

Enhanced CuAl2O4 Catalytic Activity via Alkalinization Treatment toward High CO2 Conversion during Reverse Water Gas Shift Reaction


Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalytic Activity
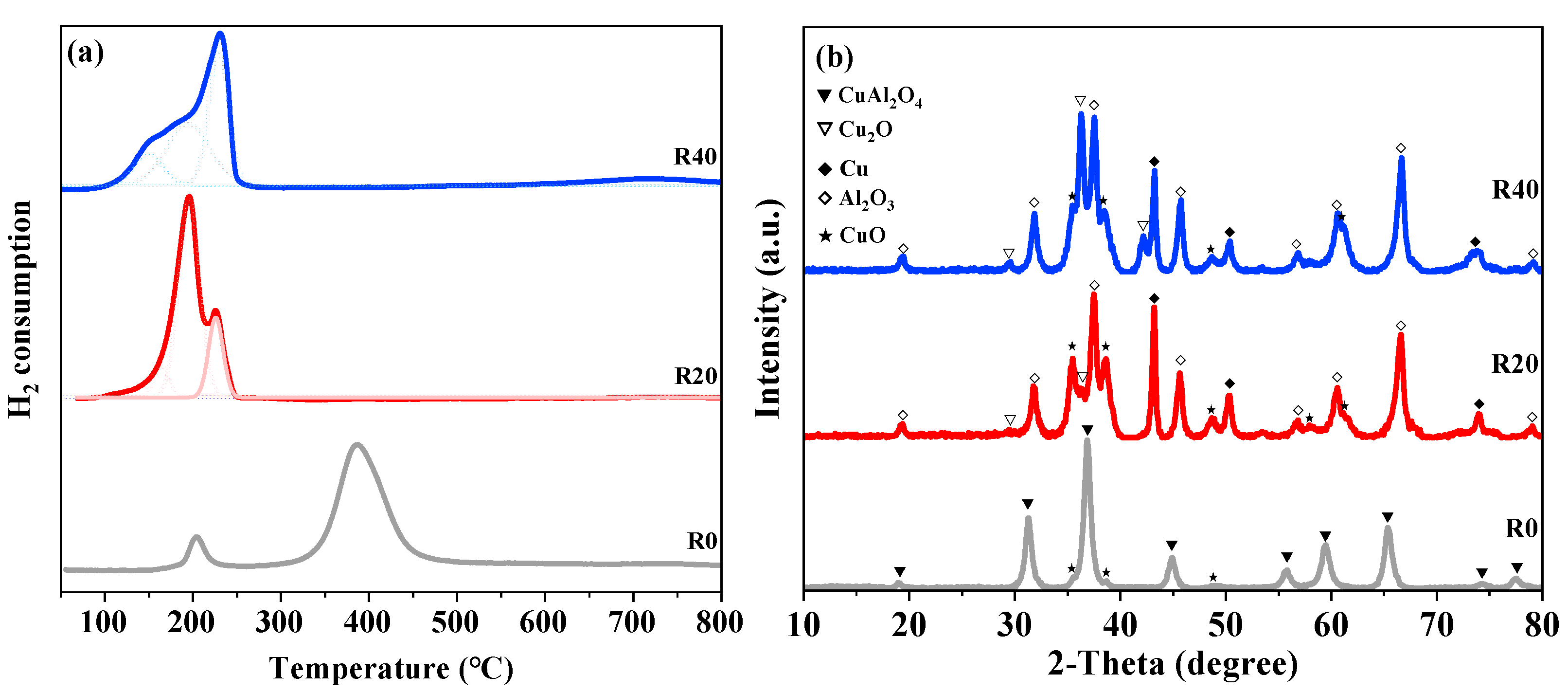
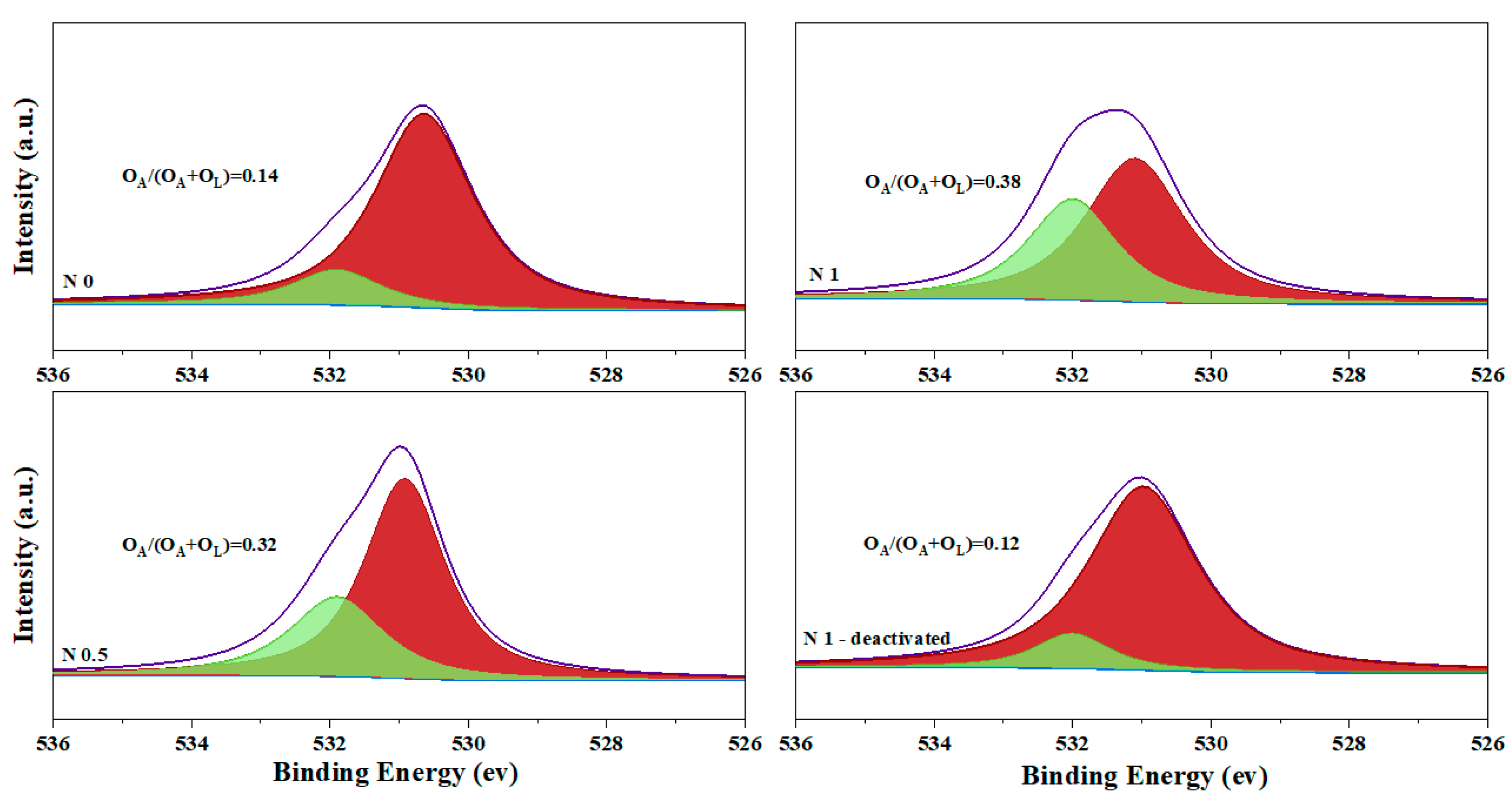
2.2. Catalyst Characterization
2.3. Alkalinization of the Catalyst
2.4. Possible Catalytic Mechanism
3. Experimental Method
3.1. Catalyst Synthesis
3.2. Characterizations
3.2.1. X-ray Diffraction (XRD)
3.2.2. H2-Temperature-Programmed Reduction (H2-TPR)
3.2.3. Scanning Electron Microscopy (SEM)
3.2.4. X-ray Photoelectron Spectra (XPS)
3.2.5. Raman Spectra
3.3. Activity Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zachos, J.C.; Dickens, G.R.; Zeebe, R.E. An Early Cenozoic Perspective on Greenhouse Warming and Carbon-Cycle Dynamics. Nature 2008, 451, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A Review on the Catalytic Conversion of CO2 Using H2 for Synthesis of CO, Methanol, and Hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Arena, F.; Barbera, K.; Italiano, G.; Bonura, G.; Spadaro, L.; Frusteri, F. Synthesis, Characterization and Activity Pattern of Cu–ZnO/ZrO2 Catalysts in the Hydrogenation of Carbon Dioxide to Methanol. J. Catal. 2007, 249, 185–194. [Google Scholar] [CrossRef]
- Liu, H.-X.; Li, S.-Q.; Wang, W.-W.; Yu, W.-Z.; Zhang, W.-J.; Ma, C.; Jia, C.-J. Partially Sintered Copper–Ceria as Excellent Catalyst for the High-Temperature Reverse Water Gas Shift Reaction. Nat. Commun. 2022, 13, 867. [Google Scholar] [CrossRef]
- Zakharova, A.; Iqbal, M.W.; Madadian, E.; Simakov, D.S.A. Reverse Microemulsion-Synthesized High-Surface-Area Cu/γ-Al2O3 Catalyst for CO2 Conversion via Reverse Water Gas Shift. ACS Appl. Mater. Interfaces 2022, 14, 22082–22094. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic Reduction of CO2 by H2 for Synthesis of CO, Methanol and Hydrocarbons: Challenges and Opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Álvarez Galván, C.; Schumann, J.; Behrens, M.; Fierro, J.L.G.; Schlögl, R.; Frei, E. Reverse Water-Gas Shift Reaction at the Cu/ZnO Interface: Influence of the Cu/Zn Ratio on Structure-Activity Correlations. Appl. Catal. B Environ. 2016, 195, 104–111. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. Effects of Oxide Carriers on Surface Functionality and Process Performance of the Cu–ZnO System in the Synthesis of Methanol via CO2 Hydrogenation. J. Catal. 2013, 300, 141–151. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhan, H.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Wang, H.; Sun, Y. Influence of Zr on the Performance of Cu/Zn/Al/Zr Catalysts via Hydrotalcite-like Precursors for CO2 Hydrogenation to Methanol. J. Catal. 2013, 298, 51–60. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F.; et al. Highly Active Copper-Ceria and Copper-Ceria-Titania Catalysts for Methanol Synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Roldan Cuenya, B.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhao, Y.; Zhao, S.; Wang, B.; Ma, X.; Gong, J. A Copper-Phyllosilicate Core-Sheath Nanoreactor for Carbon–Oxygen Hydrogenolysis Reactions. Nat. Commun. 2013, 4, 2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C. Study of Iron-Promoted Cu/SiO2 Catalyst on High Temperature Reverse Water Gas Shift Reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent Advances in Catalytic Hydrogenation of Carbon Dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.S.; Lin, J.H.; You, J.H.; Chen, C.R. Properties of Cu(Thd)2 as a Precursor to Prepare Cu/SiO2 Catalyst Using the Atomic Layer Epitaxy Technique. J. Am. Chem. Soc. 2006, 128, 15950–15951. [Google Scholar] [CrossRef]
- Maiti, S.; Llorca, J.; Dominguez, M.; Colussi, S.; Trovarelli, A.; Priolkar, K.R.; Aquilanti, G.; Gayen, A. Combustion Synthesized Copper-Ion Substituted FeAl2O4 (Cu0.1Fe0.9Al2O4): A Superior Catalyst for Methanol Steam Reforming Compared to Its Impregnated Analogue. J. Power Sources 2016, 304, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Gu, C.; Zhu, W.; Wang, X.; Yuan, X.; Cui, Z.; Wang, H.; Gao, Z. Hydrogen Production from Methanol Decomposition Using Cu-Al Spinel Catalysts. J. Clean. Prod. 2018, 183, 415–423. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Héroguel, F.; Kılıç, M.; Baranowski, C.J.; Schouwink, P.; Röthlisberger, U.; Luterbacher, J.S.; Kröcher, O. Essential Role of Oxygen Vacancies of Cu-Al and Co-Al Spinel Oxides in Their Catalytic Activity for the Reverse Water Gas Shift Reaction. Appl. Catal. B Environ. 2020, 266, 118669. [Google Scholar] [CrossRef]
- Xu, Y.; Qu, Z.; Ren, Y.; Dong, C. Enhancement of Toluene Oxidation Performance over Cu–Mn Composite Oxides by Regulating Oxygen Vacancy. Appl. Surf. Sci. 2021, 560, 149983. [Google Scholar] [CrossRef]
- Dai, B.; Zhou, G.; Ge, S.; Xie, H.; Jiao, Z.; Zhang, G.; Xiong, K. CO2 Reverse Water-Gas Shift Reaction on Mesoporous M-CeO2 Catalysts: CO2 Reverse Water-Gas Shift Reaction on Mesoporous. Can. J. Chem. Eng. 2017, 95, 634–642. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Le Monnier, B.P.; Du, Y.-P.; Héroguel, F.; Luterbacher, J.S.; Kröcher, O. Increasing the Activity of the Cu/CuAl2O4/Al2O3 Catalyst for the RWGS through Preserving the Cu2+ Ions. Chem. Commun. 2021, 57, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Cheng, X.; Peng, J.; Feng, H.; Yang, X.; Quan, L.; Jiang, L.; Tontiwachwuthikul, P. Structure–Activity Correlation Analyses of MEA + 3A1P/MAE Isomers with a Coordinative Effect Study. Ind. Eng. Chem. Res. 2022, 61, 3091–3103. [Google Scholar] [CrossRef]
- Shi, H.; Peng, J.; Cheng, X.; Yang, X.; Jin, J.; Hu, J. The CO2 Desorption Analysis of Tri-Solvent MEA+BEA+DEEA with Several Commercial Solid Acid Catalysts. Int. J. Greenh. Gas Control 2022, 116, 103647. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, J.; Liu, Q.; Gu, F.; Lu, X.; Jia, L.; Xu, G.; Zhong, Z.; Su, F. Preparation of High-Surface-Area Ni/α-Al2O3 Catalysts for Improved CO Methanation. RSC Adv. 2015, 5, 7539–7546. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Mechanism of CO Formation in Reverse Water–Gas Shift Reaction over Cu/Al2O3 Catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Chen, C.-S.; Cheng, W.-H.; Lin, S.-S. Study of Reverse Water Gas Shift Reaction by TPD, TPR and CO2 Hydrogenation over Potassium-Promoted Cu/SiO2 Catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Agzamova, P.A.; Belik, A.A.; Streltsov, S.V. Structural Stability of CuAl2O4 under Pressure. J. Phys. Condens. Matter 2021, 33, 035403. [Google Scholar] [CrossRef] [PubMed]
- Bridier, B.; López, N.; Pérez-Ramírez, J. Partial Hydrogenation of Propyne over Copper-Based Catalysts and Comparison with Nickel-Based Analogues. J. Catal. 2010, 269, 80–92. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.-W.; Shi, C.; Ma, D. Highly Dispersed Copper over β-Mo2C as an Efficient and Stable Catalyst for the Reverse Water Gas Shift (RWGS) Reaction. ACS Catal. 2017, 7, 912–918. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, B.; Xu, L.; Gao, H.; Zhang, M. Generation of Oxygen Vacancy and OH Radicals: A Comparative Study of Bi2WO6 and Bi2WO6−x Nanoplates. Chem. Cat. Chem 2015, 7, 4076–4084. [Google Scholar] [CrossRef]
- Liu, L.Z.; Wu, X.L.; Gao, F.; Shen, J.C.; Li, T.H.; Chu, P.K. Determination of Surface Oxygen Vacancy Position in SnO2 Nanocrystals by Raman Spectroscopy. Solid State Commun. 2011, 151, 811–814. [Google Scholar] [CrossRef]
- Wang, W.; Tu, Y.; Wang, L.; Liang, Y.; Shi, H. Transmission Electron Microscopy and Raman Characterization of Copper (I) Oxide Microspheres Composed of Nanoparticles. Appl. Surf. Sci. 2013, 264, 399–403. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A Study on the Effect of Support’s Reducibility on the Reverse Water-Gas Shift Reaction over Pt Catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Panagiotopoulou, P.; Kondarides, D.I.; Efstathiou, A.M. Kinetic and Mechanistic Studies of the Water–Gas Shift Reaction on Pt/TiO2 Catalyst. J. Catal. 2009, 264, 117–129. [Google Scholar] [CrossRef]
- Wang, L.C.; Tahvildar Khazaneh, M.; Widmann, D.; Behm, R.J. TAP Reactor Studies of the Oxidizing Capability of CO2 on an Au/CeO2 Catalyst—A First Step toward Identifying a Redox Mechanism in the Reverse Water–Gas Shift Reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 Hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of Synergy between Pt and Oxide Support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Tian, P.; Ford, M.E.; Chen, J.; Xu, J.; Han, Y.-F.; Wachs, I.E. Nature of Reactive Oxygen Intermediates on Copper-Promoted Iron–Chromium Oxide Catalysts during CO2 Activation. ACS Catal. 2020, 10, 7857–7863. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Santos, J.L.; Ivanova, S.; Odriozola, J.A.; Urakawa, A. Unravelling the Role of Oxygen Vacancies in the Mechanism of the Reverse Water–Gas Shift Reaction by Operando DRIFTS and Ultraviolet–Visible Spectroscopy. ACS Catal. 2018, 8, 7455–7467. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Héroguel, F.; Kılıç, M.; Baranowski, C.J.; Artiglia, L.; Röthlisberger, U.; Luterbacher, J.S.; Kröcher, O. Cu–Al Spinel as a Highly Active and Stable Catalyst for the Reverse Water Gas Shift Reaction. ACS Catal. 2019, 9, 6243–6251. [Google Scholar] [CrossRef]
- Dzuryk, S.; Rezaei, E. Intensification of the Reverse Water Gas Shift Reaction by Water-Permeable Packed-Bed Membrane Reactors. Ind. Eng. Chem. Res. 2020, 59, 18907–18920. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Temperature (°C) | Pressure (MPa) | Rate (×10−5 molCO2/gcat/s) | CO Selectivity (%) | Ref. |
|---|---|---|---|---|---|
| CuAl2O4 | 400 | 0.1 | 9.36 | 100 | This work |
| Cu/Al2O3 | 600 | 0.1 | 4.9 | 100 | [25] |
| Cu/SiO2 | 500 | 0.1 | 3.34 | 100 | [13] |
| Cu−Fe/SiO2 | 500 | 0.1 | 6.7 | 100 | [13] |
| Cu−K/SiO2 | 600 | 0.1 | 7.3 | 100 | [26] |
| Cu/CeO2 | 400 | 0.1 | 2.38 | 100 | [20] |
| Ni/CeO2 | 400 | 0.1 | 5.37 | 31.7 | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Hu, H.; Tang, S.; Pan, Z. Enhanced CuAl2O4 Catalytic Activity via Alkalinization Treatment toward High CO2 Conversion during Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1511. https://doi.org/10.3390/catal12121511
Hu M, Hu H, Tang S, Pan Z. Enhanced CuAl2O4 Catalytic Activity via Alkalinization Treatment toward High CO2 Conversion during Reverse Water Gas Shift Reaction. Catalysts. 2022; 12(12):1511. https://doi.org/10.3390/catal12121511
Chicago/Turabian StyleHu, Mian, Hongyu Hu, Suqin Tang, and Zhiyan Pan. 2022. "Enhanced CuAl2O4 Catalytic Activity via Alkalinization Treatment toward High CO2 Conversion during Reverse Water Gas Shift Reaction" Catalysts 12, no. 12: 1511. https://doi.org/10.3390/catal12121511
APA StyleHu, M., Hu, H., Tang, S., & Pan, Z. (2022). Enhanced CuAl2O4 Catalytic Activity via Alkalinization Treatment toward High CO2 Conversion during Reverse Water Gas Shift Reaction. Catalysts, 12(12), 1511. https://doi.org/10.3390/catal12121511