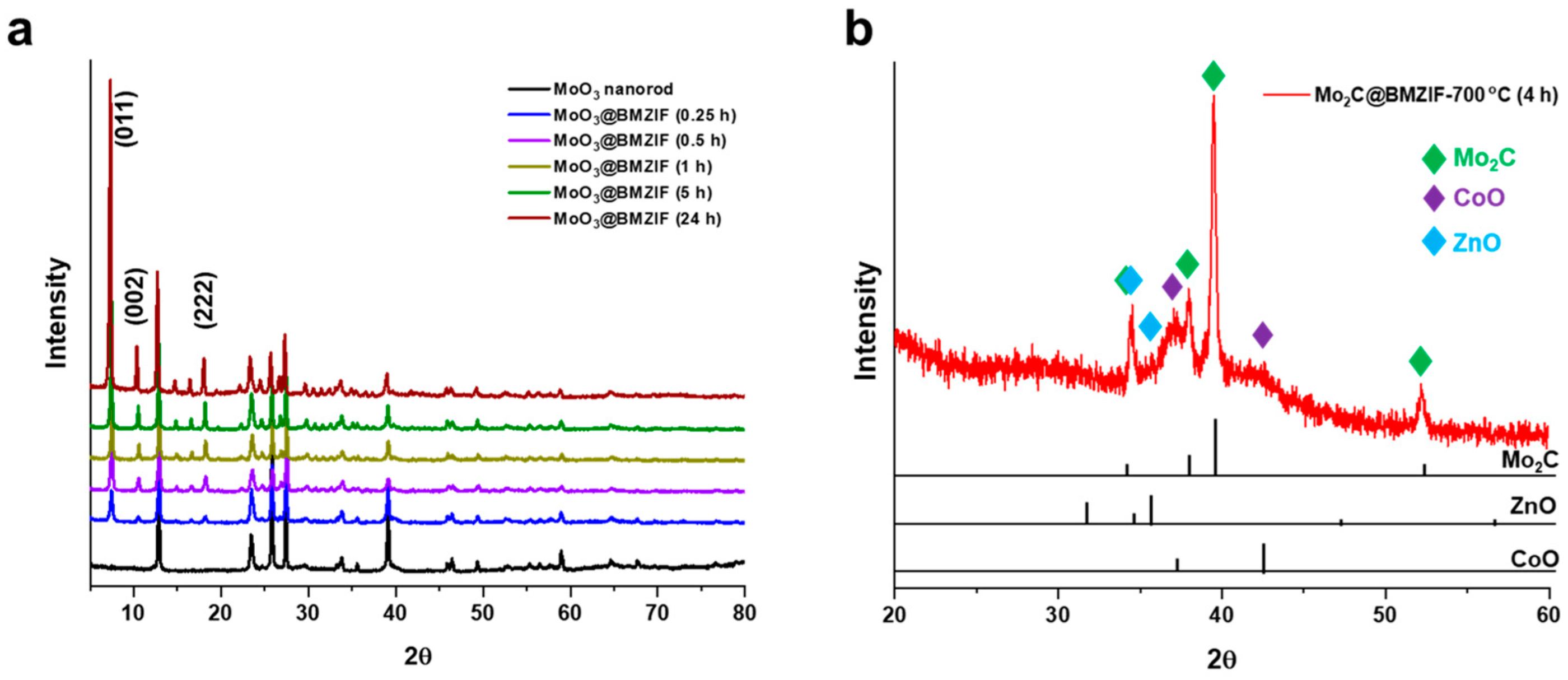
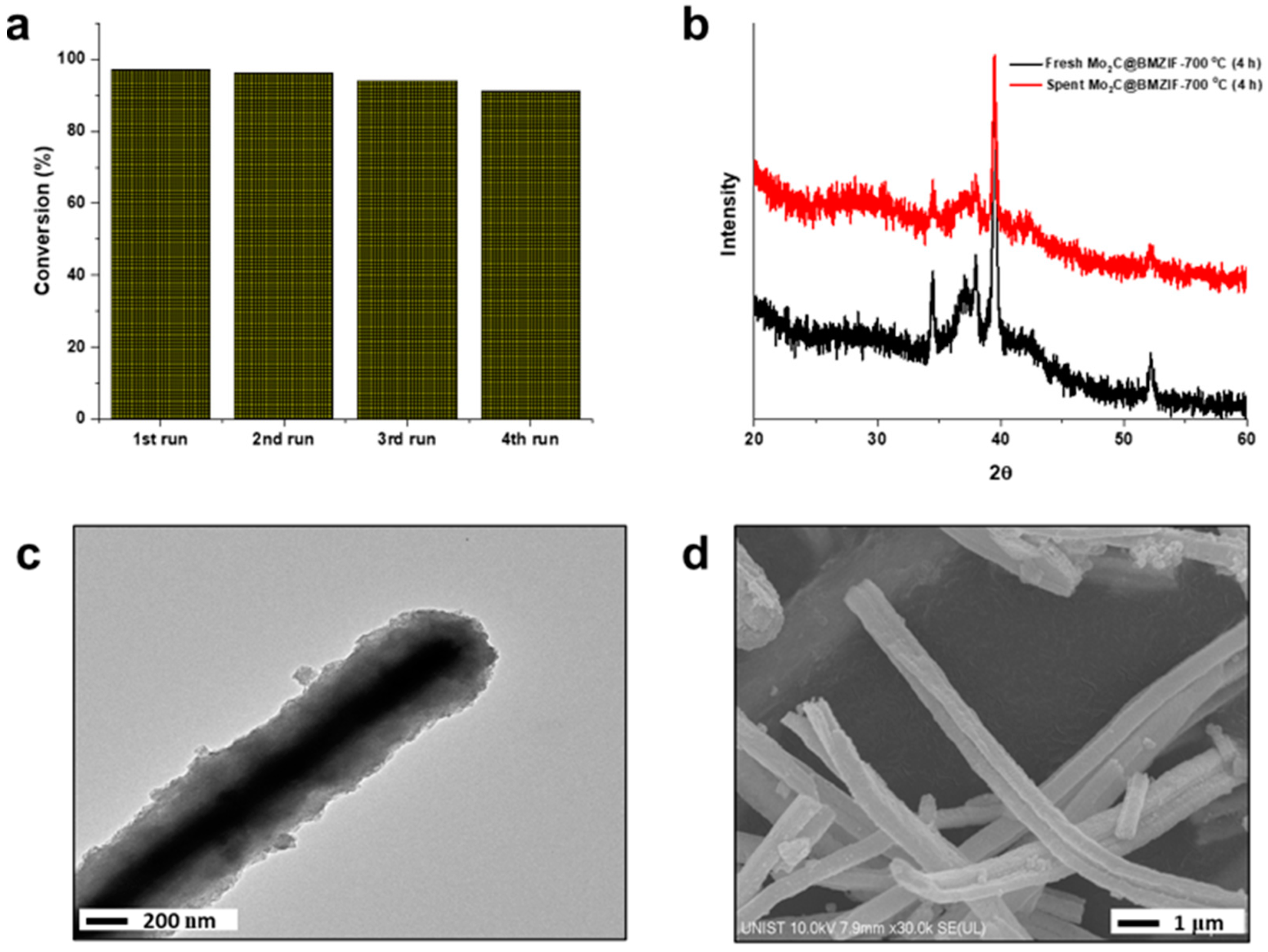
2.1. Characterization of Catalysts
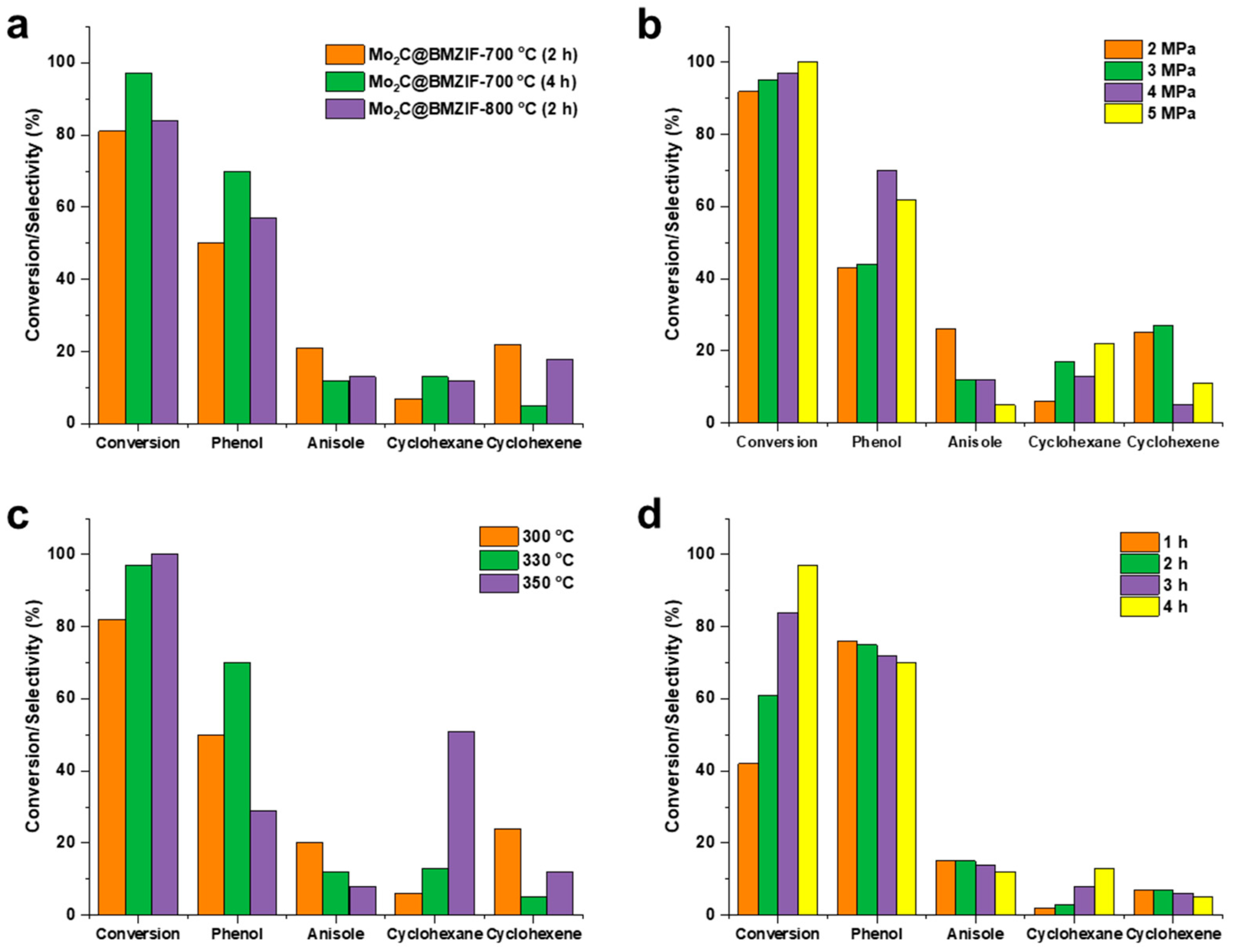
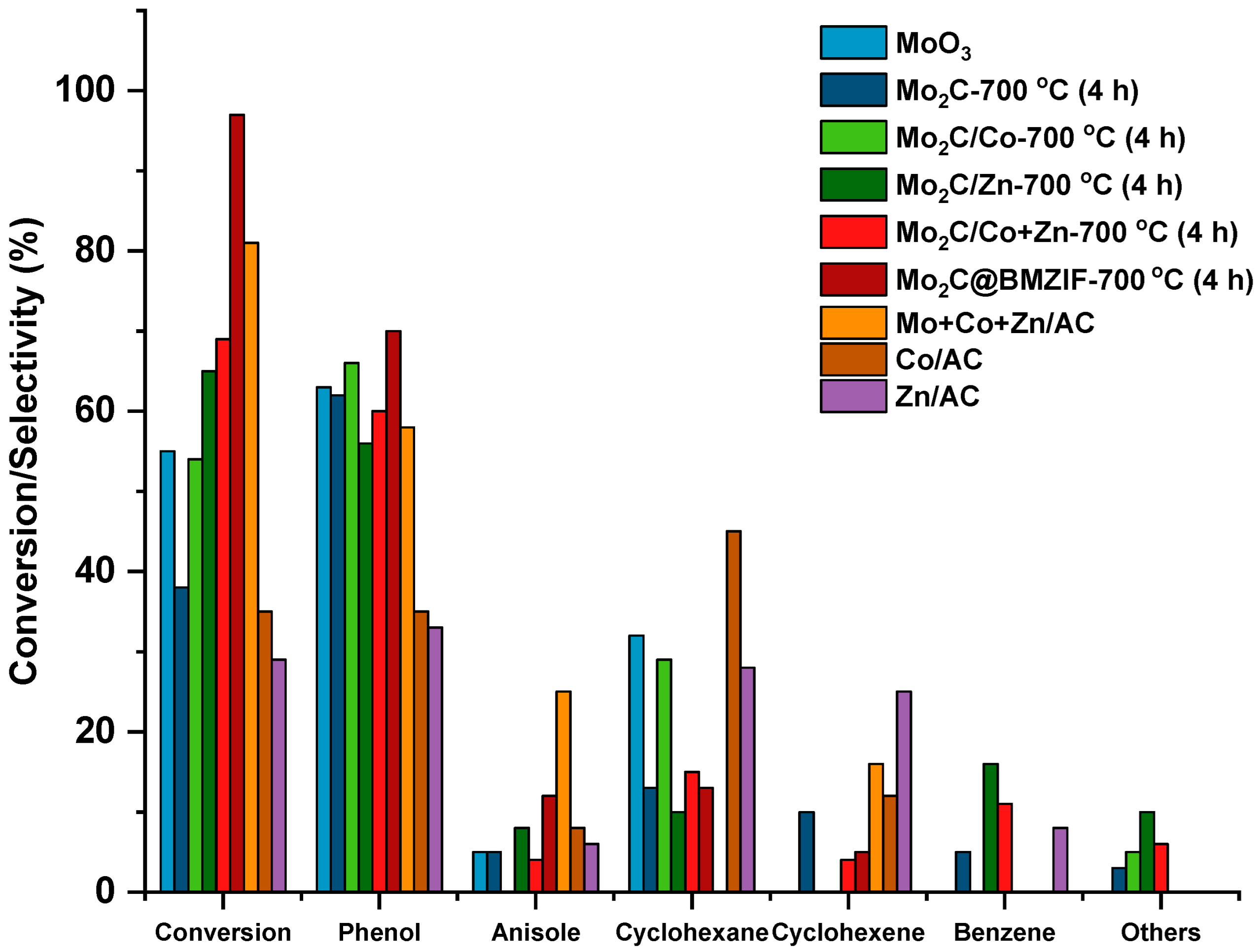
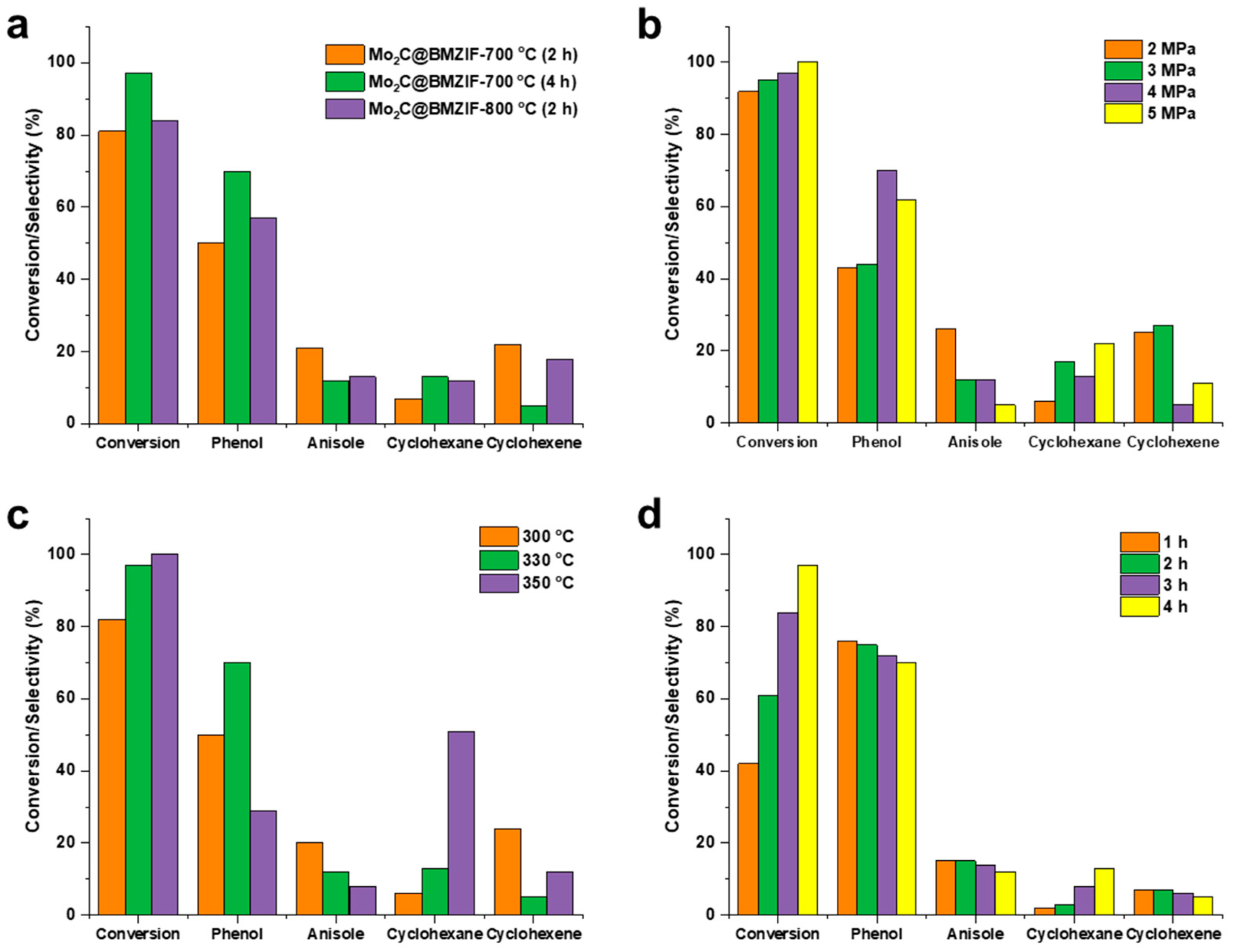
2.2. Hydrodeoxygenation of Guaiacol over Mo2C@BMZIF Catalysts
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of MoO3 Nanorods
3.3. Preparation of Composites and core@Shell Nanorods
3.4. Preparation of Reference Catalysts
3.5. Catalyst Characterization
3.6. Reaction Procedure and Product Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, S.S.; Shu, R.; Zhang, J.; Liu, H.; Yan, N. Downstream processing of lignin derived feedstock into end products. Chem. Soc. Rev. 2020, 49, 5510–5560. [Google Scholar] [CrossRef] [PubMed]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef] [Green Version]
- Tuck, C.O.; Pérez, E.; Horváth, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, W.; Renders, A.T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [PubMed]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.; Weckhuysen, B.M. Paving the way for lignin valorisation: Recent advances in bioengineering, biorefining and catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [Green Version]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Wyman, C.E. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef]
- Song, S.; Zhang, J.; Gözaydın, G.; Yan, N. Production of terephthalic acid from corn stover lignin. Angew. Chem. Int. Ed. 2019, 58, 4934–4937. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Zheng, M.; Sun, R.; Wang, A.; Wang, X.; Zhang, T. Synthesis of ethylene glycol and terephthalic acid from biomass for producing PET. Green Chem. 2016, 18, 342–359. [Google Scholar] [CrossRef]
- Zhang, Y.; Naebe, M. Lignin: A review on structure, properties, and applications as a light-colored UV absorber. ACS Sustain. Chem. Eng. 2021, 9, 1427–1442. [Google Scholar] [CrossRef]
- Terrell, E.; Dellon, L.D.; Dufour, A.; Bartolomei, E.; Broadbelt, L.J.; Garcia-Perez, M. A review on lignin liquefaction: Advanced characterization of structure and microkinetic modeling. Ind. Eng. Chem. Res. 2019, 59, 526–555. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2014, 12, 1493–1513. [Google Scholar] [CrossRef]
- Fang, R.; Dhakshinamoorthy, A.; Li, Y.; Garcia, H. Metal organic frameworks for biomass conversion. Chem. Soc. Rev. 2020, 49, 3638–3687. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, P.; Zhao, B.; Liu, K.; Kung, M.C.; Kung, H.H.; Zhang, Z.C. Selective hydrodeoxygenation of guaiacol to phenolics by Ni/anatase TiO2 catalyst formed by cross-surface migration of Ni and TiO2. ACS Catal. 2019, 9, 3551–3563. [Google Scholar] [CrossRef]
- Lee, E.H.; Park, R.S.; Kim, H.; Park, S.H.; Jung, S.C.; Jeon, J.K.; Park, Y.K. Hydrodeoxygenation of guaiacol over Pt loaded zeolitic materials. J. Ind. Eng. Chem. 2016, 37, 18–21. [Google Scholar] [CrossRef]
- Ishikawa, M.; Tamura, M.; Nakagawa, Y.; Tomishige, K. Demethoxylation of guaiacol and methoxybenzenes over carbon-supported Ru–Mn catalyst. Appl. Catal. B 2016, 182, 193–203. [Google Scholar] [CrossRef]
- Liu, X.; Jia, W.; Xu, G.; Zhang, Y.; Fu, Y. Selective hydrodeoxygenation of lignin-derived phenols to cyclohexanols over Co-based catalysts. ACS Sustain. Chem. Eng. 2017, 5, 8594–8601. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Li, C.; Nakagawa, Y.; Tamura, M.; Nakayama, A.; Tomishige, K. Hydrodeoxygenation of guaiacol to phenol over ceria-supported iron catalysts. ACS Catal. 2020, 10, 14624–14639. [Google Scholar] [CrossRef]
- Nowakowski, D.J.; Bridgwater, A.V.; Elliott, D.C.; Meier, D.; de Wild, P. Lignin fast pyrolysis: Results from an international collaboration. J. Anal. Appl. Pyrolysis 2010, 88, 53–72. [Google Scholar] [CrossRef] [Green Version]
- McGhee, W.D. Selective Introduction of Active Sites for Hydroxylation of Benzene. U.S. Patent 5, 15 September 1998. [Google Scholar]
- Lee, J.G.; Lee, S.; Lee, H.; Kurisingal, J.F.; Han, S.H.; Kim, Y.H.; An, K. Complete utilization of waste lignin: Preparation of lignin-derived carbon supports and conversion of lignin-derived guaiacol to nylon precursors. Catal. Sci. Technol. 2022, 12, 5021–5031. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Murzin, D.Y. Hydrodeoxygenation of lignin-derived phenols: From fundamental studies towards industrial applications. Catalysts 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Elliott, D.C.; Hart, T.R. Catalytic hydroprocessing of chemical models for bio-oil. Energy Fuels 2009, 23, 631–637. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O.I. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Tran, N.T.; Uemura, Y.; Chowdhury, S.; Ramli, A. Vapor-phase hydrodeoxygenation of guaiacol on Al-MCM-41 supported Ni and Co catalysts. Appl. Catal. A Gen. 2016, 512, 93–100. [Google Scholar] [CrossRef]
- Olcese, R.N.; Bettahar, M.; Petitjean, D.; Malaman, B.; Giovanella, F.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over Fe/SiO2 catalyst. Appl. Catal. B 2012, 115, 63–73. [Google Scholar] [CrossRef]
- Lup, A.N.K.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. Synergistic interaction of metal–acid sites for phenol hydrodeoxygenation over bifunctional Ag/TiO2 nanocatalyst. Chin. J. Chem. Eng. 2019, 27, 349–361. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Ren, S.; Wang, L.; Holladay, J.; Zhang, Q.; Ruan, R. Phenol and phenolics from lignocellulosic biomass by catalytic microwave pyrolysis. Bioresour. Technol. 2011, 102, 7004–7007. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, E.G.; Lee, Y.K. Active sites of Ni2P/SiO2 catalyst for hydrodeoxygenation of guaiacol: A joint XAFS and DFT study. J. Catal. 2014, 311, 144–152. [Google Scholar] [CrossRef]
- Sulman, A.; Mäki-Arvela, P.; Bomont, L.; Alda-Onggar, M.; Fedorov, V.; Russo, V.; Murzin, D.Y. Kinetic and thermodynamic analysis of guaiacol hydrodeoxygenation. Catal. Lett. 2019, 149, 2453–2467. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, J.; Lyu, P.; Nachtigall, P.; Schüth, F.; García, Á.M. The influence of water on the performance of molybdenum carbide catalysts in hydrodeoxygenation reactions: A combined theoretical and experimental study. ChemCatChem 2017, 9, 1985–1991. [Google Scholar] [CrossRef]
- Ma, R.; Cui, K.; Yang, L.; Ma, X.; Li, Y. Selective catalytic conversion of guaiacol to phenols over a molybdenum carbide catalyst. Chem. Comm. 2015, 51, 10299–10301. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic ethanolysis of Kraft lignin into high-value small-molecular chemicals over a nanostructured α-molybdenum carbide catalyst. Angew. Chem. 2014, 126, 7438–7443. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Sepúlveda, C.; Garcia, R.; Frederick, B.G.; Wheeler, M.C.; Escalona, N.; DeSisto, W.J. Guaiacol transformation over unsupported molybdenum-based nitride catalysts. Appl. Catal. A Gen. 2012, 413, 78–84. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Jastrzebski, R.; Bruijnincx, P.C.; Weckhuysen, B.M. CoMo sulfide-catalyzed hydrodeoxygenation of lignin model compounds: An extended reaction network for the conversion of monomeric and dimeric substrates. J. Catal. 2012, 285, 315–323. [Google Scholar] [CrossRef]
- Van, N.B.; Laurenti, D.; Delichère, P.; Geantet, C. Hydrodeoxygenation of guaiacol Part II: Support effect for CoMoS catalysts on HDO activity and selectivity. Appl. Catal. B 2021, 101, 246–255. [Google Scholar]
- Zhou, M.; Ge, F.; Li, J.; Xia, H.; Liu, J.; Jiang, J.; Yang, X. Catalytic Hydrodeoxygenation of Guaiacol to Cyclohexanol over Bimetallic NiMo-MOF-Derived Catalysts. Catalysts 2022, 12, 371. [Google Scholar] [CrossRef]
- Moreira, R.; Ochoa, E.; Pinilla, J.L.; Portugal, A.; Suelves, I. Liquid-phase hydrodeoxygenation of guaiacol over Mo2C supported on commercial CNF. Effects of operating conditions on conversion and product selectivity. Catalysts 2018, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Prasomsri, T.; Shetty, M.; Murugappan, K.; Román-Leshkov, Y. Insights into the catalytic activity and surface modification of MoO3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures. Energy Environ. Sci. 2014, 7, 2660–2669. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.; Weckhuysen, B.M. Carbon nanofiber supported transition-metal carbide catalysts for the hydrodeoxygenation of guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Chang, J.; Danuthai, T.; Dewiyanti, S.; Wang, C.; Borgna, A. Hydrodeoxygenation of Guaiacol over Carbon-Supported Metal Catalysts. ChemCatChem 2013, 5, 3041–3049. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, F.; Zhang, X.; Ahishakiye, R.; Xie, Y.; Shen, Y. Selective hydrodeoxygenation of guaiacol to phenolics over activated carbon supported molybdenum catalysts. Mol. Catal. 2017, 441, 28–34. [Google Scholar] [CrossRef]
- Li, R.; Shahbazi, A.; Wang, L.; Zhang, B.; Hung, A.M.; Dayton, D.C. Graphite encapsulated molybdenum carbide core/shell nanocomposite for highly selective conversion of guaiacol to phenolic compounds in methanol. Appl. Catal. A Gen. 2016, 528, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Ghampson, I.T.; Sepúlveda, C.; Garcia, R.; Radovic, L.R.; Fierro, J.G.; DeSisto, W.J.; Escalona, N. Hydrodeoxygenation of guaiacol over carbon-supported molybdenum nitride catalysts: Effects of nitriding methods and support properties. Appl. Catal. A Gen. 2012, 439, 111–124. [Google Scholar] [CrossRef]
- Sepúlveda, C.; Leiva, K.; García, R.; Radovic, L.R.; Ghampson, I.T.; DeSisto, W.J.; Escalona, N. Hydrodeoxygenation of 2-methoxyphenol over Mo2N catalysts supported on activated carbons. Catal. Today 2011, 172, 232–239. [Google Scholar] [CrossRef]
- Baddour, F.G.; Witte, V.A.; Nash, C.P.; Griffin, M.B.; Ruddy, D.A.; Schaidle, J.A. Late-transition-metal-modified β-Mo2C catalysts for enhanced hydrogenation during guaiacol deoxygenation. ACS Sustain. Chem. Eng. 2017, 5, 11433–11439. [Google Scholar] [CrossRef]
- Griboval-Constant, A.; Giraudon, J.M.; Leclercq, G.; Leclercq, L. Catalytic behaviour of cobalt or ruthenium supported molybdenum carbide catalysts for FT reaction. Appl. Catal. A Gen. 2004, 260, 35–45. [Google Scholar] [CrossRef]
- Lewandowski, M.; Szymańska-Kolasa, A.; Da Costa, P.; Sayag, C. Catalytic performances of platinum doped molybdenum carbide for simultaneous hydrodenitrogenation and hydrodesulfurization. Catal. Today 2007, 119, 31–34. [Google Scholar] [CrossRef]
- Schweitzer, N.M.; Schaidle, J.A.; Ezekoye, O.K.; Pan, X.; Linic, S.; Thompson, L.T. High activity carbide supported catalysts for water gas shift. J. Am. Chem. Soc. 2011, 133, 2378–2381. [Google Scholar] [CrossRef]
- Schaidle, J.A.; Lausche, A.C.; Thompson, L.T. Effects of sulfur on Mo2C and Pt/Mo2C catalysts: Water gas shift reaction. J. Catal. 2010, 272, 235–245. [Google Scholar] [CrossRef]
- Perret, N.; Wang, X.; Delannoy, L.; Potvin, C.; Louis, C.; Keane, M.A. Enhanced selective nitroarene hydrogenation over Au supported on β-Mo2C and β-Mo2C/Al2O3. J. Catal. 2012, 286, 172–183. [Google Scholar] [CrossRef]
- Xiang, M.; Li, D.; Xiao, H.; Zhang, J.; Li, W.; Zhong, B.; Sun, Y. K/Ni/β-Mo2C: A highly active and selective catalyst for higher alcohols synthesis from CO hydrogenation. Catal. Today 2008, 131, 489–495. [Google Scholar] [CrossRef]
- Chakraborty, G.; Park, I.H.; Medishetty, R.; Vittal, J.J. Two-dimensional metal-organic framework materials: Synthesis, structures, properties and applications. Chem. Rev. 2021, 121, 3751–3891. [Google Scholar] [CrossRef]
- Herbst, A.; Janiak, C. MOF catalysts in biomass upgrading towards value-added fine chemicals. Cryst. Eng. Comm. 2017, 19, 4092–4117. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.T.; Matsagar, B.M.; Wu, K.C.W. Metal–organic framework (MOF)-derived effective solid catalysts for valorization of lignocellulosic biomass. ACS Sustain. Chem. Eng. 2018, 6, 13628–13643. [Google Scholar] [CrossRef]
- Xia, B.Y.; Yan, Y.; Li, N.; Wu, H.B.; Lou, X.W.D.; Wang, X. A metal–organic framework-derived bifunctional oxygen electrocatalyst. Nat. Energy 2016, 1, 15006. [Google Scholar] [CrossRef]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to metal–organic frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Deng, Y.; Dong, Y.; Wang, G.; Sun, K.; Shi, X.; Zheng, L.; Liao, S. Well-defined ZIF-derived Fe–N codoped carbon nanoframes as efficient oxygen reduction catalysts. ACS Appl. Mater. Interfaces 2017, 9, 9699–9709. [Google Scholar] [CrossRef]
- Xu, Y.; Shan, W.; Liang, X.; Gao, X.; Li, W.; Li, H.; Qiu, X. Cobalt nanoparticles encapsulated in nitrogen-doped carbon shells: Efficient and stable catalyst for nitrobenzene reduction. Ind. Eng. Chem. Res. 2020, 59, 4367–4376. [Google Scholar] [CrossRef]
- Zhou, K.; Mousavi, B.; Luo, Z.; Phatanasri, S.; Chaemchuen, S.; Verpoort, F. Characterization and properties of Zn/Co zeolitic imidazolate frameworks vs. ZIF-8 and ZIF-67. J. Mater. Chem. A 2017, 5, 952–957. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Wang, C.; Wu, Z.Y.; Xiong, Y.; Xu, Q.; Yu, S.H.; Jiang, H.L. From bimetallic metal-organic framework to porous carbon: High surface area and multicomponent active dopants for excellent electrocatalysis. Adv. Mater. 2015, 27, 5010–5016. [Google Scholar] [CrossRef]
- Huang, M.; Mi, K.; Zhang, J.; Liu, H.; Yu, T.; Yuan, A.; Xiong, S. MOF-derived bi-metal embedded N-doped carbon polyhedral nanocages with enhanced lithium storage. J. Mater. Chem. A 2017, 5, 266–274. [Google Scholar] [CrossRef]
- Lee, J.G.; Yoon, S.; Yang, E.; Lee, J.H.; Song, K.; Moon, H.R.; An, K. Structural evolution of ZIF-67-derived catalysts for furfural hydrogenation. J. Catal. 2020, 392, 302–312. [Google Scholar] [CrossRef]
- Tian, W.; Hu, H.; Wang, Y.; Li, P.; Liu, J.; Liu, J.; Wu, M. Metal–organic frameworks mediated synthesis of one-dimensional molybdenum-based/carbon composites for enhanced lithium storage. ACS Nano 2018, 12, 1990–2000. [Google Scholar] [CrossRef]
- Ma, F.X.; Wu, H.B.; Xia, B.Y.; Xu, C.Y.; Lou, X.W. Hierarchical β-Mo2C nanotubes organized by ultrathin nanosheets as a highly efficient electrocatalyst for hydrogen production. Angew. Chem. Int. Ed. 2015, 54, 15395–15399. [Google Scholar] [CrossRef]
- Qi, B.; Ni, X.; Li, D.; Zheng, H. A facile non-hydrothermal fabrication of uniform α-MoO3 nanowires in high yield. Chem. Lett. 2008, 37, 336–337. [Google Scholar] [CrossRef]
- Reddy, C.V.S.; Walker Jr, E.H.; Wen, C.; Mho, S.I. Hydrothermal synthesis of MoO3 nanobelts utilizing poly (ethylene glycol). J. Power Sources 2008, 183, 330–333. [Google Scholar] [CrossRef]
- Li, W.; Cheng, F.; Tao, Z.; Chen, J. Vapor-transportation preparation and reversible lithium intercalation/deintercalation of α-MoO3 microrods. J. Phys. Chem. B 2006, 110, 119–124. [Google Scholar] [CrossRef]
- Tri, T.M.; Candy, J.P.; Gallezot, P.; Massardier, J.; Prlmet, M.; Vedrine, J.C.; Imelik, B. PtMo bimetallic catalysts supported on Y-zeolite: I. Characterization of the catalysts. J. Catal. 1983, 79, 396–409. [Google Scholar] [CrossRef]
- Tokarz-Sobieraj, R.; Hermann, K.; Witko, M.; Blume, A.; Mestl, G.; Schlögl, R. Properties of oxygen sites at the MoO3 (010) surface: Density functional theory cluster studies and photoemission experiments. Surf. Sci. 2001, 489, 107–125. [Google Scholar] [CrossRef]
- Chen, J.S.; Cheah, Y.L.; Madhavi, S.; Lou, X.W. Fast synthesis of α-MoO3 nanorods with controlled aspect ratios and their enhanced lithium storage capabilities. J. Phys. Chem. C 2010, 114, 8675–8678. [Google Scholar] [CrossRef]
- Zhong, H.X.; Wang, J.; Zhang, Y.W.; Xu, W.L.; Xing, W.; Xu, D.; Zhang, X.B. ZIF-8 derived graphene-based nitrogen-doped porous carbon sheets as highly efficient and durable oxygen reduction electrocatalysts. Angew. Chem. Int. Ed. 2014, 53, 14235–14239. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, G.; Wang, H. Hybrids of Mo 2 C nanoparticles anchored on graphene sheets as anode materials for high performance lithium-ion batteries. J. Mater. Chem. A 2015, 3, 17403–17411. [Google Scholar] [CrossRef]
- Smyth, C.M.; Addou, R.; McDonnell, S.; Hinkle, C.L.; Wallace, R.M. Contact metal–MoS2 interfacial reactions and potential implications on MoS2-based device performance. J. Phys. Chem. C 2016, 120, 14719–14729. [Google Scholar] [CrossRef]
- Oshikawa, K.; Nagai, M.; Omi, S. Characterization of molybdenum carbides for methane reforming by TPR, XRD, and XPS. J. Phys. Chem. B 2001, 105, 9124–9131. [Google Scholar] [CrossRef]
- Ledoux, M.J.; Huu, C.P.; Guille, J.; Dunlop, H. Compared activities of platinum and high specific surface area Mo2C and WC catalysts for reforming reactions: I. Catalyst activation and stabilization: Reaction of n-hexane. J. Catal. 1992, 134, 383–398. [Google Scholar] [CrossRef]
- Kumar, P.; Singh, M.; Reddy, G.B. Oxidized core–shell MoO2–MoS2 nanostructured thin films for hydrogen evolution. ACS Appl. Nano Mater. 2019, 3, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Liu, J.; Wang, Y.; Liu, Y.; Wang, X.; Nam, K.W.; Qiu, J. Synthesis of ultrathin hollow carbon shell from petroleum asphalt for high-performance anode material in lithium-ion batteries. Chem. Eng. J. 2016, 286, 632–639. [Google Scholar] [CrossRef]
- Murugappan, K.; Anderson, E.M.; Teschner, D.; Jones, T.E.; Skorupska, K.; Román-Leshkov, Y. Operando NAP-XPS unveils differences in MoO3 and Mo2C during hydrodeoxygenation. Nat. Catal. 2018, 1, 960–967. [Google Scholar] [CrossRef]
- Zhong, M.; Yang, D.; Xie, C.; Zhang, Z.; Zhou, Z.; Bu, X.H. Yolk–Shell MnO@ZnMn2O4/N–C Nanorods Derived from α-MnO2/ZIF-8 as Anode Materials for Lithium Ion Batteries. Small 2016, 12, 5564–5571. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, S.; Wang, S.; He, Y.; Liu, Y.; Wang, J.; Lv, Y. Low temperature hydrodeoxygenation of guaiacol into cyclohexane over Ni/SiO2 catalyst combined with Hβ zeolite. RSC Adv. 2019, 9, 3868–3876. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, K.; Krishna, J.J.; Anjana, S.; Selvam, P.; Vinu, R. Hydrodeoxygenation kinetics of syringol, guaiacol and phenol over H-ZSM-5. Catal. Commun. 2021, 148, 106164. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, W.; Jin, L.; Yang, X.; Xu, F.; Zhu, J.; Huang, W. Direct XPS evidence for charge transfer from a reduced rutile TiO2 (110) surface to Au clusters. J. Phys. Chem. C 2007, 111, 12434–12439. [Google Scholar] [CrossRef]
- Tran, C.C.; Han, Y.; Garcia-Perez, M.; Kaliaguine, S. Synergistic effect of Mo–W carbides on selective hydrodeoxygenation of guaiacol to oxygen-free aromatic hydrocarbons. Catal. Sci. Technol. 2019, 9, 1387–1397. [Google Scholar] [CrossRef]
- Liu, X.; Song, Y.; Geng, W.; Li, H.; Xiao, L.; Wu, W. Cu-Mo2C/MCM-41: An efficient catalyst for the selective synthesis of methanol from CO2. Catalysts 2016, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Kurisingal, J.F.; Babu, R.; Kim, S.H.; Li, Y.X.; Chang, J.S.; Cho, S.J.; Park, D.W. Microwave-induced synthesis of a bimetallic charge-transfer metal organic framework: A promising host for the chemical fixation of CO2. Catal. Sci. Technol. 2018, 8, 591–600. [Google Scholar] [CrossRef]
- Velicky, M.; Donnelly, G.E.; Hendren, W.R.; McFarland, S.; Scullion, D.; DeBenedetti, W.J.; Huang, F. Mechanism of gold-assisted exfoliation of centimeter-sized transition-metal dichalcogenide monolayers. ACS Nano 2018, 12, 10463–10472. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Sepulveda, C.; Cruces, K.; García-Fierro, J.L.; Ghampson, I.T.; Escalona, N. Conversion of guaiacol over metal carbides supported on activated carbon catalysts. Catal. Today 2020, 356, 376–383. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurisingal, J.F.; Lee, S.; Lee, J.G.; An, K. Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol. Catalysts 2022, 12, 1605. https://doi.org/10.3390/catal12121605
Kurisingal JF, Lee S, Lee JG, An K. Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol. Catalysts. 2022; 12(12):1605. https://doi.org/10.3390/catal12121605
Chicago/Turabian StyleKurisingal, Jintu Francis, Shinjae Lee, Jun Gyeong Lee, and Kwangjin An. 2022. "Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol" Catalysts 12, no. 12: 1605. https://doi.org/10.3390/catal12121605
APA StyleKurisingal, J. F., Lee, S., Lee, J. G., & An, K. (2022). Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol. Catalysts, 12(12), 1605. https://doi.org/10.3390/catal12121605