
Kinetic Resolution in Transannular Morita-Baylis-Hillman Reaction: An Approximation to the Synthesis of Sesquiterpenes from Guaiane Family

 ,
,
Abstract
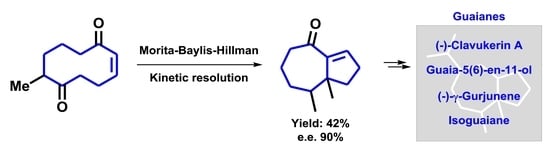
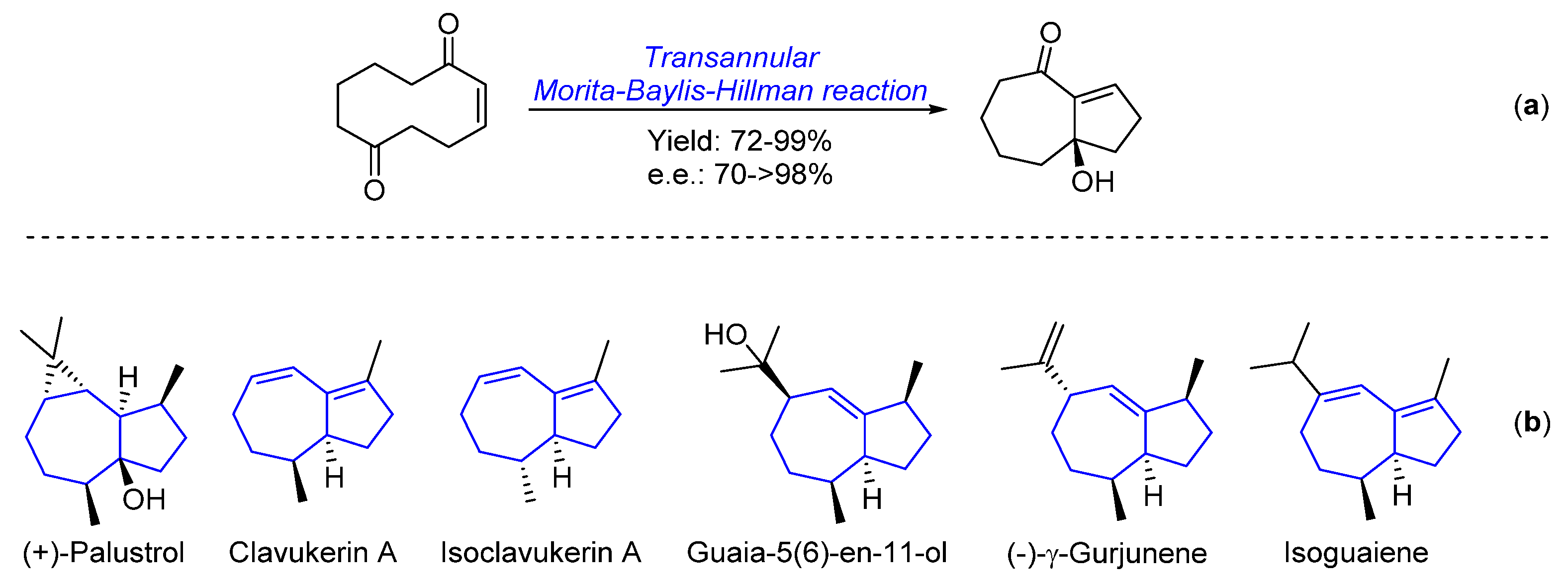
1. Introduction
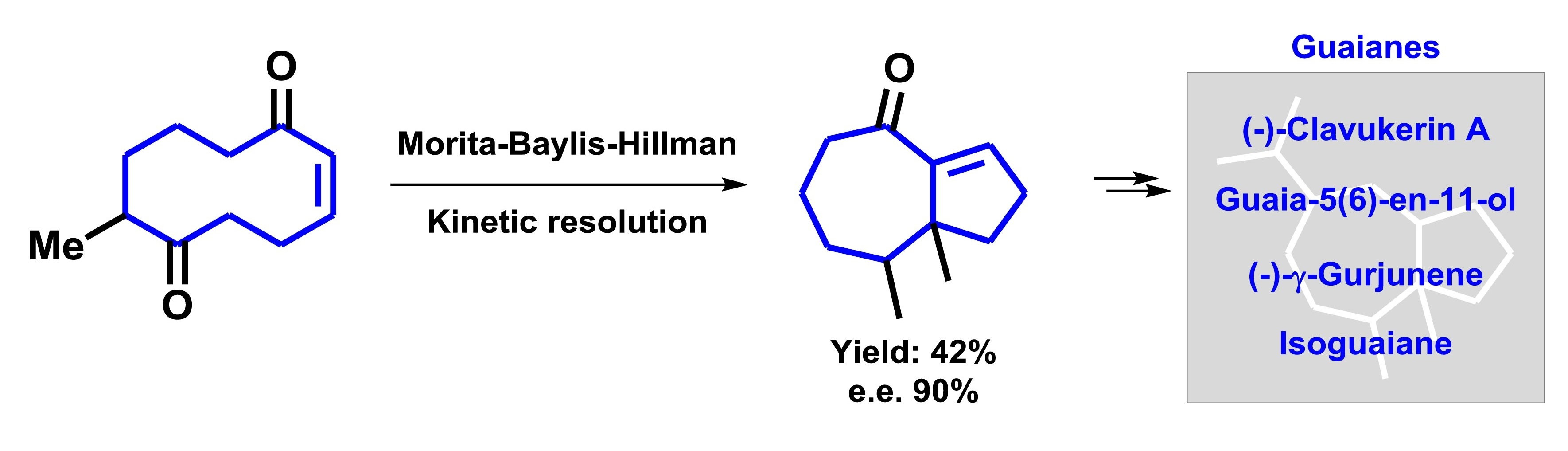
2. Results and Discussion
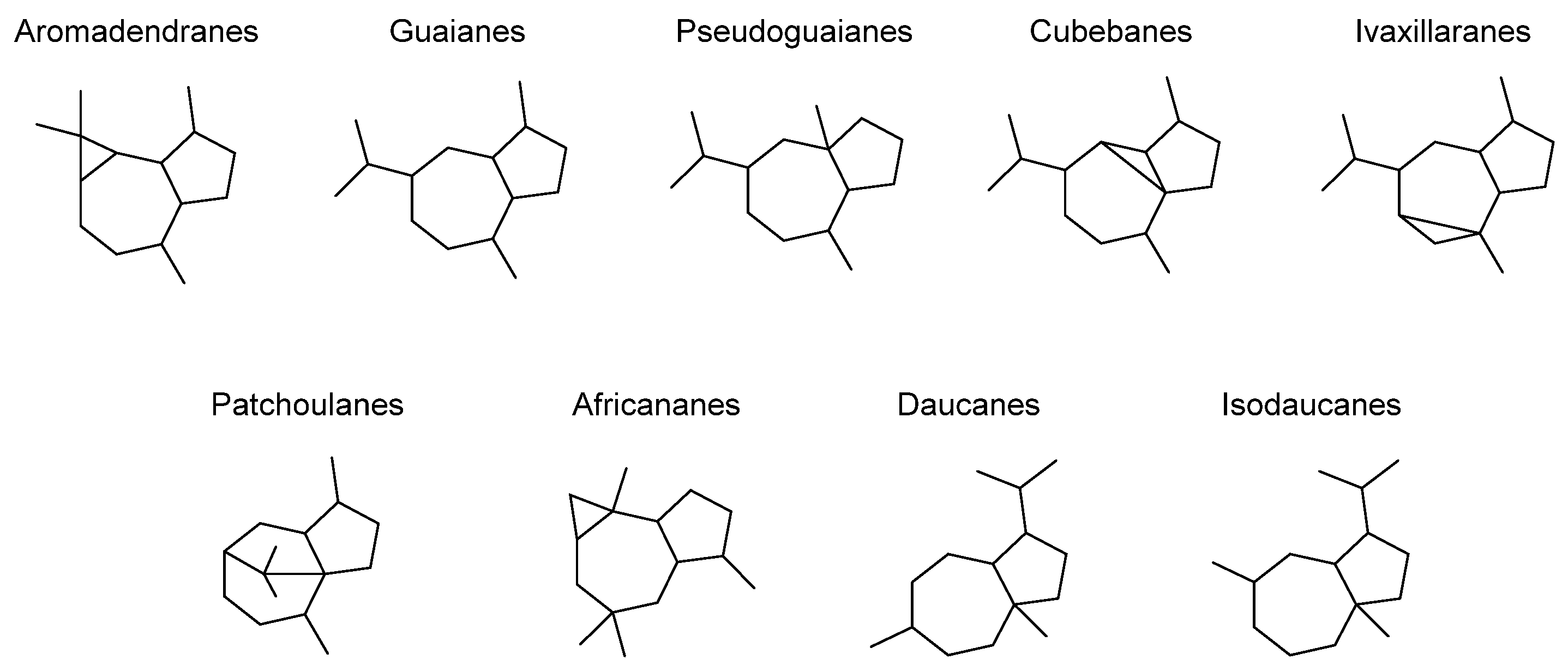
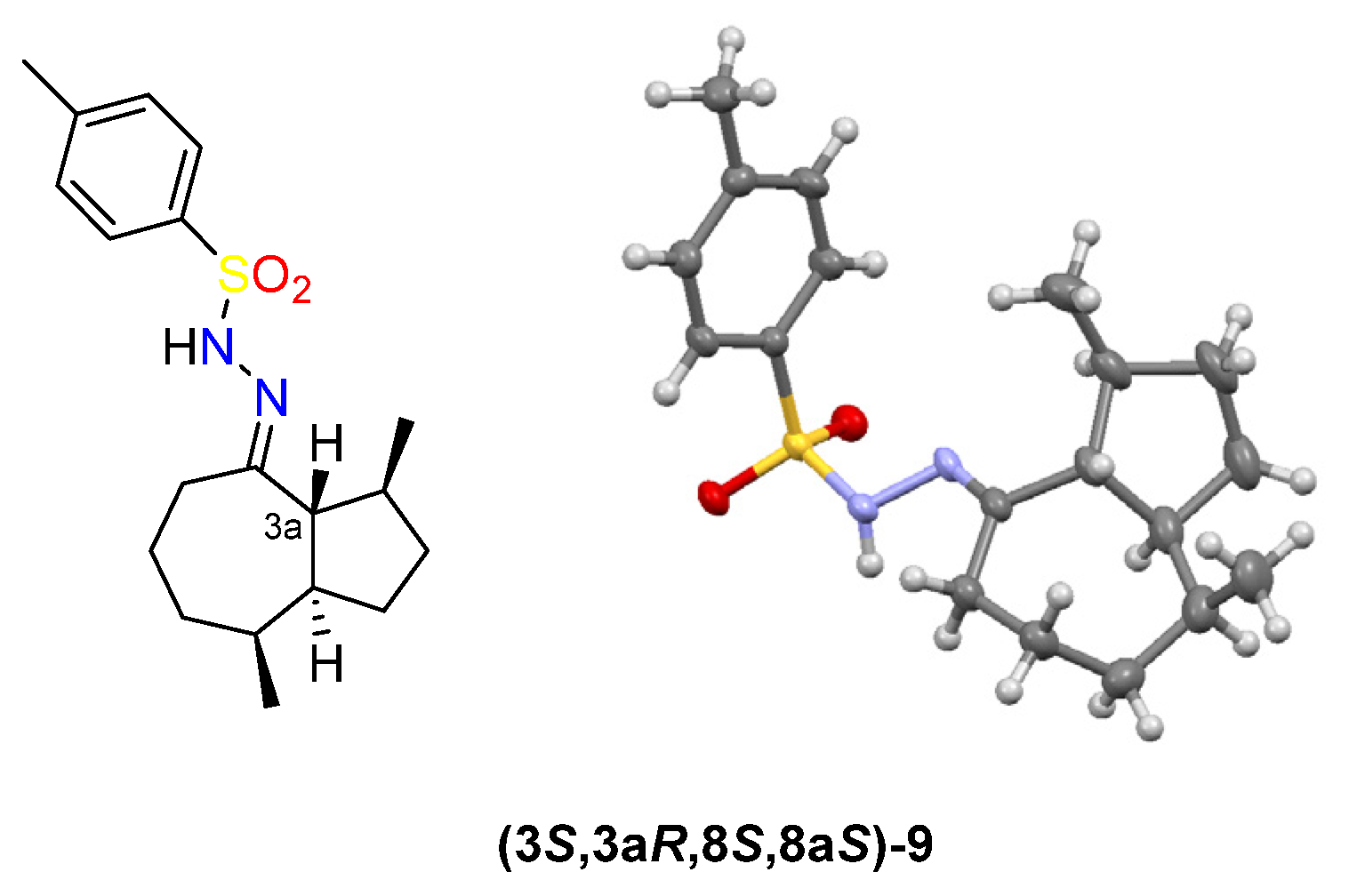
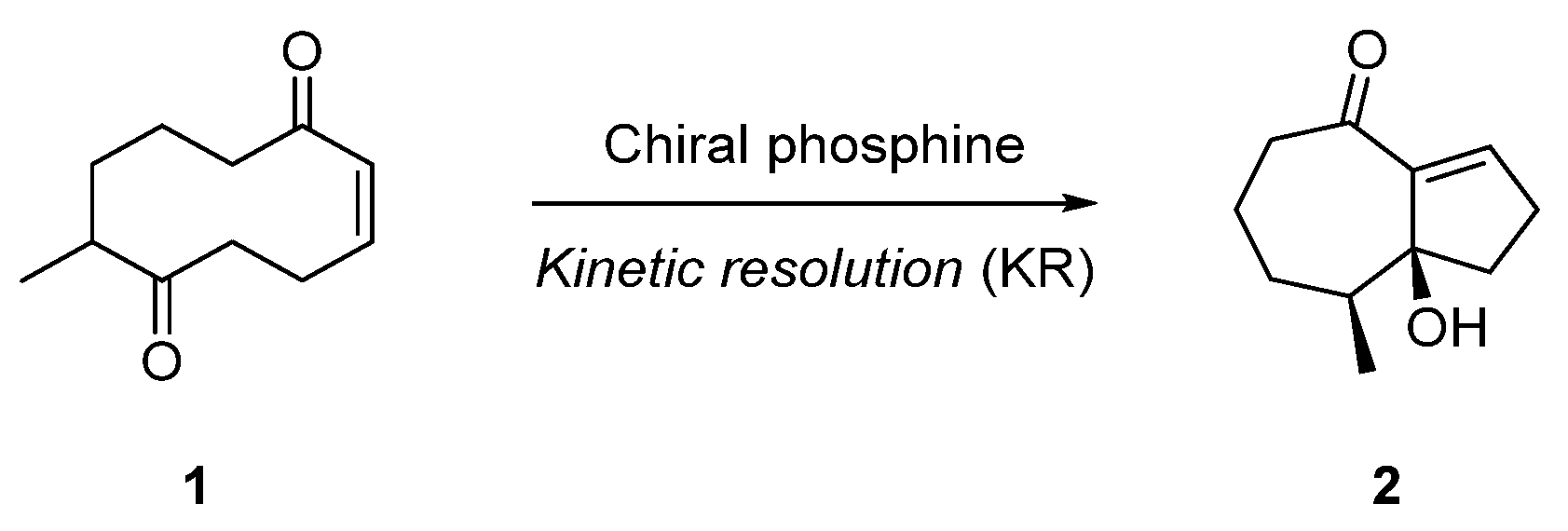
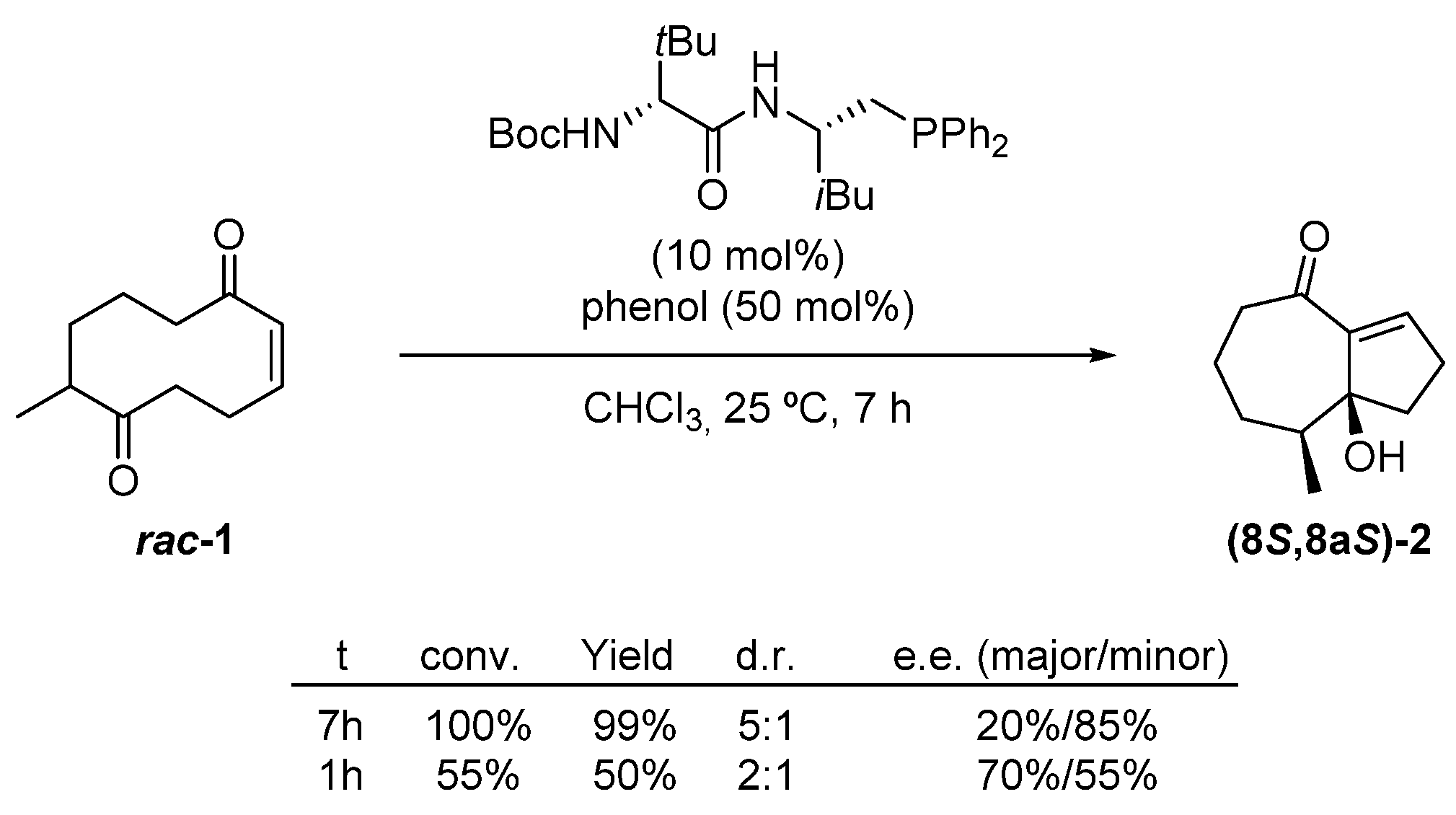
2.1. Transannular Kinetic Resolution
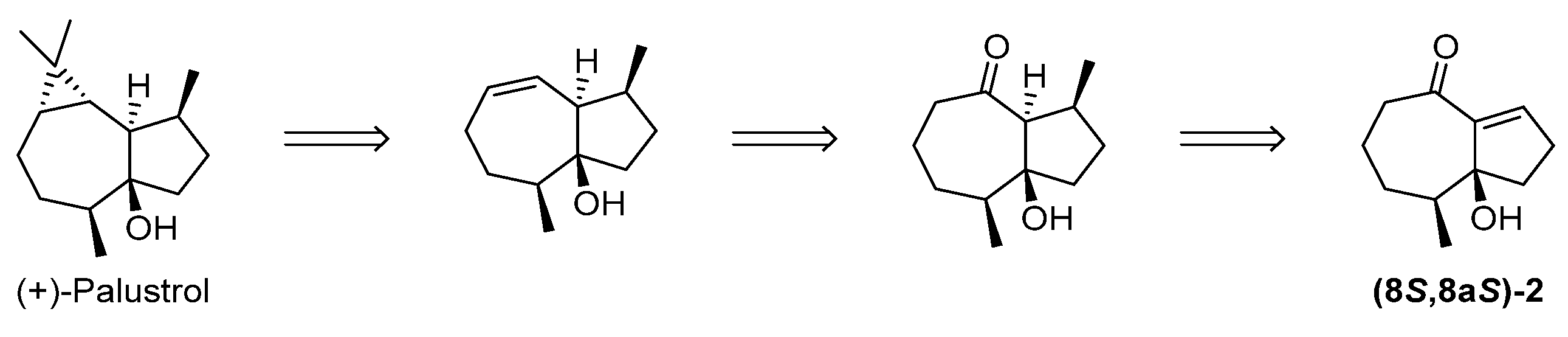
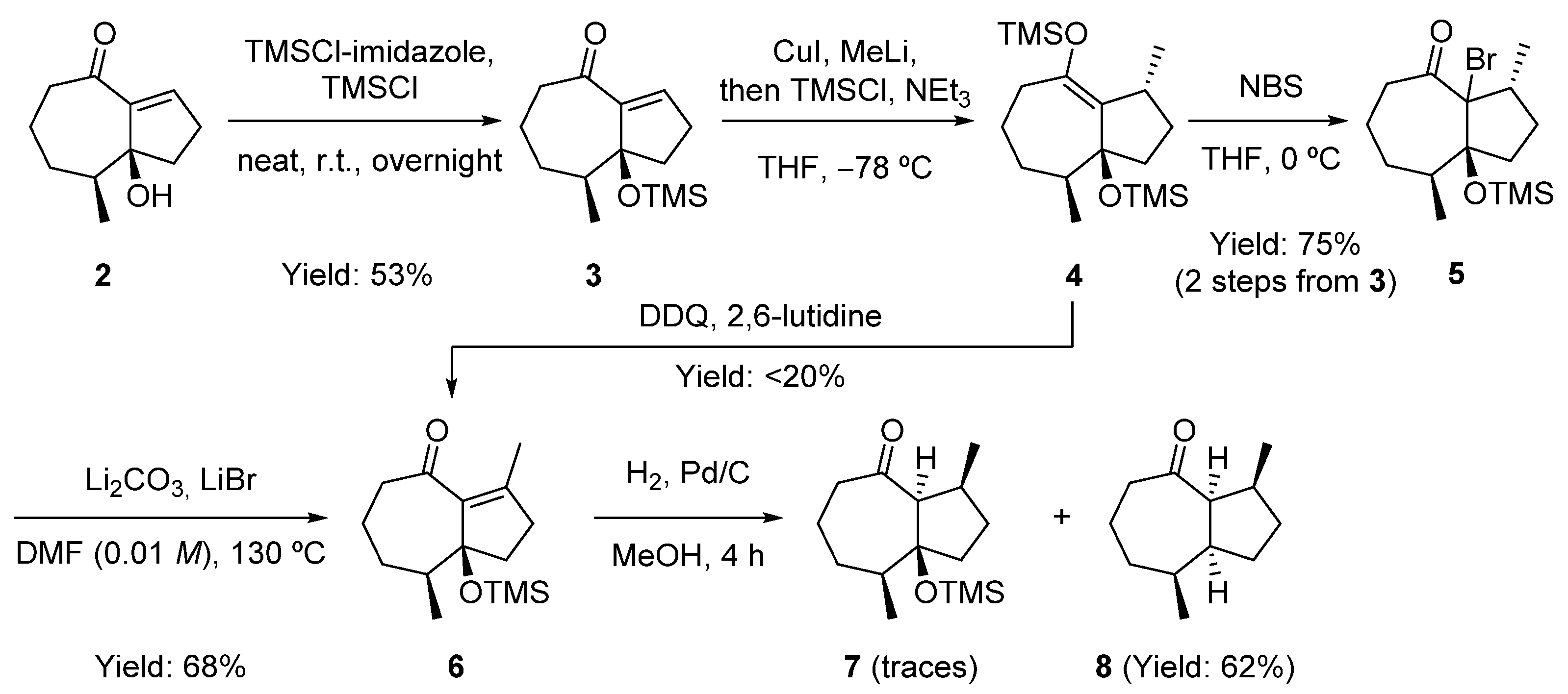
2.2. Preparation of an Advanced Intermediate in the Synthesis of Palustrol
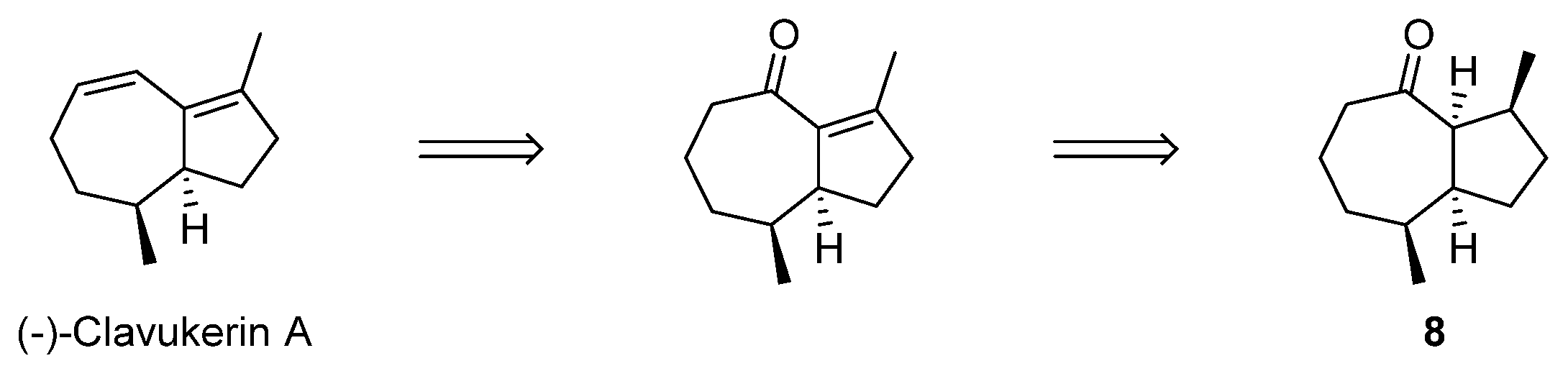
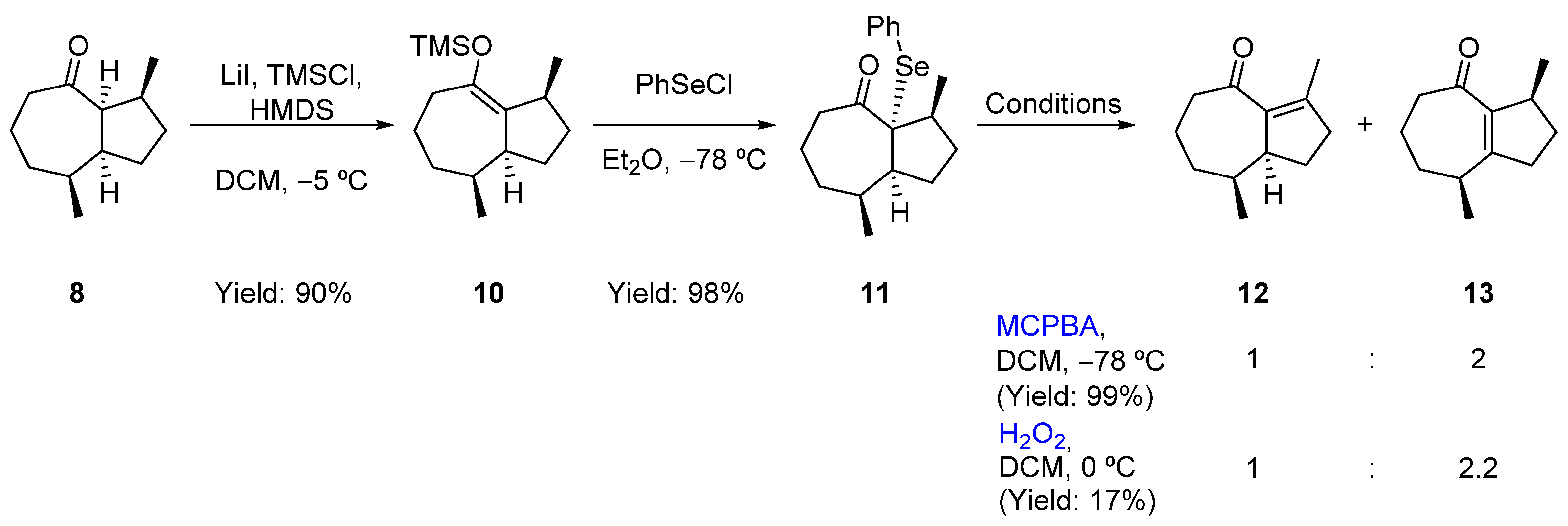
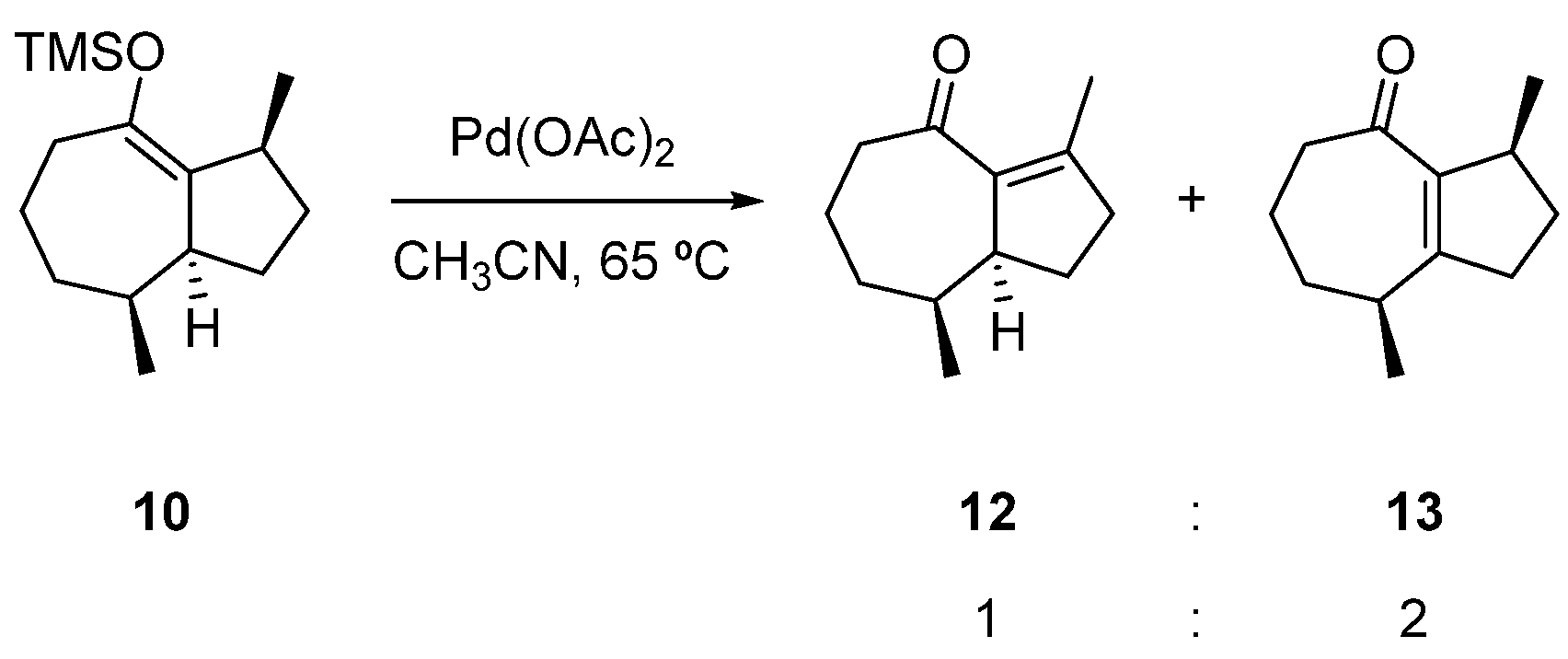
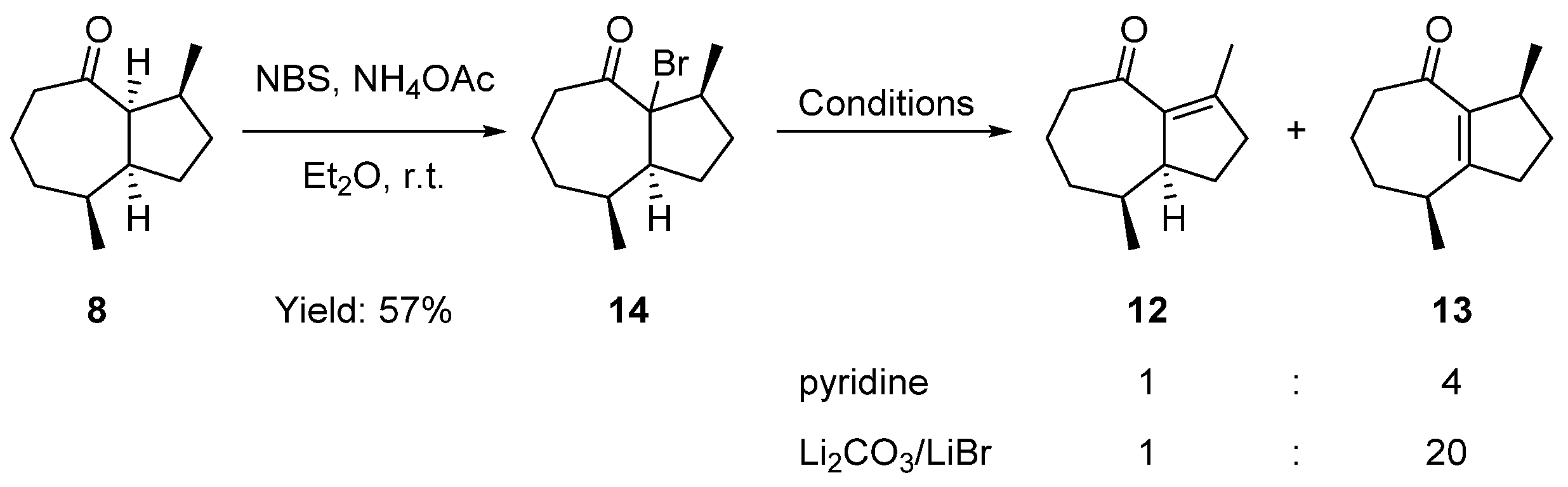
2.3. Formal Total Synthesis of Clavukerin A
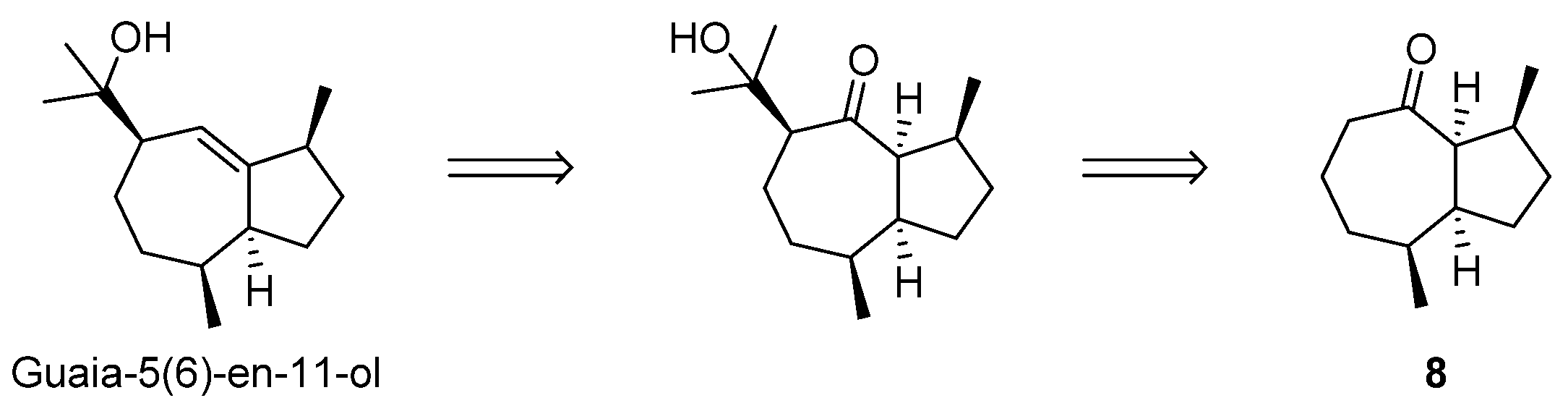
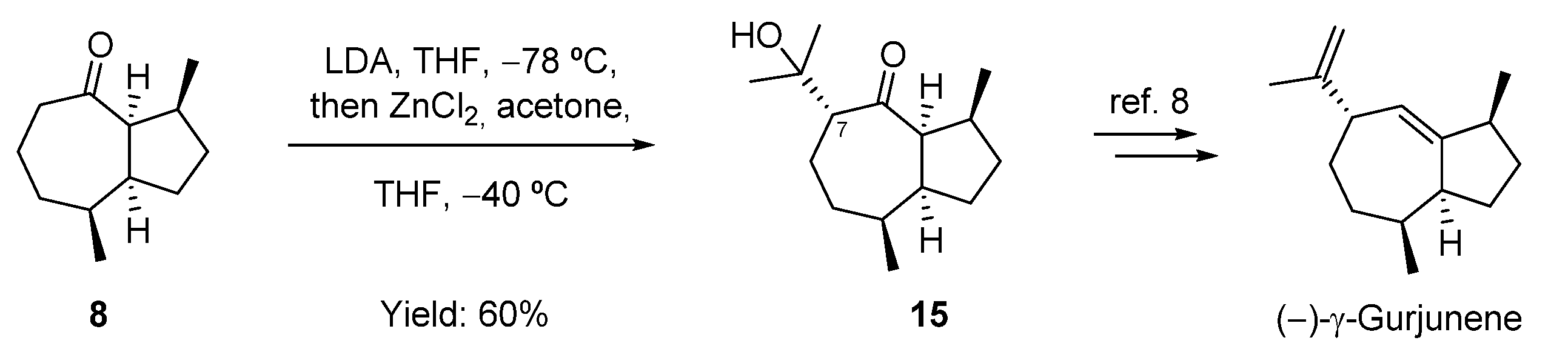
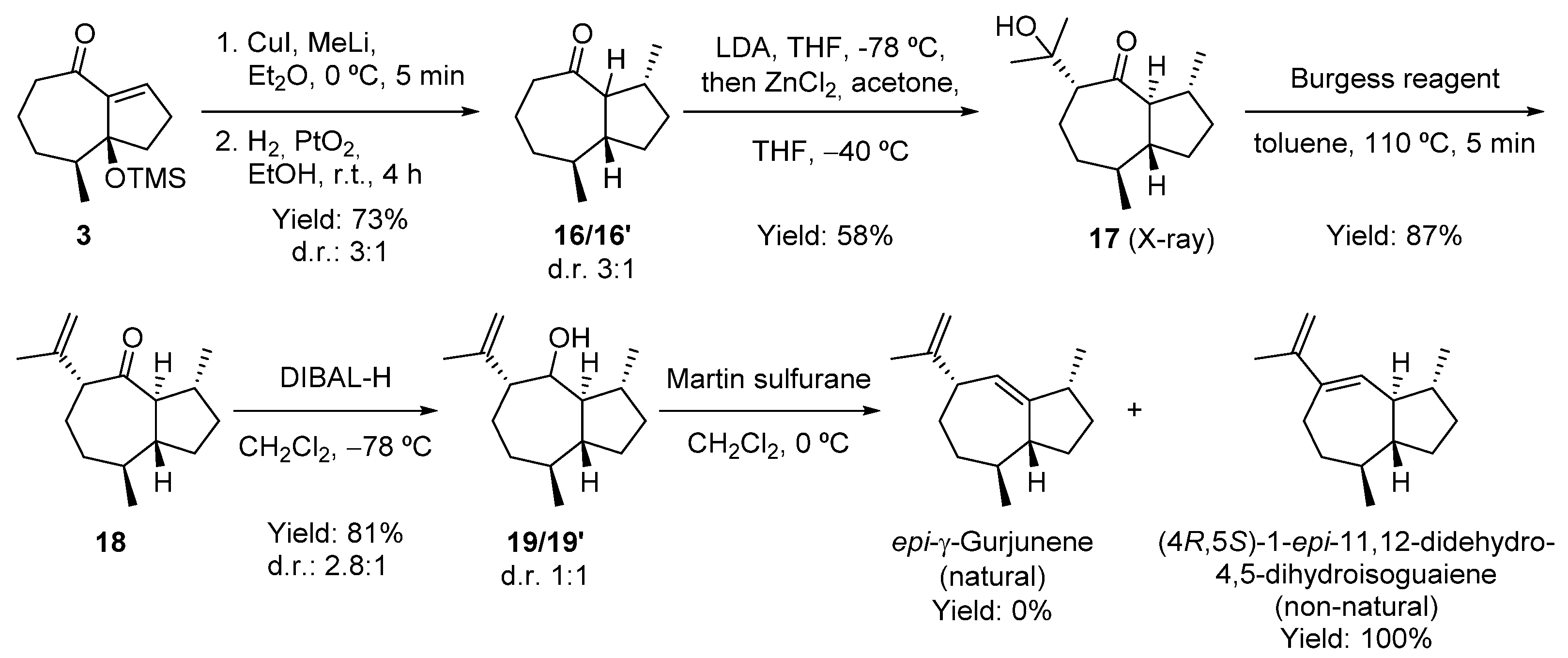
2.4. Attempt to the Synthesis Guaia-5(6)-en-11-ol; Synthesis of (−)-γ-Gurjunene and Non-Natural 1-epi-11,12-didehydro-4,5-dihydroisoguaiane
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, G. Chemistry of Terpenoids and Carotenoids; Discovery Publishing Home: New Dehli, India, 2007. [Google Scholar]
- Zhang, L.; Demain, A.L. Natural Products: Drug Discovery and Therapeutic Medicine; Human Press: Totowa, NJ, USA, 2005. [Google Scholar]
- Thomson, R.H. The Chemistry of Natural Products; Blackie Academic & Professional: Glasgow, Scotland, 1993. [Google Scholar]
- Pirrung, M.C.; Morehead, A.T.; Young, B.G. The Total Synthesis of Natural Products: Bicyclic and Tricyclic Sesquiterpenes; John Wiley & Sons: Atlanta, GA, USA, 2009. [Google Scholar]
- Overton, K.H. Terpenoids and Steroids; RSC Publishing: London, UK, 1975. [Google Scholar]
- Kornprobst, J.-M. Encyclopedia of Marine Natural Products; Wiley-Blackwell: Oxford, UK, 2010. [Google Scholar]
- Bideau, F.L.; Kousara, M.; Chen, L.; Wei, L.; Dumas, F. Tricyclic Sesquiterpenes from Marine Origin. Chem. Rev. 2017, 117, 6110–6159. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine Pharmacology in 2009–2011: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-Inflammatory, Antiprotozoal, Antituberculosis, and Antiviral Activities; Affecting the Immune and Nervous Systems, and other Miscellaneous Mechanisms of Action. Mar. Drugs 2013, 11, 2510–2573. [Google Scholar]
- Wright, A.D.; König, G.M.; Angerhofer, C.K.; Greenidge, P.; Linden, A.; Desqueyroux-Faundez, R. Antimalarial Activity: The Search for Marine-Derived Natural Products with Selective Antimalarial Activity. J. Nat. Prod. 1996, 59, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 1998, 15, 73–92. [Google Scholar] [CrossRef]
- Mato, R.; Manzano, R.; Reyes, E.; Carrillo, L.; Uria, U.; Vicario, J.L. Catalytic Enantioselective Transannular Morita–Baylis–Hillman Reaction. J. Am. Chem. Soc. 2019, 141, 9495–9499. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.N.; Cramer, N. Biomimetic Synthesis of (+)-Ledene, (+)-Viridiflorol, (−)-Palustrol, (+)-Spathulenol, and Psiguadial A, C, and D via the Platform Terpene (+)-Bicyclogermacrene. Chem. Eur. J. 2014, 20, 10654–10660. [Google Scholar] [CrossRef]
- Barthel, A.; Kaden, F.; Jäger, A.; Metz, P. Enantioselective Synthesis of Guaianolides in the Osmitopsin Family by Domino Metathesis. Org. Lett. 2016, 18, 3298–3301. [Google Scholar] [CrossRef] [PubMed]
- Knüppel, S.; Rogachev, V.O.; Metz, P. A Concise Catalytic Route to the Marine Sesquiterpenoids (−)-Clavukerin A and (−)-Isoclavukerin A. Eur. J. Org. Chem. 2010, 6145–6148. [Google Scholar] [CrossRef]
- Srikrishna, A.; Pardehi, V.H.; Satyanarayana, G. Enantioselective formal total syntheses of Clavukerin A and Isoclavukerin A via a ring-closing metathesis reaction. Tetrahedron Asymmetry 2010, 21, 746–750. [Google Scholar] [CrossRef]
- Grimm, E.L.; Methot, J.-L.; Shamji, M. Total synthesis of ( )-Clavukerin A. Pure Appl. Chem. 2003, 75, 231–234. [Google Scholar] [CrossRef][Green Version]
- Friese, J.C.; Krause, S.; Schäfer, H.J. Formal total synthesis of the trinorguaiane sesquiterpenes (+/−)-Clavukerin A and (+/−)-Isoclavukerin. Tetrahedron Lett. 2002, 43, 2683–2685. [Google Scholar] [CrossRef]
- Lee, E.; Yoon, C.H. 8-endo Cyclization of (alkoxycarbonyl)methyl radicals: Stereoselective synthesis of (−)-Clavukerin A and (−)-11-hydroxyguaiene. Tetrahedron Lett. 1996, 37, 5929–5930. [Google Scholar] [CrossRef]
- Shimizu, I.; Ishikawa, T. Stereoselective synthesis of (±)-Clavukerin A and (±)-Isoclavukerin A based on palladium-catalyzed reductive cleavage of alkenylcyclopropanes with formic acid. Tetrahedron Lett. 1994, 35, 1905–1908. [Google Scholar] [CrossRef]
- Honda, T.; Ishige, H.; Nagase, H. Chiral synthesis of a trinorguaiane sesquiterpene, Clavukerin A. J. Chem. Soc. Perkin Trans 1994, 3305–3310. [Google Scholar] [CrossRef]
- Kim, S.K.; Pak, C.S. Total synthesis of (.+-.)-Clavukerin A: A new trinorguaiane sesquiterpene. Biomimetic synthesis of (.+-.)-clavularin A from (.+-.)-Clavukerin A. J. Org. Chem. 1991, 56, 6829–6832. [Google Scholar] [CrossRef]
- Asaoka, M.; Kosaka, T.; Itahana, H.; Takei, H. Total Synthesis of Clavukerin A and Its Epimer. Chem. Lett. 1991, 1295–1298. [Google Scholar] [CrossRef]
- Trost, B.M.; Higuchi, R.I. On the Diastereoselectivity of Intramolecular Pd-Catalyzed TMM Cycloadditions. An Asymmetric Synthesis of the Perhydroazulene (−)-Isoclavukerin A. J. Am. Chem. Soc. 1996, 118, 10094–10105. [Google Scholar] [CrossRef]
- Huang, A.-C.; Sumby, C.J.; Tiekink, E.R.T.; Taylor, D.K. Synthesis of guaia-4(5)-en-11-ol, guaia-5(6)-en-11-ol, aciphyllene, 1-epi-melicodenones C and E, and other guaiane-type sesquiterpenoids via the diastereoselective epoxidation of guaiol. J. Nat. Prod. 2014, 77, 2522–2536. [Google Scholar] [CrossRef] [PubMed]
- Rienäcker, R.; Graefe, J. Catalytic Transformations of Sesquiterpene Hydrocarbons on Alkali Metal/Aluminum Oxide. Angew. Chem. Int. Ed. Engl. 1985, 24, 320–321. [Google Scholar] [CrossRef]
- Wang, Y.; Darweesh, A.F.; Zimdars, P.; Metz, P. An efficient synthesis of the guaiane sesquiterpene (−)-isoguaiene by domino metathesis. Belstein J. Org. Chem. 2019, 15, 858–862. [Google Scholar] [CrossRef]
- Blay, G.; Garcia, B.; Molina, E.; Pedro, J.R. Total Syntheses of Four Stereoisomers of 4α-Hydroxy-1β,7β-peroxy- 10βH-guaia-5-ene. Org. Lett. 2005, 7, 3291–3294. [Google Scholar] [CrossRef]
- Sasaki, S.; Sutoh, K.; Shimizu, Y.; Kato, K.; Yoshifuji, M. Oxidation of tris(2,4,6-triisopropylphenyl)phosphine and arsine. Tetrahedron Lett. 2014, 55, 322–325. [Google Scholar] [CrossRef]
- Hilliard, C.R.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Synthesis, purification, and characterization of phosphine oxides and their hydrogen peroxide adducts. Dalton Trans. 2012, 41, 1742–1754. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, P.; Slawin, A.M.Z.; Smith, M.B.; Woolins, J.D. Palladium(II) and Platinum(II) Complexes of the Heterodifunctional Ligand Ph2PNHP(O)Ph2. Inorg. Chem. 1996, 35, 3675–3682. [Google Scholar] [CrossRef]
- Whitaker, C.M.; Kott, K.L.; McMahon, R.J. Synthesis and Solid-State Structure of Substituted Arylphosphine Oxides. J. Org. Chem. 1995, 60, 3499–3508. [Google Scholar] [CrossRef]
- Relles, H.M.; Schluenz, R.W. Chemical Transformations with Regenerable, Polymer-Supported Trisubstituted Phosphine Dichlorides. Efficacious Incorporation of Phosphorus Reagents on Polymer Supports. J. Am. Chem. Soc. 1974, 96, 6469–6475. [Google Scholar] [CrossRef]
- Kociensky, P.J. Protecting Groups; Thieme: Stuttgart, Germany, 2005. [Google Scholar]
- Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Kerwin, S.M.; Paul, A.G.; Heathcock, C.H. Quassinoid synthesis. 2. Preparation of a tetracyclic intermediate having the bruceantin tetrahydrofuran ring. J. Org. Chem. 1987, 52, 1686–1695. [Google Scholar] [CrossRef]
- Kumar, J.S.R.; O’Sullivan, M.; Resiman, S.E.; Hulford, C.A.; Ovaska, T.V. Facile approach to the bicyclo[5.3.0]decane ring system; efficient synthesis of (±)-7-epi-β-bulnesene. Tetrahedron Lett. 2002, 43, 1939–1941. [Google Scholar] [CrossRef]
- Banwell, M.G.; Hockless, D.C.R.; McLeod, M.D. Chemoenzymatic total syntheses of the sesquiterpene (−)-patchoulenone. New J. Chem. 2003, 27, 50–59. [Google Scholar] [CrossRef]
- Bertz, S.H.; Miao, G.; Rossiter, B.E.; Snyder, J.P. New Copper Chemistry. 25. Effect of TMSCl on the Conjugate Addition of Organocuprates to alpha.-Enones: A New Mechanism. J. Am. Chem. Soc. 1995, 117, 11023–11024. [Google Scholar] [CrossRef]
- Reuss, R.H.; Hassner, A. Synthetic methods. IV. Halogenation of carbonyl compounds via silyl enol ethers. J. Org. Chem. 1974, 39, 1785–1787. [Google Scholar] [CrossRef]
- Jeganathan, A.; Richardson, S.K.; Watt, D.S. Manganese Triacetate Oxidation of 3β, 3aβ, 6-Trimethyl-3a, 7aβ-dihydro-2(3H), 5(4H)-benzofurandione. Synth. Commun. 1989, 19, 1091–1100. [Google Scholar] [CrossRef]
- Roosen, P.C.; Vanderwal, C.D. Investigations into an Anionic Oxy-Cope/Transannular Conjugate Addition Approach to 7,20-Diisocyanoadociane. Org. Lett. 2014, 16, 4368–4371. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Katoh, S.-I.; Kato, T.; Urabe, D.; Inoue, M. Total Synthesis of Resiniferatoxin Enabled by Radical-Mediated Three-Component Coupling and 7-endo Cyclization. J. Am. Chem. Soc. 2017, 139, 16420–16429. [Google Scholar] [CrossRef] [PubMed]
- Shenvi, R.A.; Guerrero, C.A.; Shi, J.; Li, C.-C.; Baran, P.S. Synthesis of (+)-cortistatin A. J. Am. Chem. Soc. 2008, 130, 7241–7243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ikawa, T.; Hattori, K.; Sajiki, H.; Hirota, K. Solvent-modulated Pd/C-catalyzed deprotection of silyl ethers and chemoselective hydrogenation. Tetrahedron 2004, 60, 6901–6911. [Google Scholar] [CrossRef]
- Mahoney, W.S.; Brestensky, D.M.; Stryker, J.M.J. Selective hydride-mediated conjugate reduction of .alpha.,.beta.-unsaturated carbonyl compounds using [(Ph3P)CuH]6. Am. Chem. Soc. 1988, 110, 291. [Google Scholar] [CrossRef]
- Meiries, S.; Bartoli, A.; Decostanzi, M.; Parrain, J.-L.; Commeiras, L. Directed studies towards the total synthesis of (+)-13-deoxytedanolide: Simple and convenient synthesis of the C8–C16 fragment. Org. Biomol. Chem. 2013, 11, 4882–4890. [Google Scholar] [CrossRef]
- Ojima, I.; Kogure, T. Reduction of carbonyl compounds via hydrosilylation. 4. Highly regioselective reductions of. alpha.,. beta.-unsaturated carbonyl compounds. Organometallics 1982, 1, 1390–1399. [Google Scholar] [CrossRef]
- Ito, H.; Ishizuka, T.; Arimoto, K.; Miura, K.; Hosomi, A. Generation of a reducing reagent from copper (I) salt and hydrosilane. New practical method for conjugate reduction. Tetrahedron Lett. 1997, 38, 8887–8890. [Google Scholar] [CrossRef]
- Miller, R.D.; McKean, D.R. The Facile Silylation of Aldehydes and Ketones using Trimethylsilyl Iodide: An Exceptionally Simple Procedure for the Generation of Thermodynamically Equilibrated Trimethylsilylenol Ethers. Synthesis 1979, 730–732. [Google Scholar] [CrossRef]
- Ryu, I.; Murai, S.; Niwa, I.; Sonoda, N. A Convenient Synthesis of α-Phenylseleno Ketones and Aldehydes from Enol Silyl Ethers and Phenylselenenyl Bromide. Synthesis 1977, 12, 874–876. [Google Scholar] [CrossRef]
- Kamikubo, T.; Ogasawara, K. Preparation of (+)-Tricyclo[6.2.1.02,7]undec-2(7)-en-3-one and Its Conversion into (+)-epi-β-Santalene. Chem. Lett. 1995, 2, 95–96. [Google Scholar] [CrossRef]
- Fernández, B.; Pérez, J.A.M.; Granja, J.R.; Castedo, L.; Mouriño, A. Synthesis of hydrindan derivatives related to vitamin D. J. Org. Chem. 1992, 57, 3173–3178. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Younh, M.W.; Lauer, R.F. Reactions of selenoxides: Thermal syn-elimination and H218O exchange. Tetrahedron Lett. 1973, 22, 1979–1982. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Lauer, R.F.; Teranishi, A.Y. Electrophilic and nucleophilic organoselenium reagents. New routes to .alpha.,.beta.-unsaturated carbonyl compounds. J. Am. Chem. Soc. 1973, 95, 6137–6139. [Google Scholar] [CrossRef]
- Reich, H.J.; Renga, J.M.; Reich, I.L. Organoselenium chemistry. Conversion of ketones to enones by selenoxide syn elimination. J. Am. Chem. Soc. 1975, 97, 5434–5447. [Google Scholar] [CrossRef]
- Angeles, A.R.; Waters, S.P.; Danishefsky, S.J. Total Syntheses of (+)- and (−)-Peribysin E. J. Am. Chem. Soc. 2008, 130, 13765–13770. [Google Scholar] [CrossRef]
- Uchida, K.; Yokoshima, S.; Kan, T.; Fukuyama, T. Total Synthesis of (±)-Morphine. Org. Lett. 2006, 8, 5311–5313. [Google Scholar] [CrossRef]
- Hu, X.-D.; Tu, Y.Q.; Zhang, E.; Gao, S.; Wang, S.; Wang, A.; Fan, C.-A.; Wang, M. Total Synthesis of (±)-Galanthamine. Org. Lett. 2006, 8, 1823–1825. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Hirao, T.; Saegusa, T. Synthesis of .alpha.,.beta.-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers. J. Org. Chem. 1978, 43, 1011–1013. [Google Scholar] [CrossRef]
- Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. A mild and efficient procedure for α-bromination of ketones using N-bromosuccinimide catalysed by ammonium acetate. Chem. Commun. 2004, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Ushakov, D.B.; Navickas, V.; Ströbele, M.; Maichle-Mössmer, C.; Sasse, F.; Maier, M.E. Total Synthesis and Biological Evaluation of (−)-9-Deoxy-englerin A. Org. Lett. 2011, 13, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; Wijnberg, J.B.P.A.; Stork, G.A.; Groot, A. The synthesis of (−)-kessane, starting from natural (+)-aromadendrene-II. Tetrahedron 1991, 47, 4409–4416. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 7th ed.; Elsevier: Oxford, UK, 2012. [Google Scholar]
- Williams, D.B.G.; Lawton, M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Still, W.C.; Kahn, H.; Mitra, A.J. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
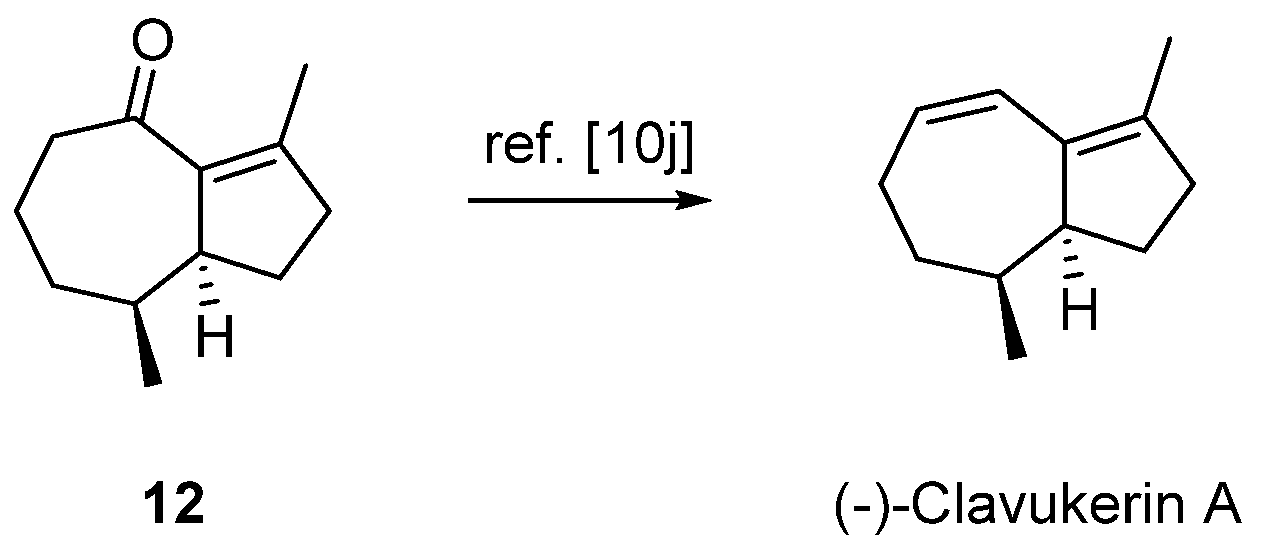{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | T (°C) | Time (min) | Recovered 1 (%) | e.e. 1 (%) 2 | Yield 2 (%) | e.e. 2 (%) 2 |
|---|---|---|---|---|---|---|---|
| 1 | CHCl3 | 25 | 40 | 61 | 50 | 35 | 88 |
| 2 | CHCl3 | 25 | 45 | 62 | 54 | 37 | 88 |
| 3 | CHCl3 | 25 | 50 | 57 | 54 | 39 | 88 |
| 4 | CHCl3 | 25 | 55 | 50 | 66 | 42 | 86 |
| 5 | CCl4 | 5 | 20 | - | - | 46 | 90 |
| 6 3 | CCl4 | 5 | 30 | 56 | 76 | 42 | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mato, R.; Manzano, R.; Reyes, E.; Prieto, L.; Uria, U.; Carrillo, L.; Vicario, J.L. Kinetic Resolution in Transannular Morita-Baylis-Hillman Reaction: An Approximation to the Synthesis of Sesquiterpenes from Guaiane Family. Catalysts 2022, 12, 67. https://doi.org/10.3390/catal12010067
Mato R, Manzano R, Reyes E, Prieto L, Uria U, Carrillo L, Vicario JL. Kinetic Resolution in Transannular Morita-Baylis-Hillman Reaction: An Approximation to the Synthesis of Sesquiterpenes from Guaiane Family. Catalysts. 2022; 12(1):67. https://doi.org/10.3390/catal12010067
Chicago/Turabian StyleMato, Raquel, Rubén Manzano, Efraím Reyes, Liher Prieto, Uxue Uria, Luisa Carrillo, and Jose L. Vicario. 2022. "Kinetic Resolution in Transannular Morita-Baylis-Hillman Reaction: An Approximation to the Synthesis of Sesquiterpenes from Guaiane Family" Catalysts 12, no. 1: 67. https://doi.org/10.3390/catal12010067
APA StyleMato, R., Manzano, R., Reyes, E., Prieto, L., Uria, U., Carrillo, L., & Vicario, J. L. (2022). Kinetic Resolution in Transannular Morita-Baylis-Hillman Reaction: An Approximation to the Synthesis of Sesquiterpenes from Guaiane Family. Catalysts, 12(1), 67. https://doi.org/10.3390/catal12010067