
Photocatalytic Transformations of the Resveratrol Derivative in Microflow Reactor

 ,
,  ,
,  , ,
, ,  , , and
, , and
Abstract
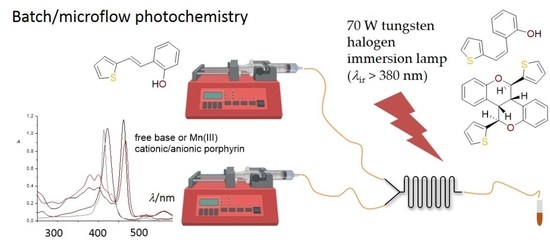
:
1. Introduction
2. Results and Discussion
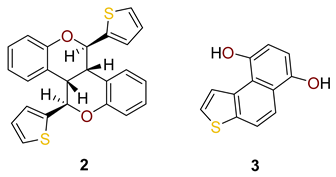
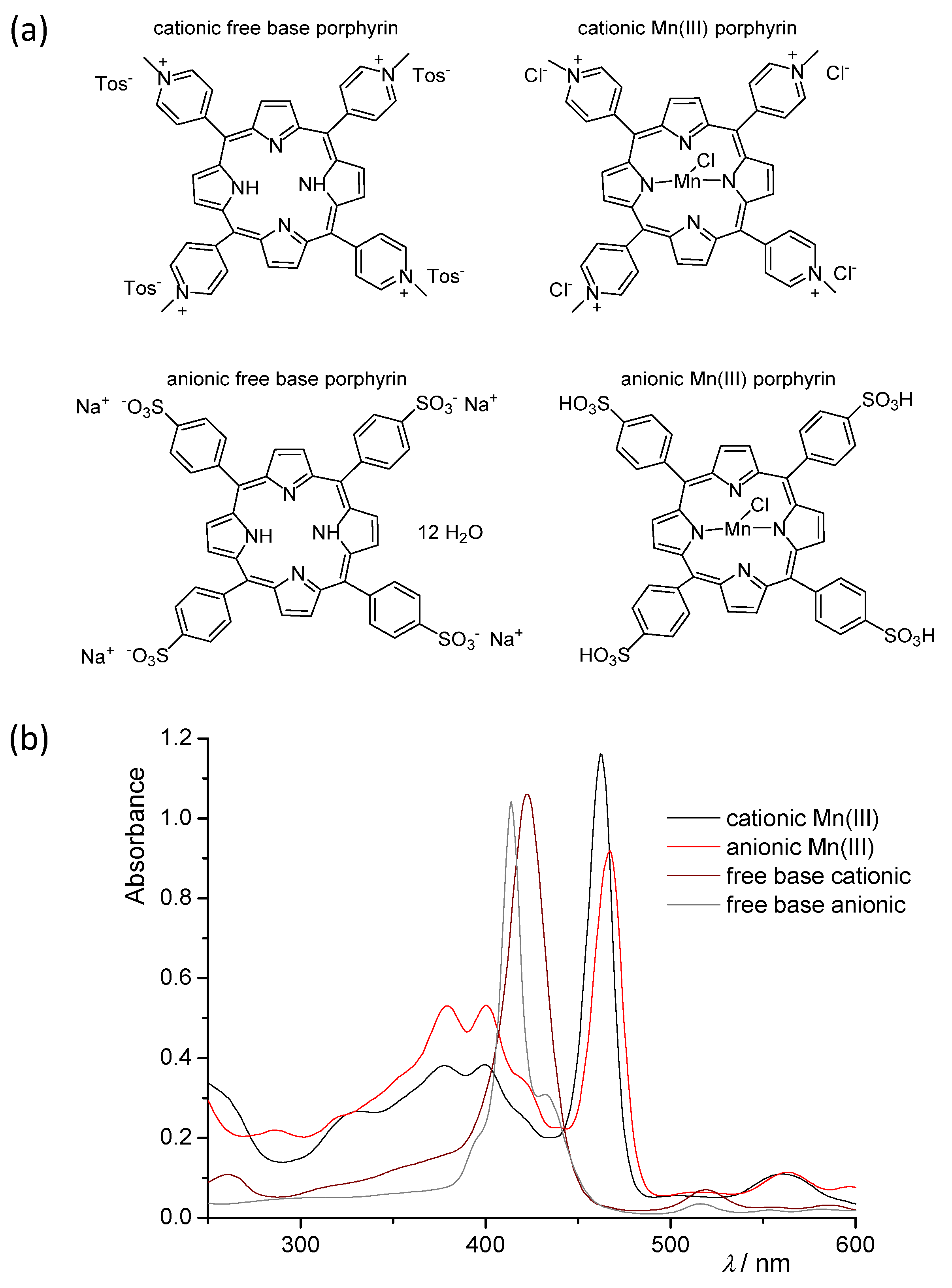
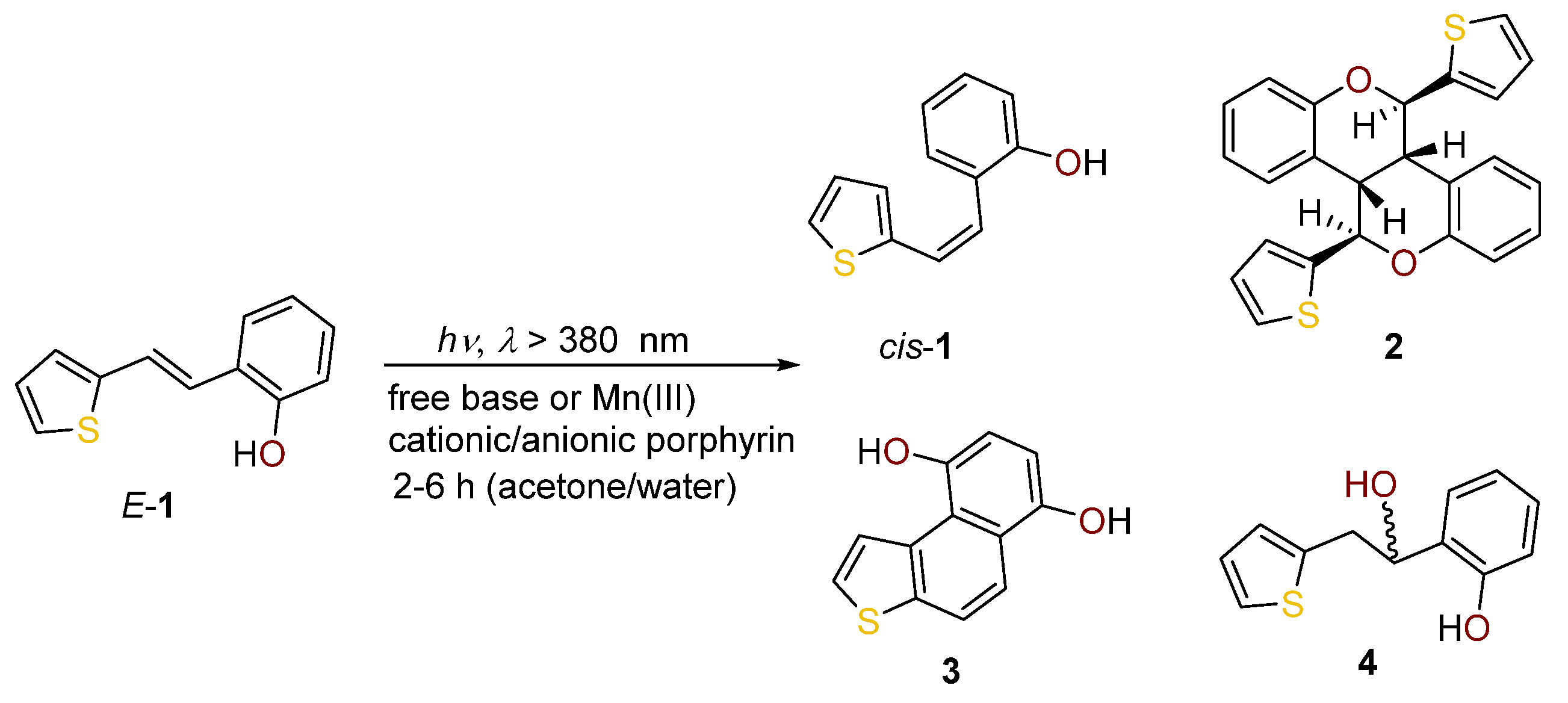
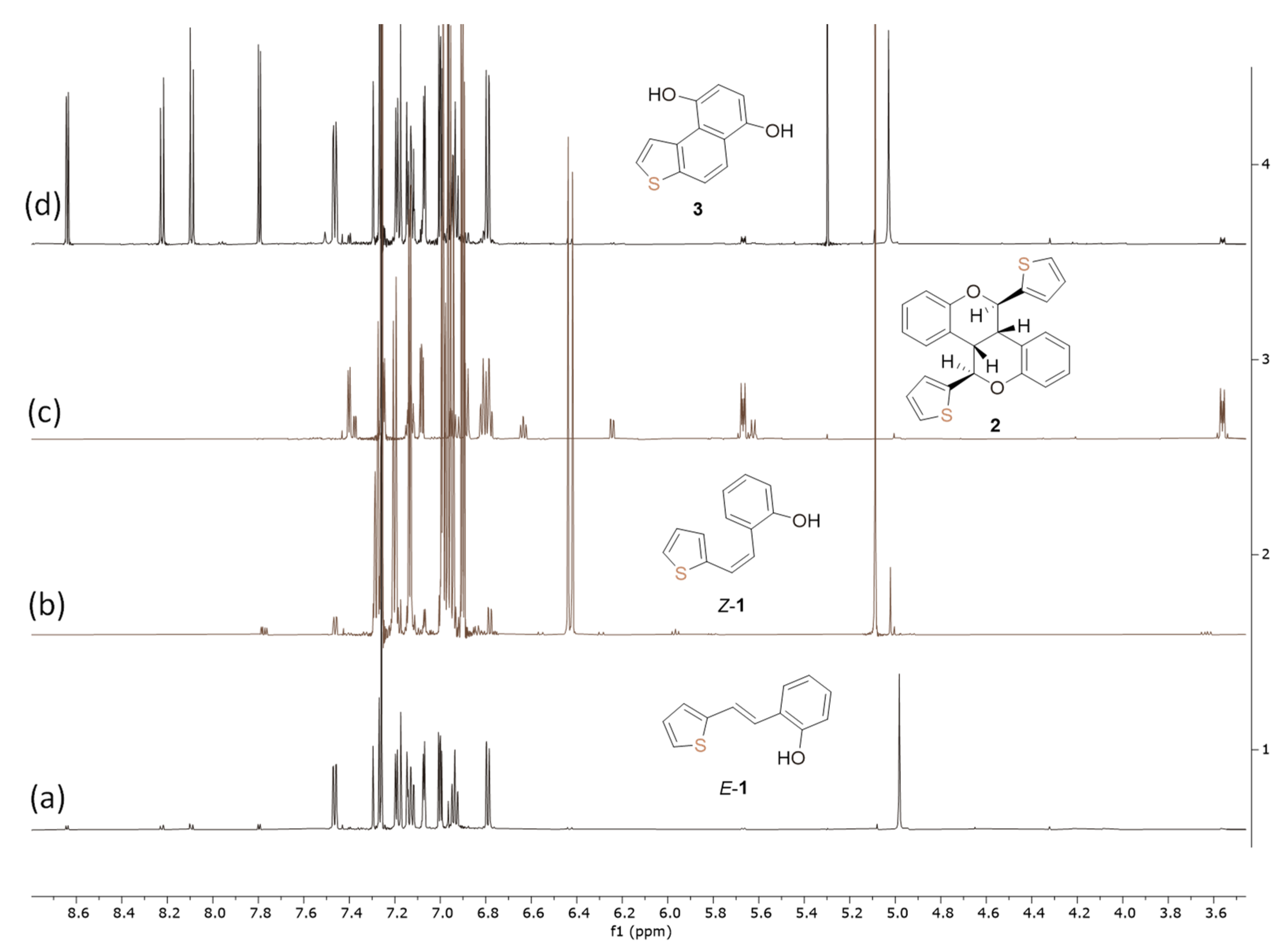
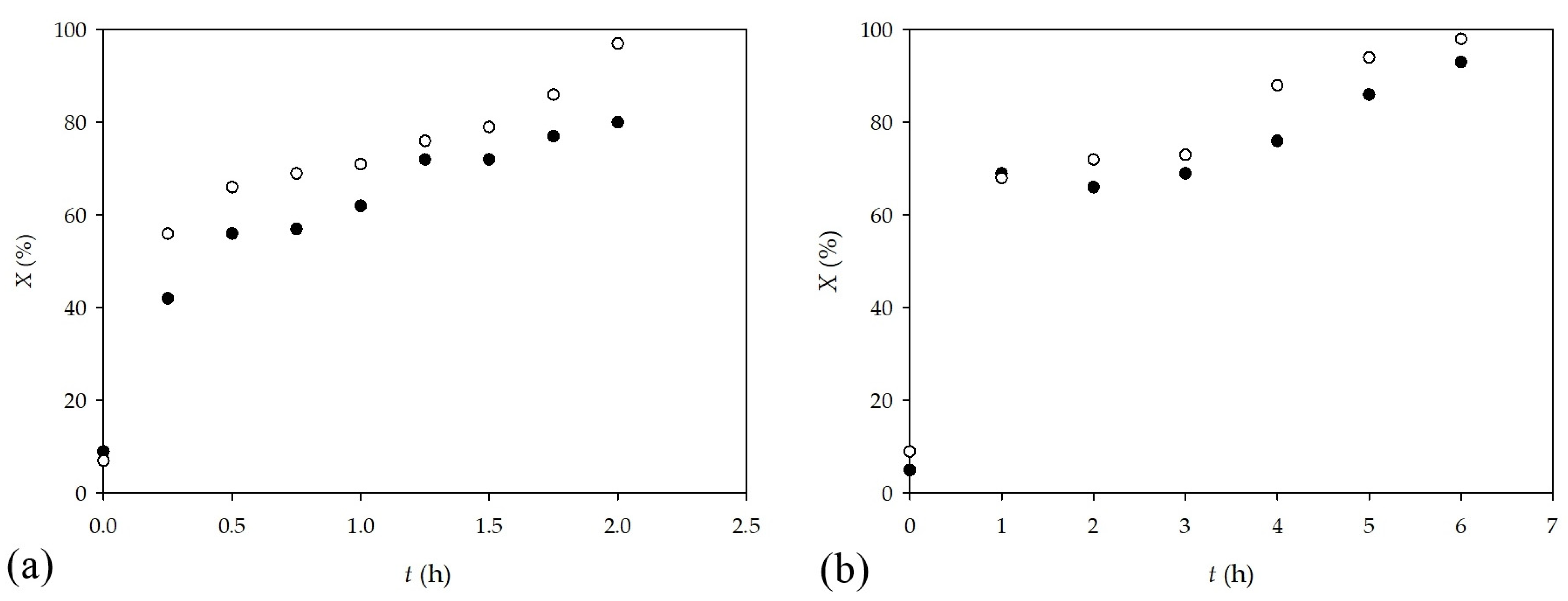
2.1. Synthesis in a Batch Reactor and Identification of Photocatalytic Transformation Products
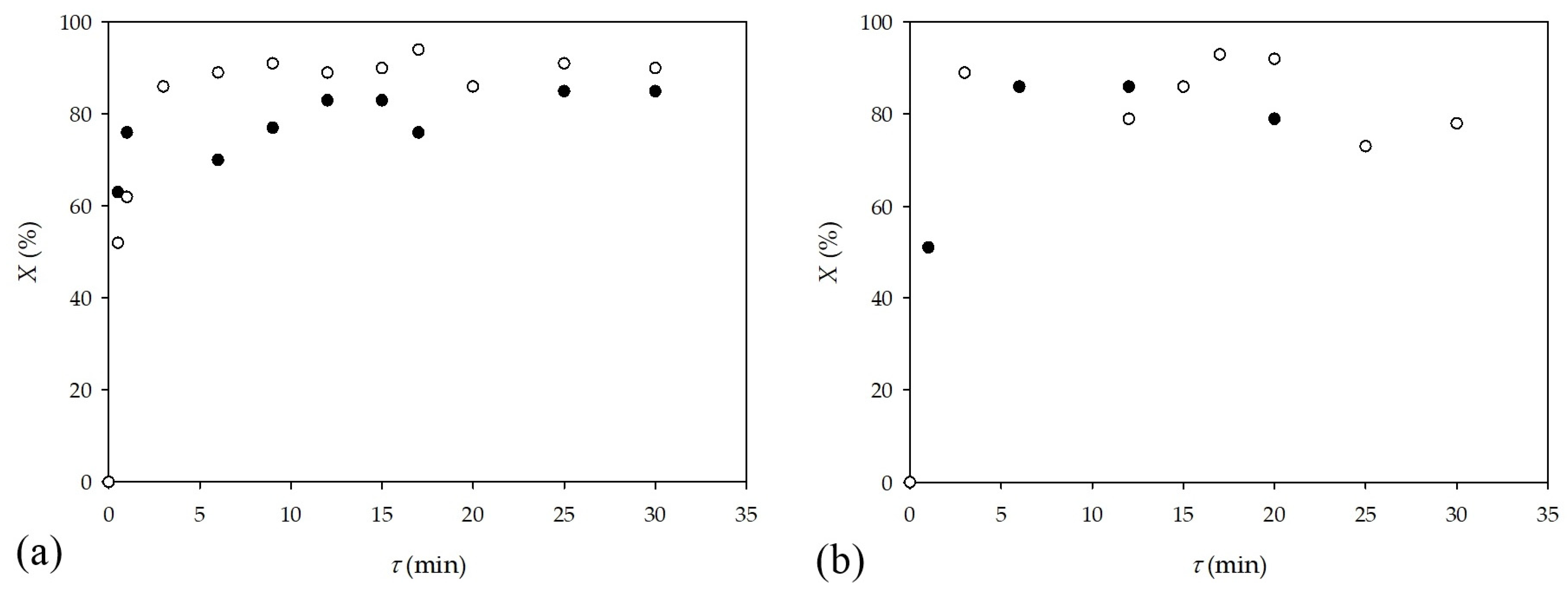
2.2. Synthesis in a Microflow Reactor
3. Materials and Methods
3.1. General
3.2. Typical Experimental Procedure for the Synthesis of E-1

3.3. Typical Experimental Procedure for the Photocatalytic Reactions in a Batch Reactor
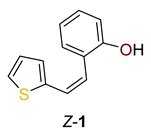
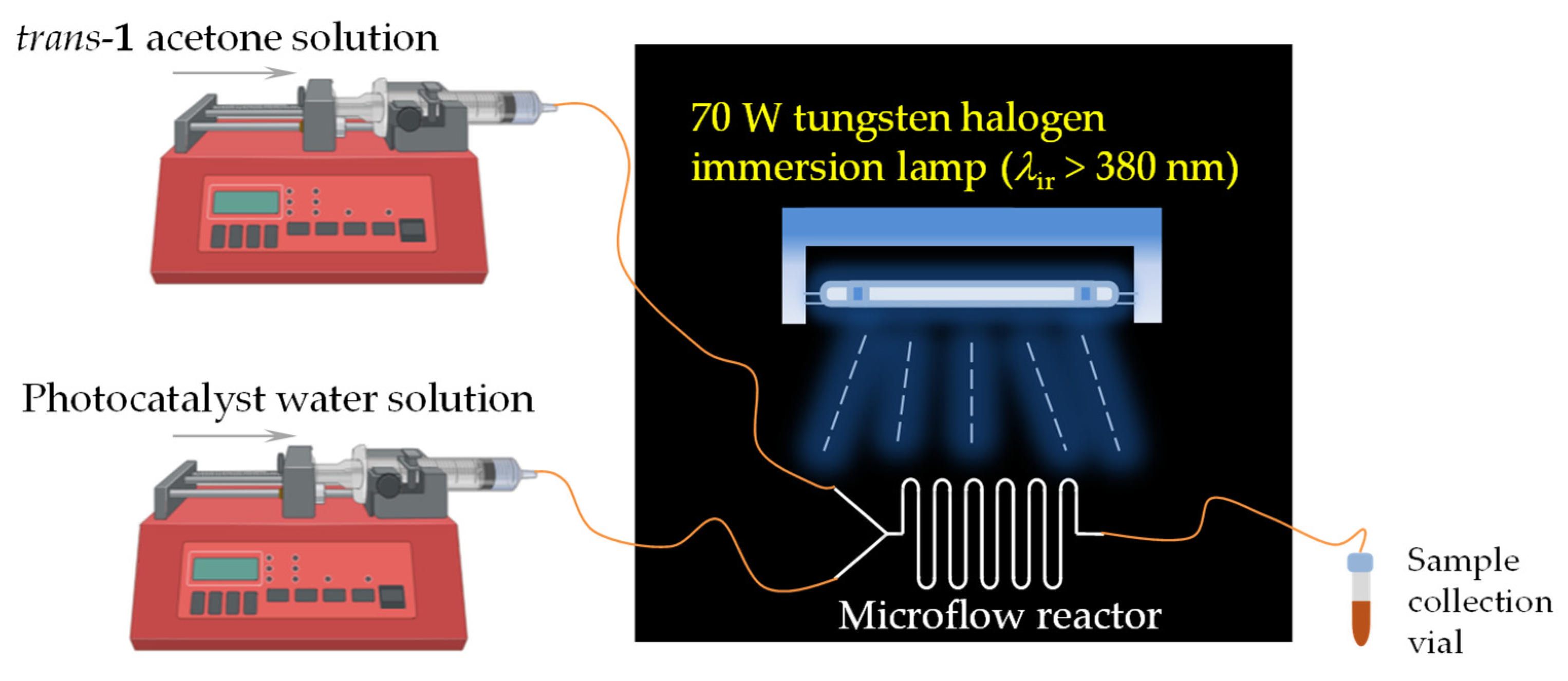
3.4. Experimental Procedure for Photocatalytic Reactions in Microflow Reactors
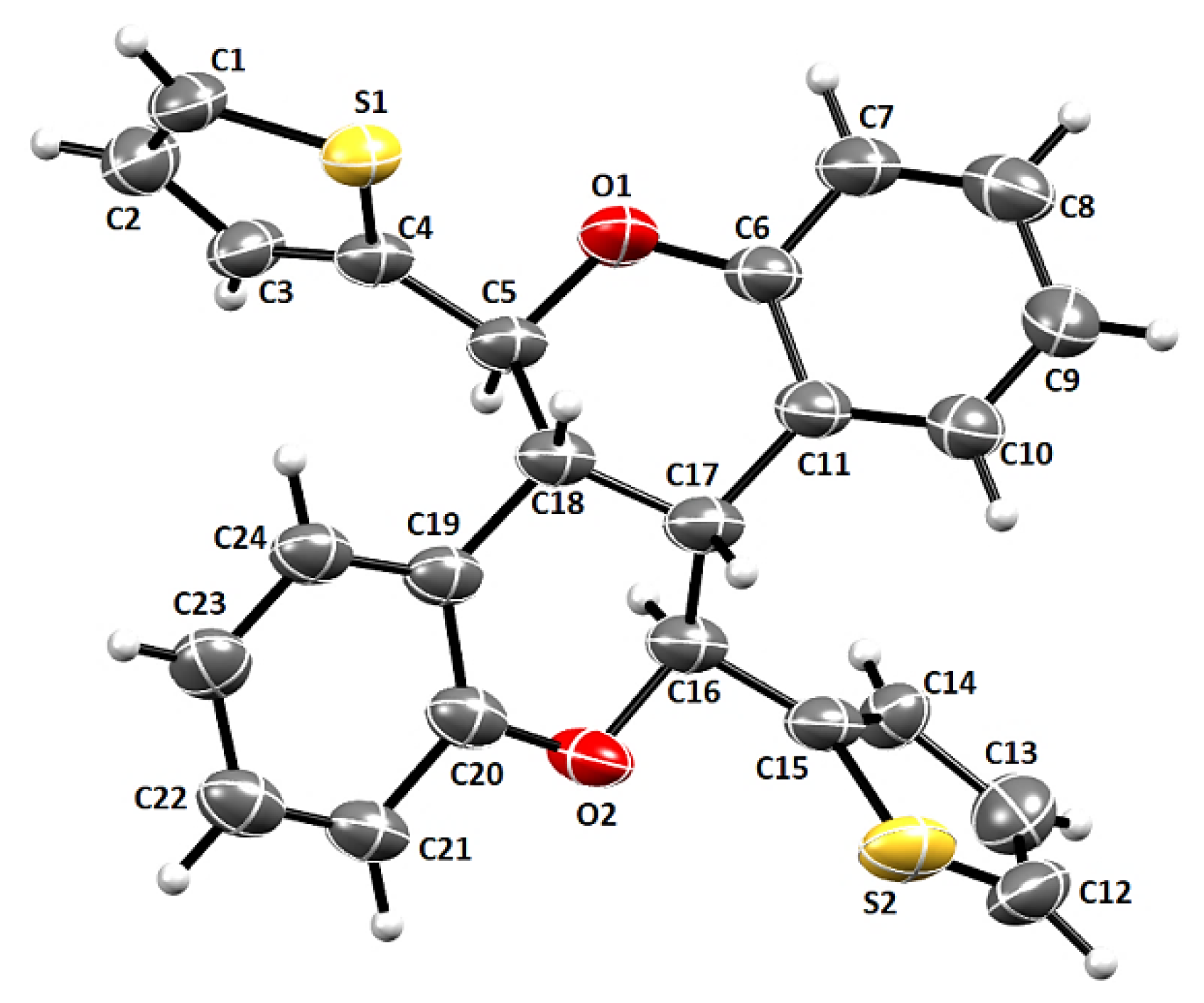
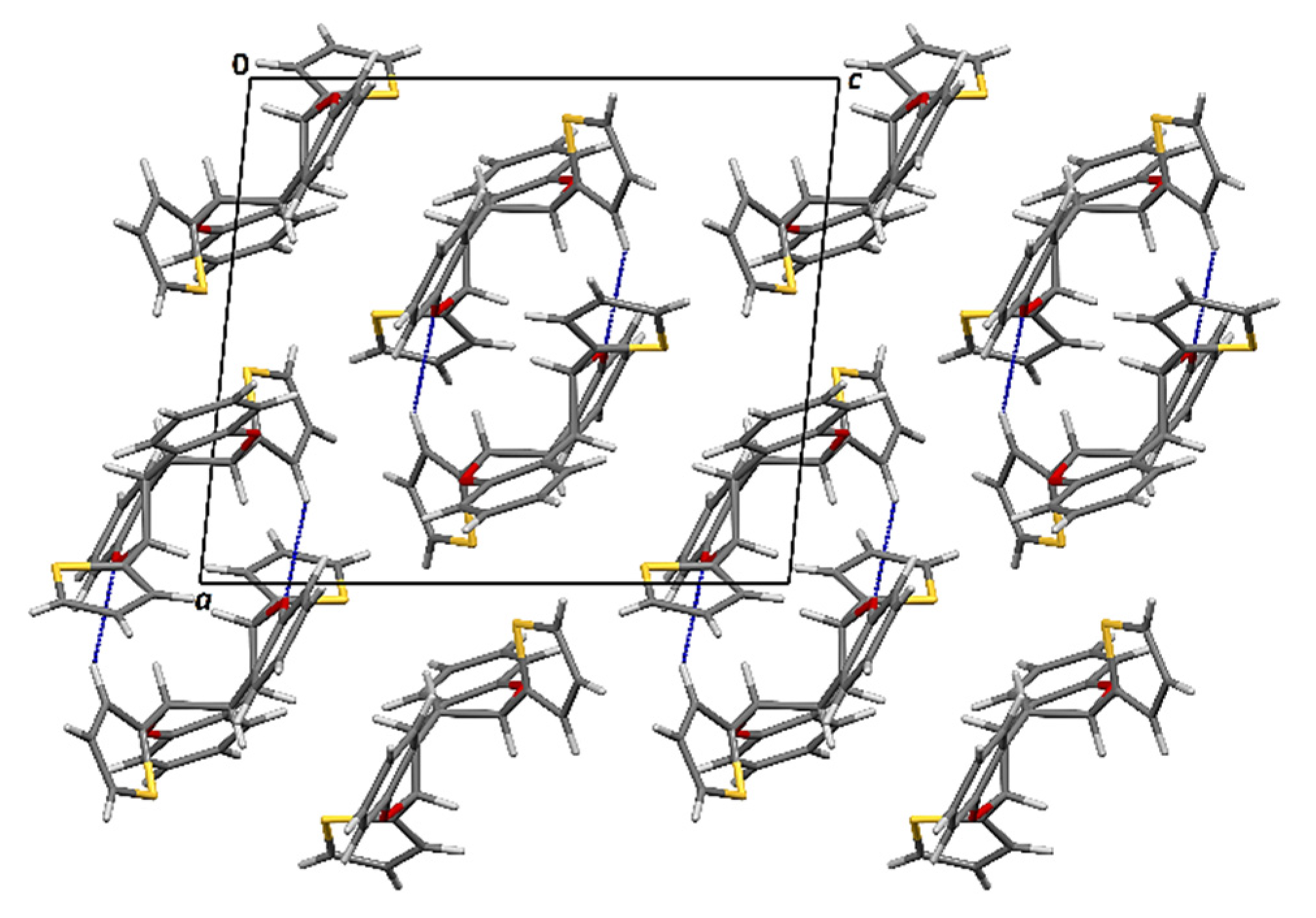
3.5. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noël, T.; Naber, J.R.; Hartman, R.L.; Mcmullen, J.P.; Jensen, K.F.; Buchwald, S.L. Palladium-catalyzed amination reactions in flow: Overcoming the challenges of clogging via acoustic irradiation. Chem. Sci. 2011, 2, 287–290. [Google Scholar] [CrossRef]
- Rashmi Pradhan, S.; Colmenares-Quintero, R.F.; Colmenares Quintero, J.C. Designing Microflowreactors for photocatalysis using sonochemistry: A systematic review article. Molecules 2019, 24, 3315. [Google Scholar] [CrossRef] [Green Version]
- Sambiagio, C.; Noël, T. Flow photochemistry: Shine some light on those tubes! Trends Chem. 2019, 2, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Oelgemoeller, M. Highlights of photochemical reactions in microflow reactors. Chem. Eng. Technol. 2012, 35, 1144–1152. [Google Scholar] [CrossRef]
- Chandrasekhar, D.; Borra, S.; Kapure, J.S.; Shivaji, G.S.; Srinivasulu, G.; Maurya, R.A. Visible-light photoredox catalysis: Direct synthesis of fused-carbolines through an oxidation/[3 + 2]cycloaddition/oxidative aromatization reaction cascade in batch and flow microreactors. Org. Chem. Front. 2015, 2, 1308–1312. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; Oliveira, K.T. Porphyrins as catalysts in scalable organic reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.C.; Silva, L.O.; Andrade Bartolomeu, A.; Brocksom, T.J.; Oliveira, K.T. Recent applications of porphyrins as photocatalysts in organic synthesis: Batch and continuous flow approaches. Beilstein J. Org. Chem. 2020, 16, 917–955. [Google Scholar] [CrossRef] [PubMed]
- Šagud, I.; Škorić, I. Photocatalytic oxygenation by water-soluble metalloporphyrins as a pathway to functionalized polycycles. Int. J. Photoenergy 2018, 2018, 1017957. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.H.; Han, J.W.; Jung, J.; Nakagawa, T.; Lee, Y.M.; Nam, W.; Fukuzumi, S. Photocatalytic oxygenation reactions with a cobalt porphyrin complex using water as an oxygen source and dioxygen as an oxidant. J. Am. Chem. Soc. 2019, 141, 9155–9159. [Google Scholar] [CrossRef]
- Vuk, D.; Horváth, O.; Škorić, I. New functionalized polycycles obtained by photocatalytic oxygenation using Mn(III) porphyrins in basic media. Catalysts 2019, 9, 304. [Google Scholar] [CrossRef]
- Mlakić, M.; Šalić, A.; Bačić, M.; Zelić, B.; Šagud, I.; Horváth, O.; Škorić, I. Photocatalytic oxygenation of heterostilbenes—Batch versus microflow reactor. Catalysts 2021, 11, 395. [Google Scholar] [CrossRef]
- Zhang, R.; Horner, J.H.; Newcomb, M. Laser flash photolysis generation and kinetic studies of porphyrin−manganese−oxo intermediates. Rate constants for oxidations effected by porphyrin−Mn(V)−oxo species and apparent disproportionation equilibrium constants for porphyrin−Mn(IV)−oxo species. J. Am. Chem. Soc. 2005, 127, 6573–6582. [Google Scholar] [CrossRef]
- Newcomb, M.; Zhang, R.; Pan, Z.; Harischandra, D.; Chandrasena, R.; Horner, J.; Martinez, E., II. Laser flash photolysis production of metal-oxo derivatives and direct kinetic studies of their oxidation reactions. Catal. Today 2006, 117, 98–104. [Google Scholar] [CrossRef]
- Zhang, R.; Newcomb, M. Laser Flash Photolysis generation of high-valent transition metal−oxo species: Insights from kinetic studies in real time. Acc. Chem. Res. 2008, 41, 468–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglia, R.A.; Zaragoza, J.P.T.; Goldberg, D.P. Biomimetic reactivity of oxygen-derived manganese and iron porphyrinoid complexes. Chem. Rev. 2017, 117, 13320–13352. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Ohkubo, K.; Goldberg, D.P.; Fukuzumi, S. Photocatalytic oxygenation of 10-methyl-9,10-dihydroacridine by O2 with manganese porphyrins. J. Phys. Chem. A 2014, 118, 6223–6229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quina, F.H.; Medeiros Silva, G.T. The photophysics of photosensitization: A brief overview. J. Photochem. Photobiol. 2021, 7, 100042. [Google Scholar] [CrossRef]
- Buglak, A.A.; Filatov, M.A.; Hussain, M.A.; Sugimoto, M. Singlet oxygen generation by porphyrins and metalloporphyrins revisited: A quantitative structure-property relationship (QSPR) study. J. Photochem. Photobiol. A 2020, 403, 112833. [Google Scholar] [CrossRef]
- Kou, J.; Dou, D.; Yang, L. Porphyrin photosensitizers in photodynamic therapy and its applications. Oncotarget 2017, 8, 81591–81603. [Google Scholar] [CrossRef] [Green Version]
- Prashanthi, S.; Kumar, P.H.; Siva, D.; Lanke, S.R.; Rao, V.J.; Basak, S.; Bangal, P.R. Photochemical E(trans)–Z(Z)–E isomerization of an amphiphilic cholest-5-en-3β-yl(E)-9-anthraceneprop-2-enoate on solid substrate. J. Phys. Chem. C 2011, 115, 20682–20688. [Google Scholar] [CrossRef]
- Isokuortti, J.; Kuntze, K.; Virkki, M.; Ahmed, Z.; Vuorimaa-Laukkanen, E.; Filatov, M.A.; Turshatov, A.; Laaksonen, T.; Priimagi, A.; Durandin, N.A. Expanding excitation wavelengths for azobenzene photoswitching into the near-infrared range via endothermic triplet energy transfer. Chem. Sci. 2021, 12, 7504–7509. [Google Scholar] [CrossRef]
- Mlakić, M.; Rajič, L.; Ljubić, A.; Vušak, V.; Zelić, B.; Gojun, M.; Odak, I.; Čule, I.; Šagud, I.; Šalić, A.; et al. Synthesis of new heterocyclic resveratrol analogues in milli- and microreactors: Intensification of the Wittig reaction. J. Flow Chem. 2022. [Google Scholar] [CrossRef]
- Gülçin, I. Antioxidant properties of resveratrol: A structure–activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Fauconneau, B.; Waffo-Teguo, P.; Huguet, F.; Barrier, L.; Decendit, A.; Merillon, J.M. Comparative study of radical scavenger and antioxidant properties of phenolic compounds from vitis vinifera cell cultures using in vitro tests. Life Sci. 1997, 61, 2103–2110. [Google Scholar] [CrossRef]
- Tsuji, K.; Ichikawa, K.; Yamatoto, H.; Tokunaga, J. Solubilities of oxygen and nitrogen in acetone-water mixed solvent. Kagaku Kogaku Ronbunshu 1987, 13, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Erikson, P.R.; McNeill, K. Triplet-state dissolved organic matter quantum yields and lifetimes from direct observation of aromatic amine oxidation. Environ. Sci. Technol. 2017, 51, 13151–13160. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, D65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. It ORTEP-3 for Windows—A Version of It ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H/Å | H···A/Å | D···A/Å | D–H···A/º | Symm. Op. on A | |
|---|---|---|---|---|---|
| C3–H3∙∙∙O2 | 0.93 | 2.66 | 3.479(4) | 147 | x, y, z |
| Compound | Time, h | E-1 | Z-1 | 2 | 3 | 4 | Undefined |
|---|---|---|---|---|---|---|---|
| % | |||||||
| free-base cationic porphyrin | 2 | 8 | 8 | 15 | 6 | 3 | 60 |
| free-base anionic porphyrin | 2 | - | 8 | 11 | - | - | 81 |
| cationic Mn(III) porphyrin | 6 | 6 | 62 | - | 8 | 2 | 22 |
| anionic Mn(III) porphyrin | 6 | traces | 25 | 5 | 4 | 2 | 64 |
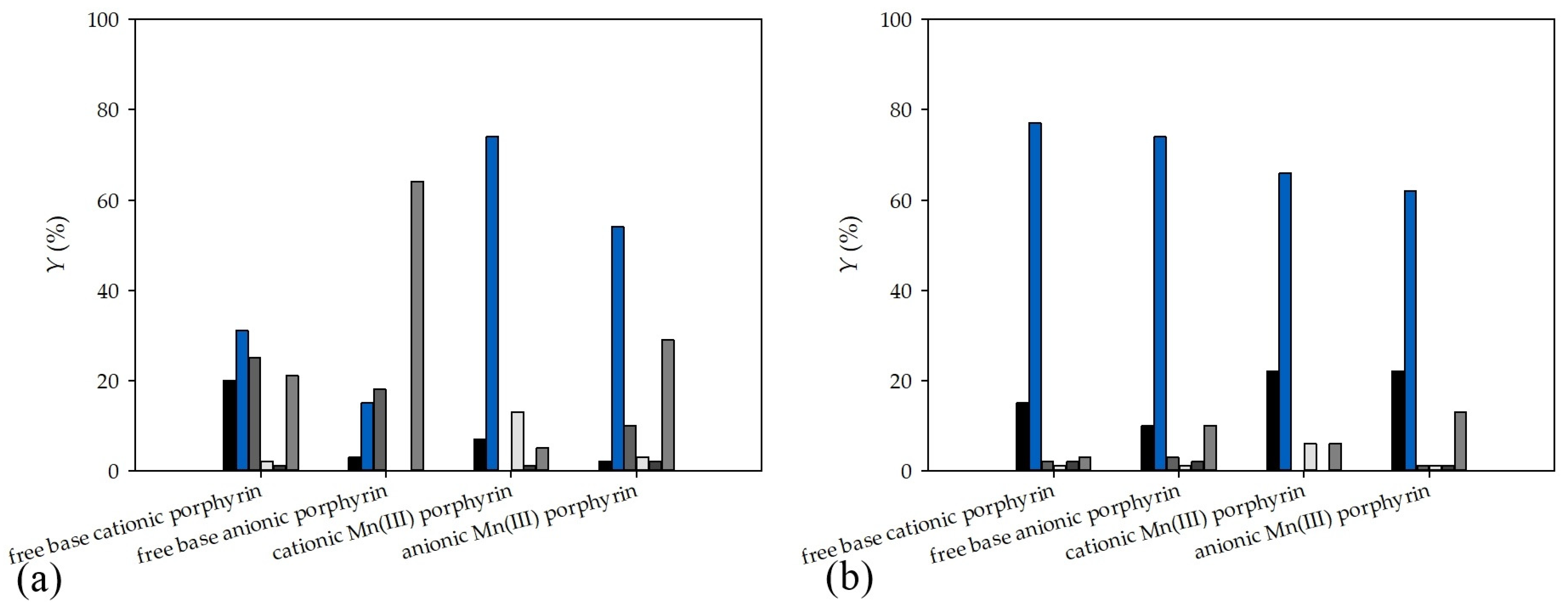
| Compound | Reactor | Time, h | E-1 | Z-1 | 2 | 3 | 4 | Unidenti-Fied | Productivity, μmol mL−1 min−1 | Ratio of Productivities |
|---|---|---|---|---|---|---|---|---|---|---|
| % | ||||||||||
| free-base cationic porphyrin | Batch | t = 2 | 20 | 31 | 25 | 2 | 1 | 21 | 0.0550 | 1/4.25 |
| Microflow | τ = 0.5 | 15 | 77 | 2 | 1 | 2 | 3 | 0.2338 | ||
| free-base anionic porphyrin | Batch | t = 2 | 3 | 15 | 18 | - | - | 64 | 0.0667 | 1/3.71 |
| Microflow | τ = 0.5 | 10 | 74 | 3 | 1 | 2 | 10 | 0.2475 | ||
| cationic Mn(III) porphyrin | Batch | t = 6 | 7 | 74 | - | 13 | 1 | 5 | 0.0213 | 1/10.07 |
| Microflow | τ = 0.5 | 22 | 66 | - | 6 | - | 6 | 0.2145 | ||
| anionic Mn(III) porphyrin) | Batch | t = 6 | 2 | 54 | 10 | 3 | 2 | 29 | 0.0225 | 1/9.51 |
| Microflow | τ = 0.5 | 22 | 62 | 1 | 1 | 1 | 13 | 0.2140 | ||
| Compound | 2 |
|---|---|
| Empirical formula | C24H18O2S1.8 |
| Formula wt./g mol−1 | 393.96 |
| Crystal dimensions/mm | 0.3 × 0.25 × 0.1 |
| Space group | P21/n |
| a/Å | 12.3164(7) |
| b/Å | 11.2022(11) |
| c/Å | 14.2889(9) |
| α/° | 90 |
| β/° | 95.702(5) |
| γ/° | 90 |
| Z | 4 |
| V/Å3 | 1961.7(3) |
| Dcalc/g cm−3 | 1.334 |
| µ/mm−1 | 2.355 |
| Θ range/° | 4.523–80.415 |
| T(K) | 293(2) |
| Radiation wavelength | 1.54184 (CuKα) |
| Diffractometer type | XtaLAB Synergy, Dualflex, HyPix |
| Range of h, k, l | −15 > h > 15; −13 > k > 14; −18 > l > 16 |
| Reflections collected | 15177 |
| Independent reflections | 4003 |
| Observed reflections (I ≥ 2σ) | 1866 |
| Rint | 0.1331 |
| R (F) | 0.0951 |
| Rw (F2) | 0.3112 |
| No. of parameters, restraints | 255, 0 |
| Goodness of fit | 0.952 |
| Δρmax, Δρmin (eÅ−3) | 0.443; –0.289 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakić, M.; Ljubić, A.; Šalić, A.; Zelić, B.; Horváth, O.; Milašinović, V.; Gojun, M.; Molčanov, K.; Škorić, I. Photocatalytic Transformations of the Resveratrol Derivative in Microflow Reactor. Catalysts 2022, 12, 1510. https://doi.org/10.3390/catal12121510
Mlakić M, Ljubić A, Šalić A, Zelić B, Horváth O, Milašinović V, Gojun M, Molčanov K, Škorić I. Photocatalytic Transformations of the Resveratrol Derivative in Microflow Reactor. Catalysts. 2022; 12(12):1510. https://doi.org/10.3390/catal12121510
Chicago/Turabian StyleMlakić, Milena, Anabela Ljubić, Anita Šalić, Bruno Zelić, Ottó Horváth, Valentina Milašinović, Martin Gojun, Krešimir Molčanov, and Irena Škorić. 2022. "Photocatalytic Transformations of the Resveratrol Derivative in Microflow Reactor" Catalysts 12, no. 12: 1510. https://doi.org/10.3390/catal12121510
APA StyleMlakić, M., Ljubić, A., Šalić, A., Zelić, B., Horváth, O., Milašinović, V., Gojun, M., Molčanov, K., & Škorić, I. (2022). Photocatalytic Transformations of the Resveratrol Derivative in Microflow Reactor. Catalysts, 12(12), 1510. https://doi.org/10.3390/catal12121510