
Boron-Based Lewis Acid Catalysis: Challenges and Perspectives



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
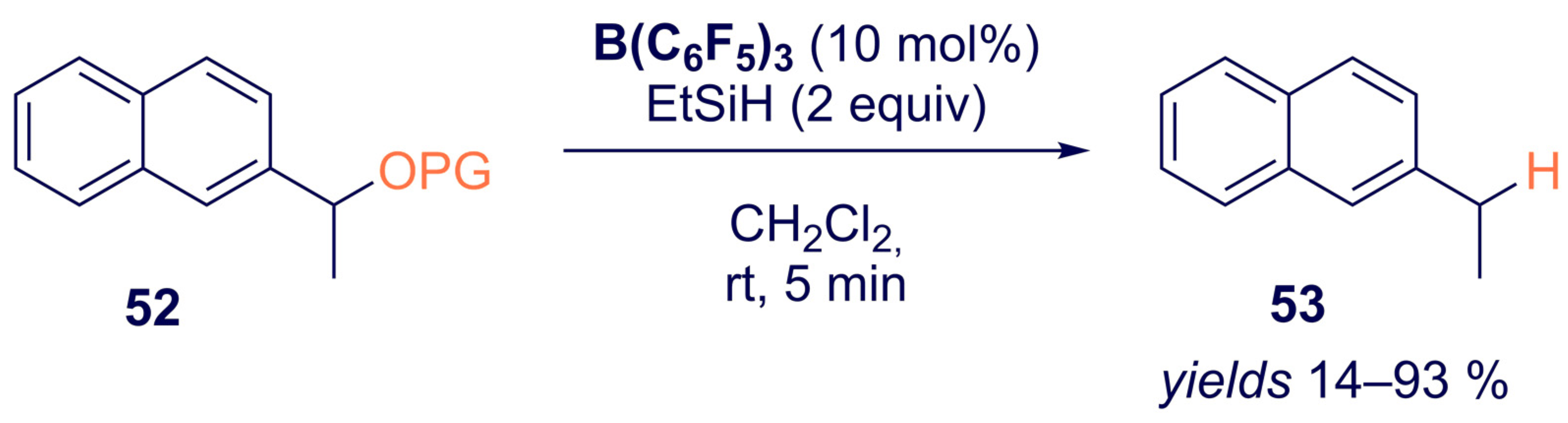{kind=link}
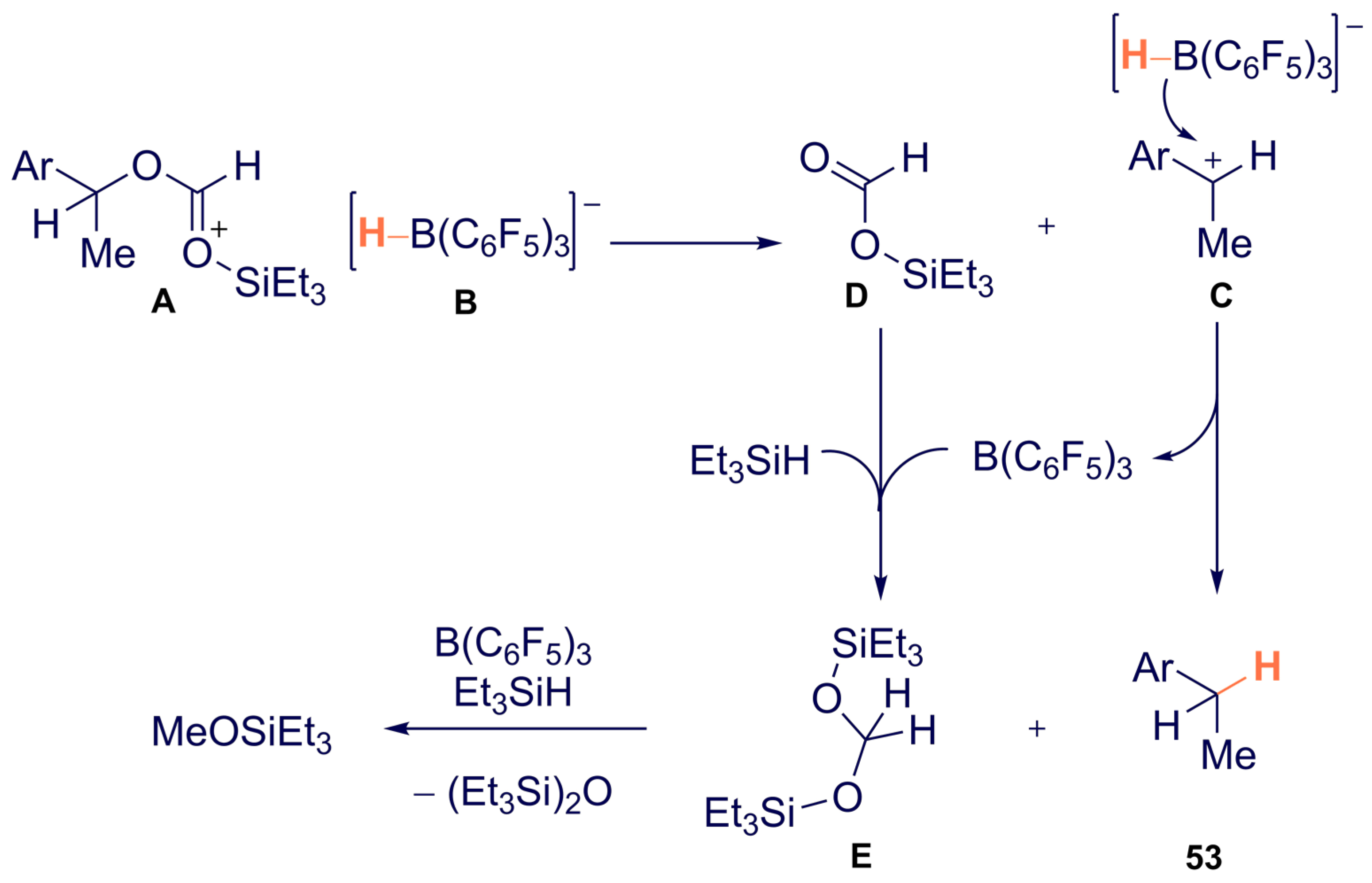{kind=link}
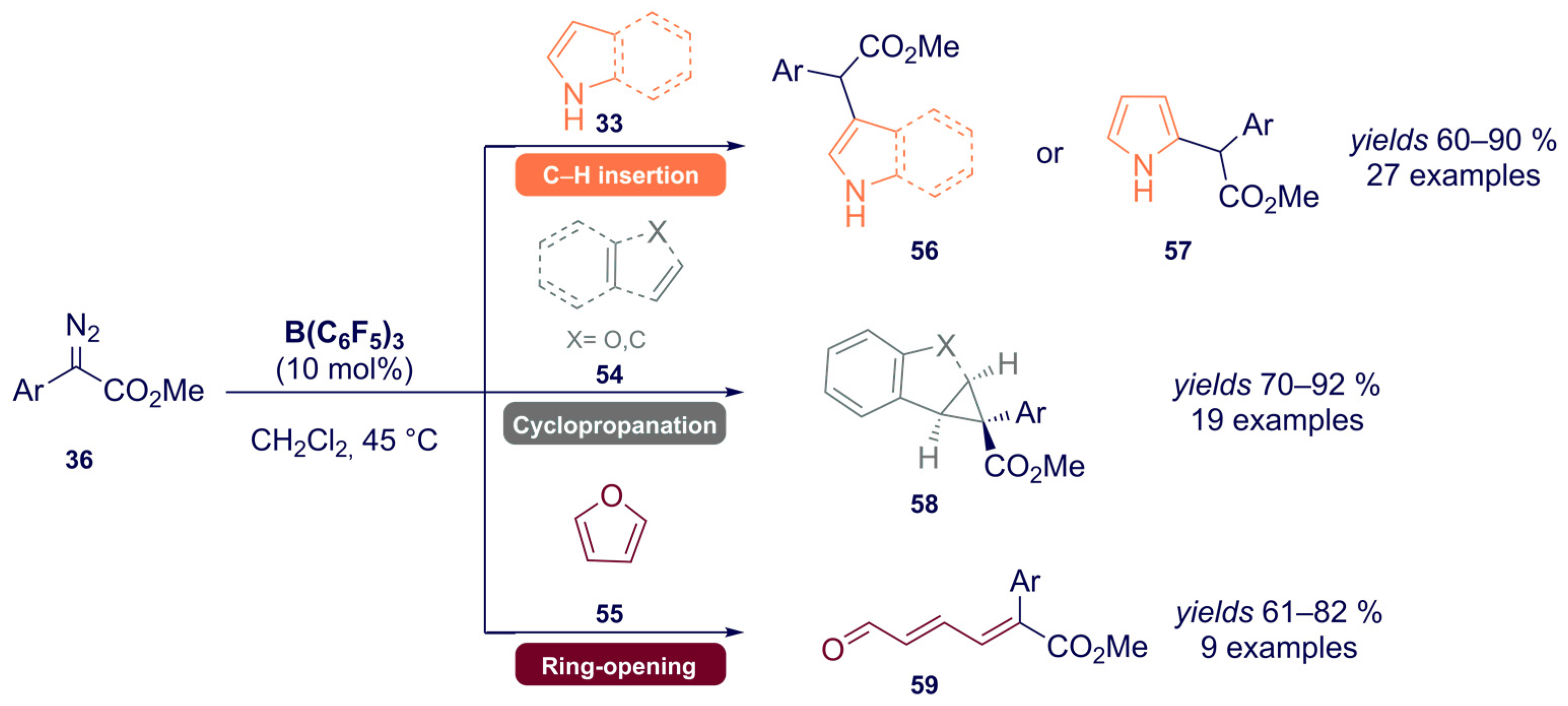{kind=link}
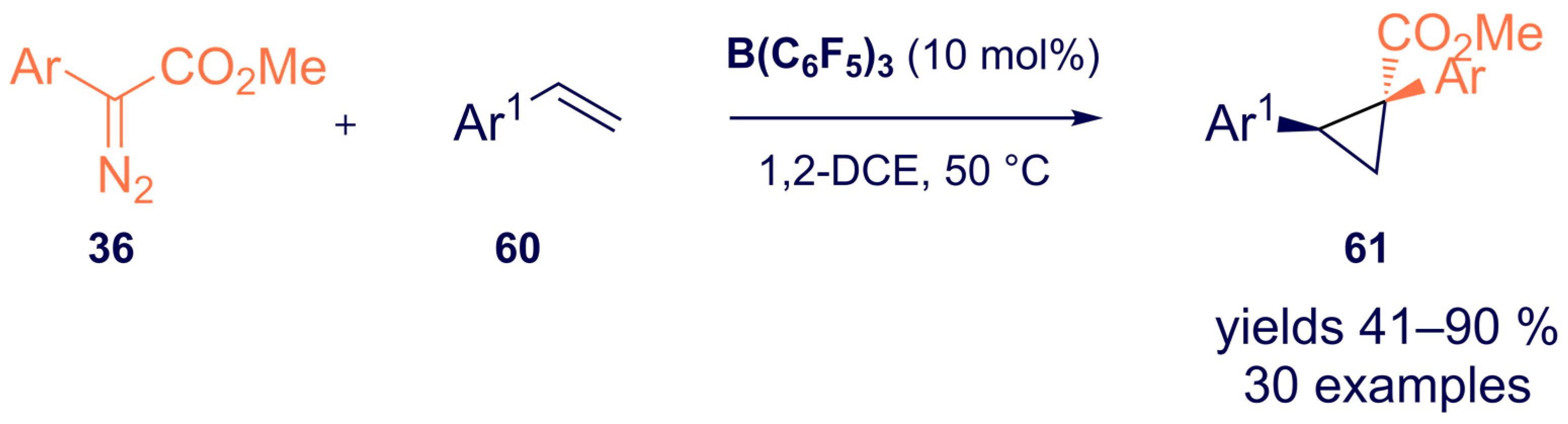{kind=link}
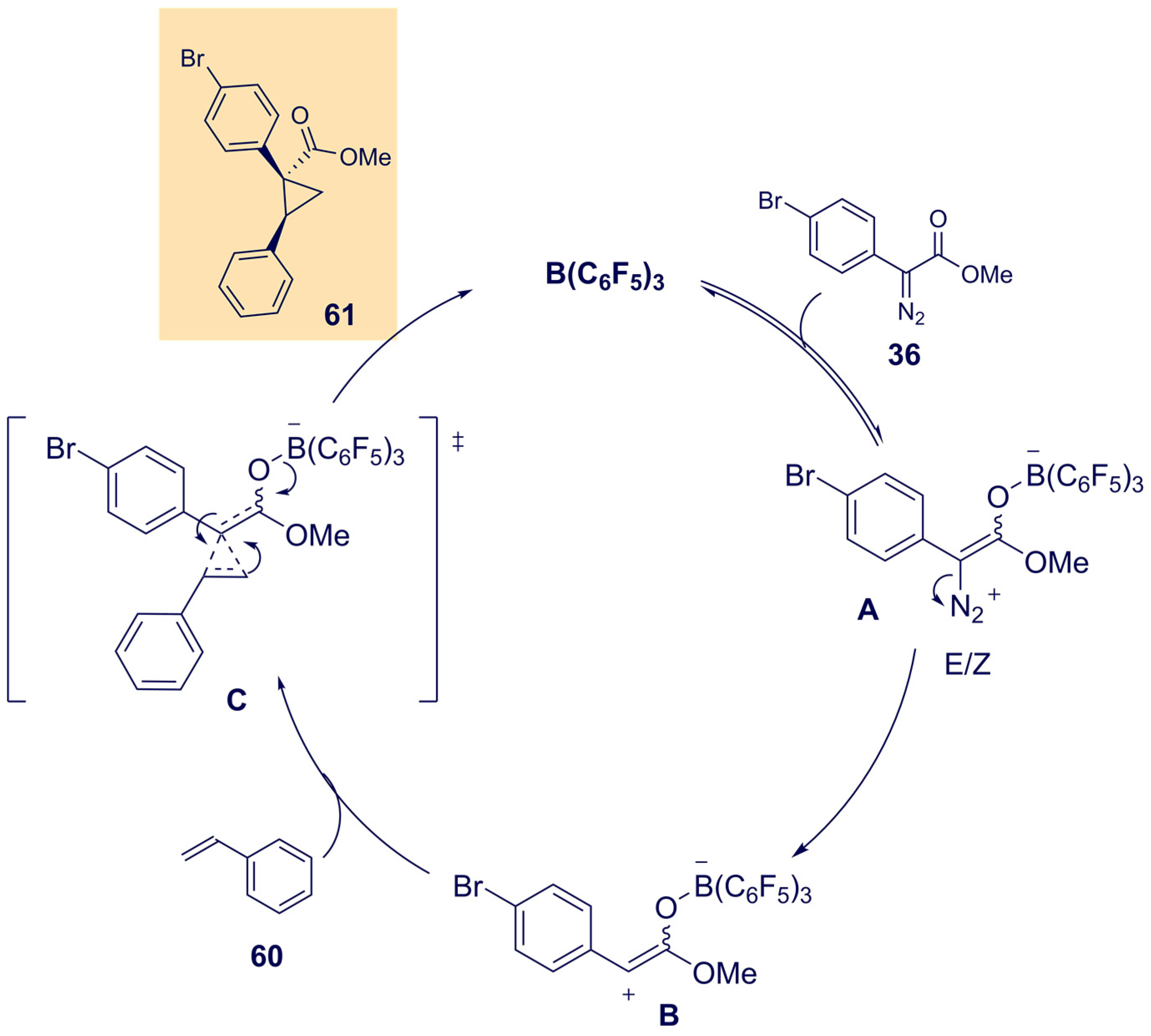{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
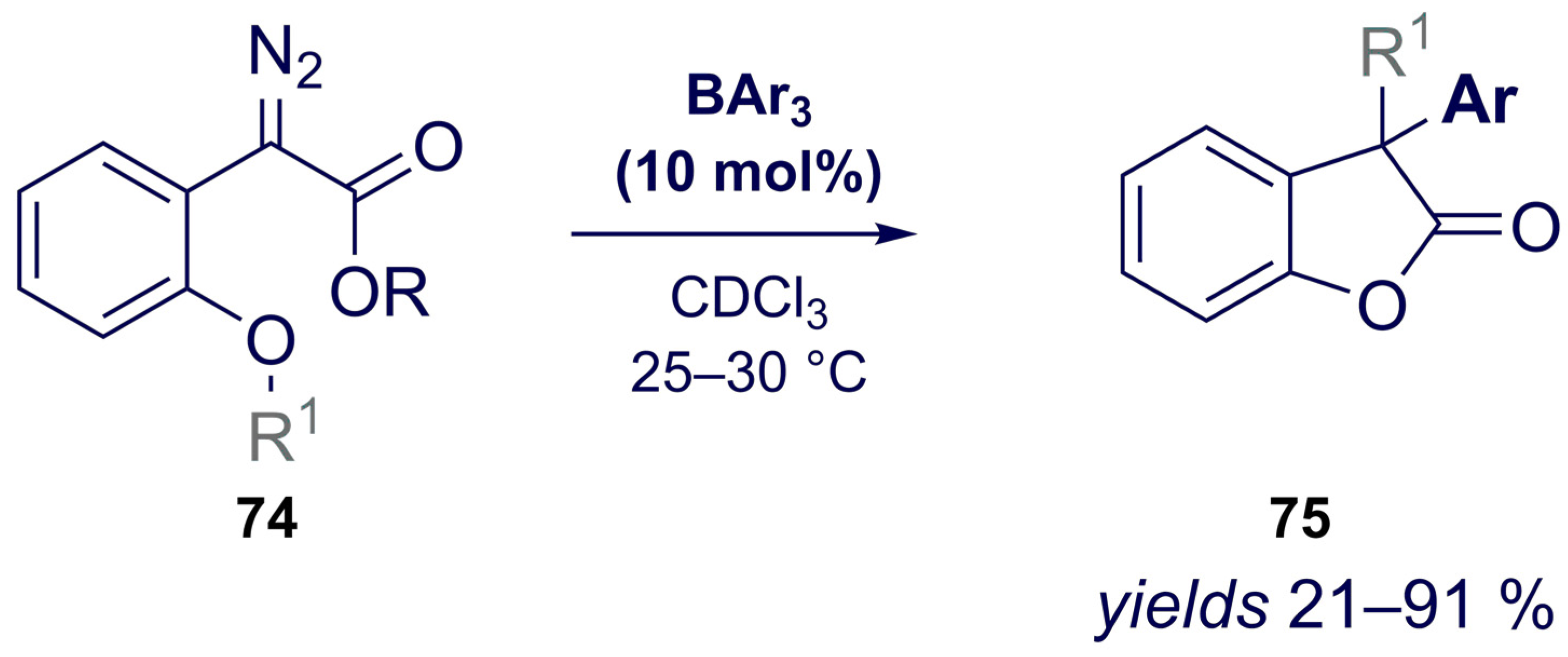{kind=link}
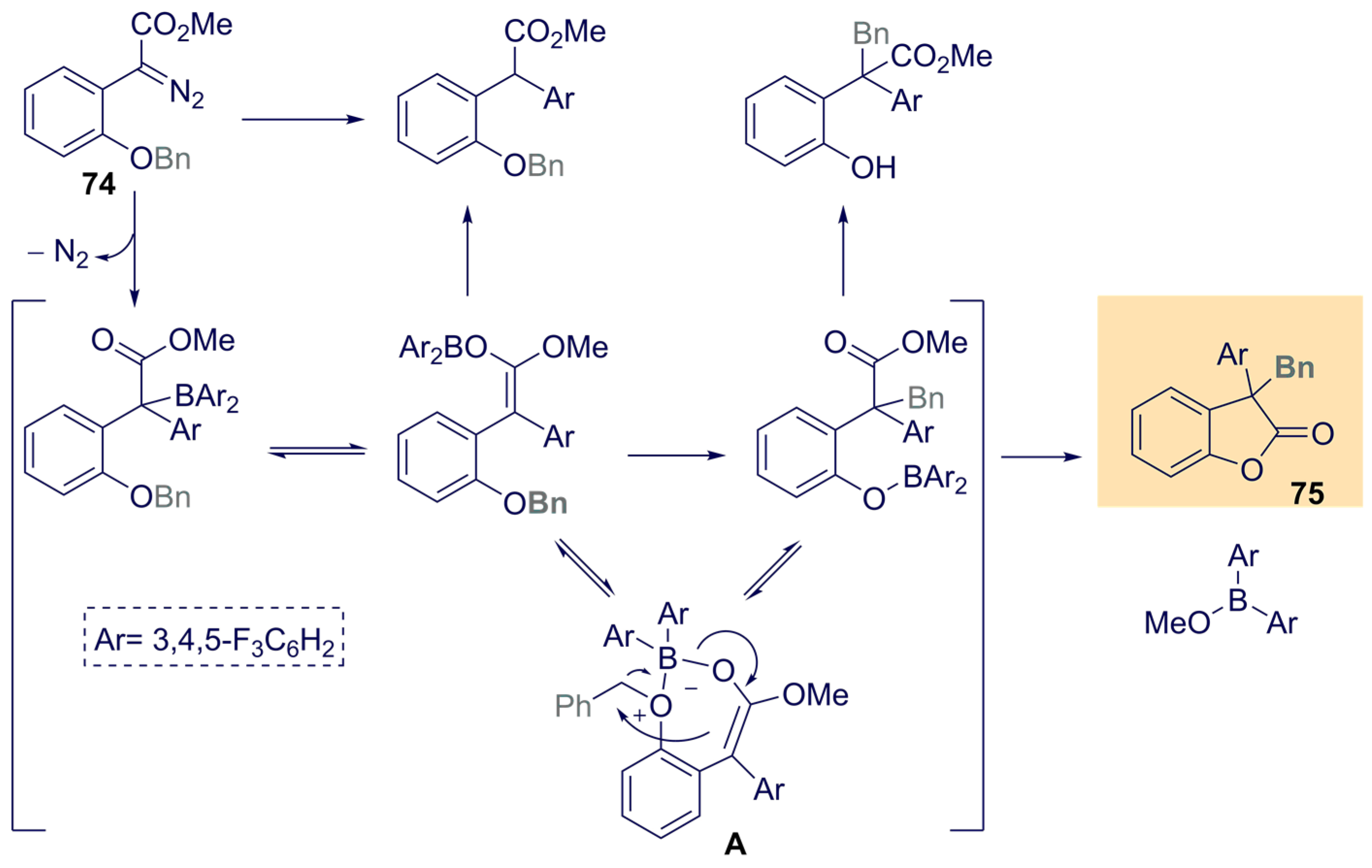{kind=link}
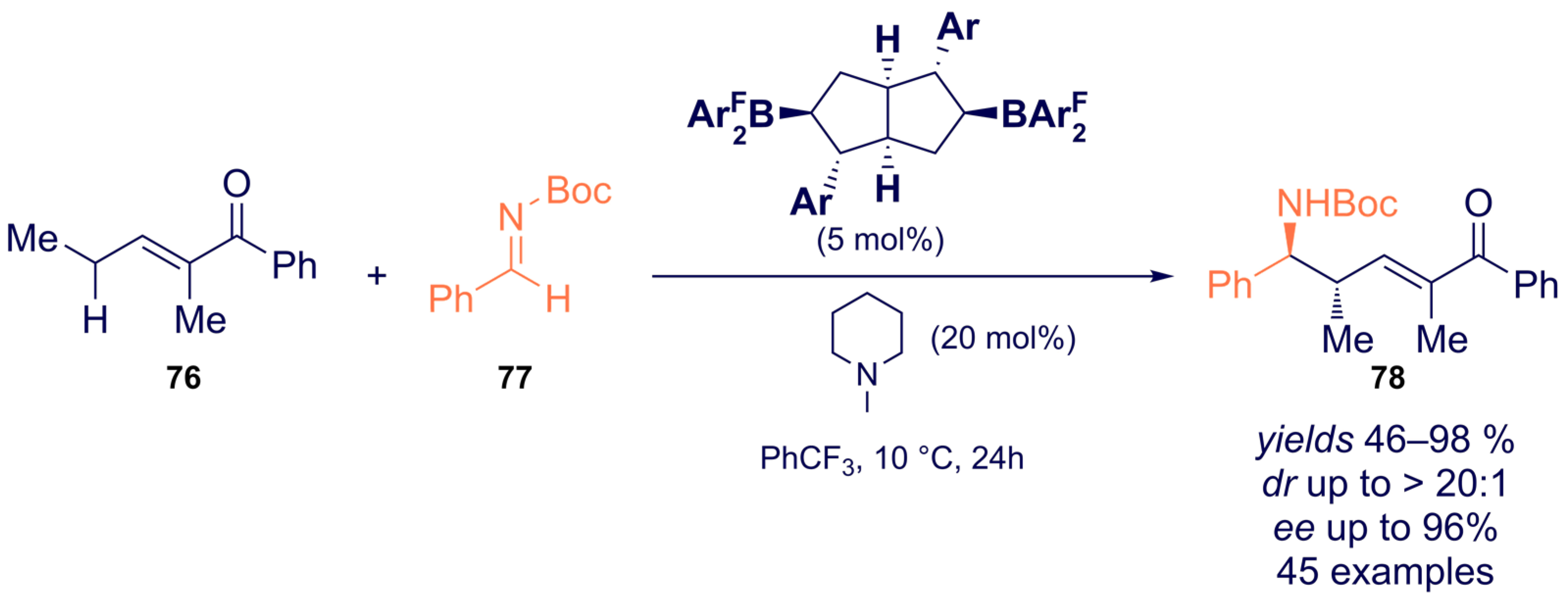{kind=link}
{kind=link}
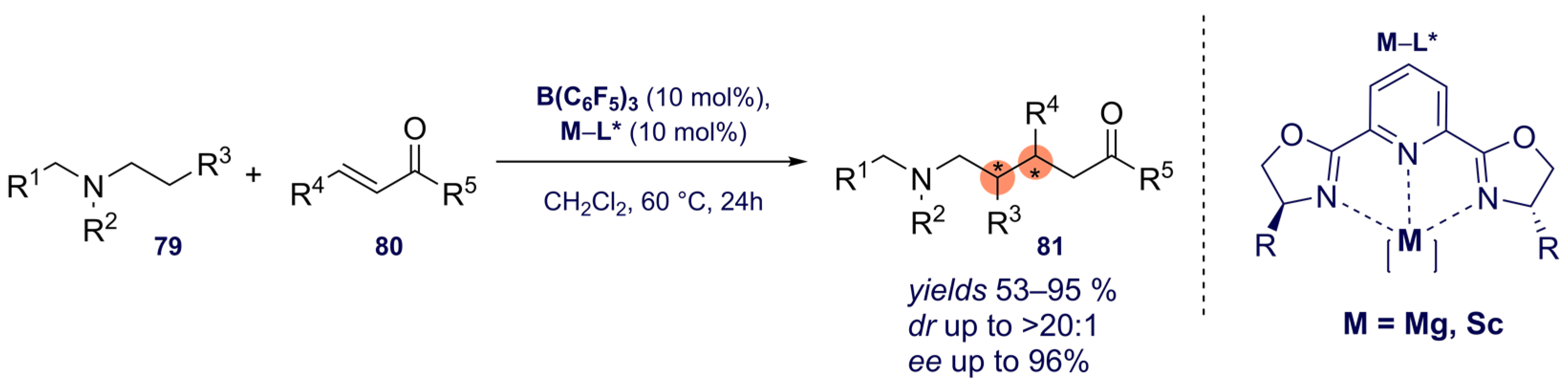{kind=link}
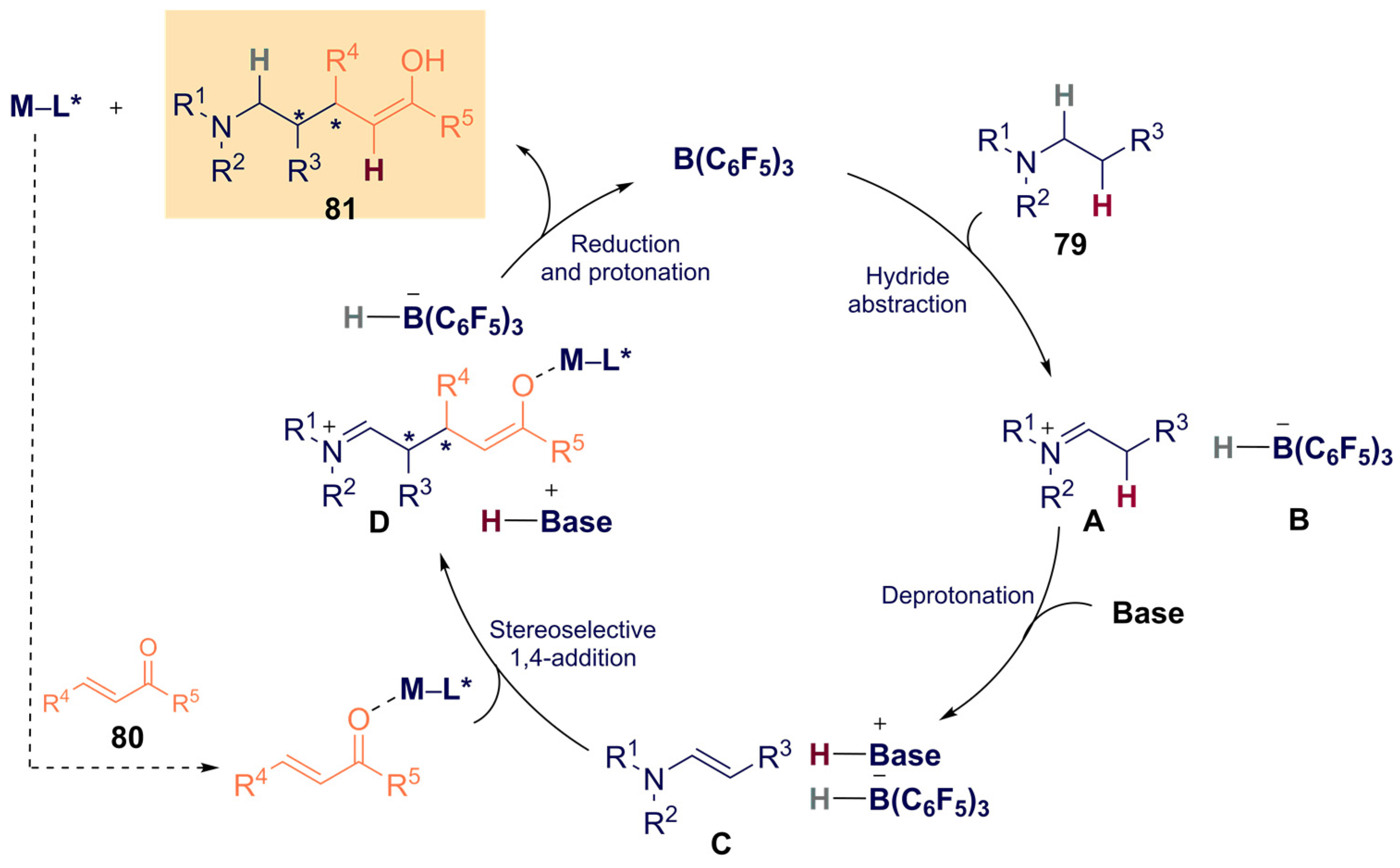{kind=link}
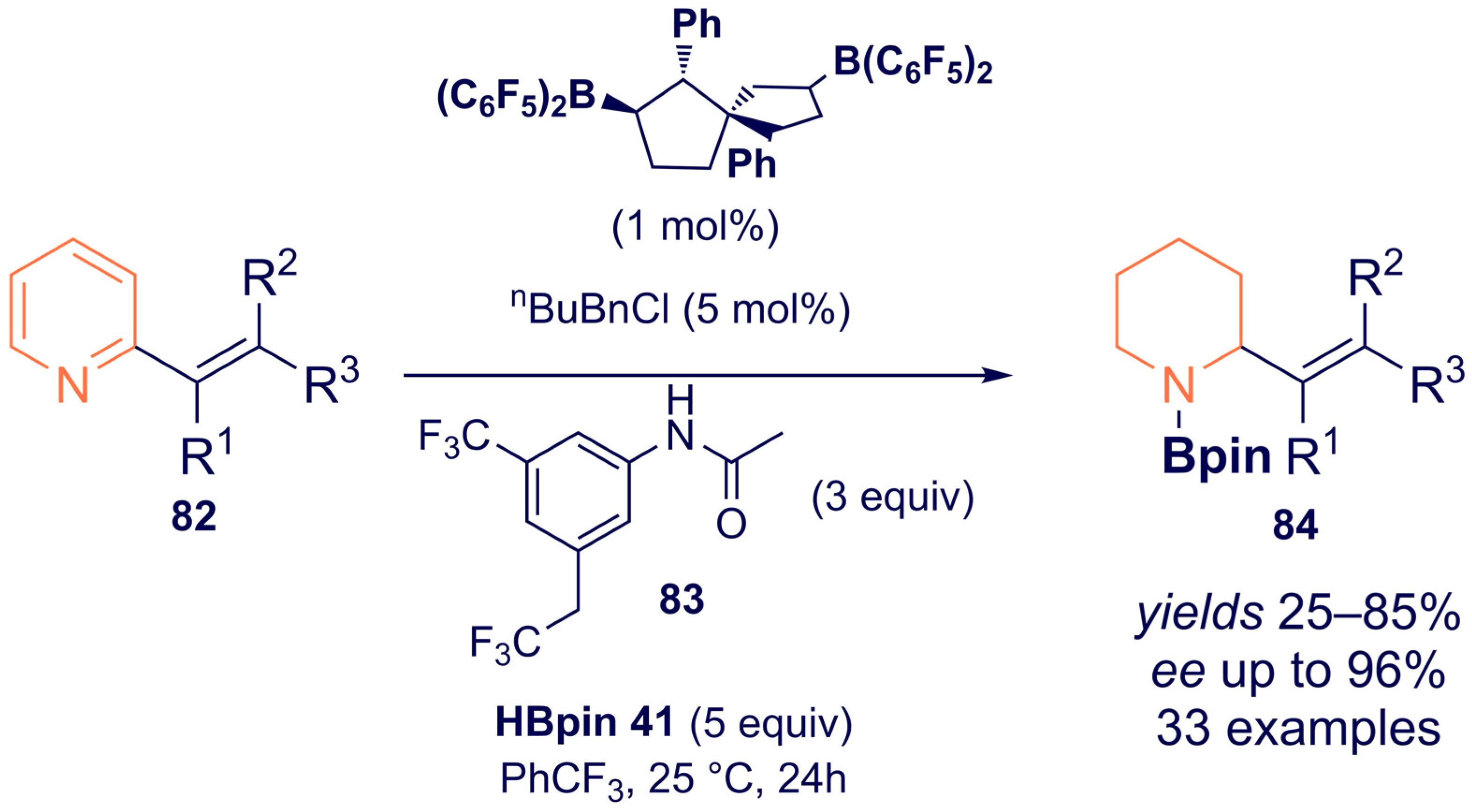{kind=link}
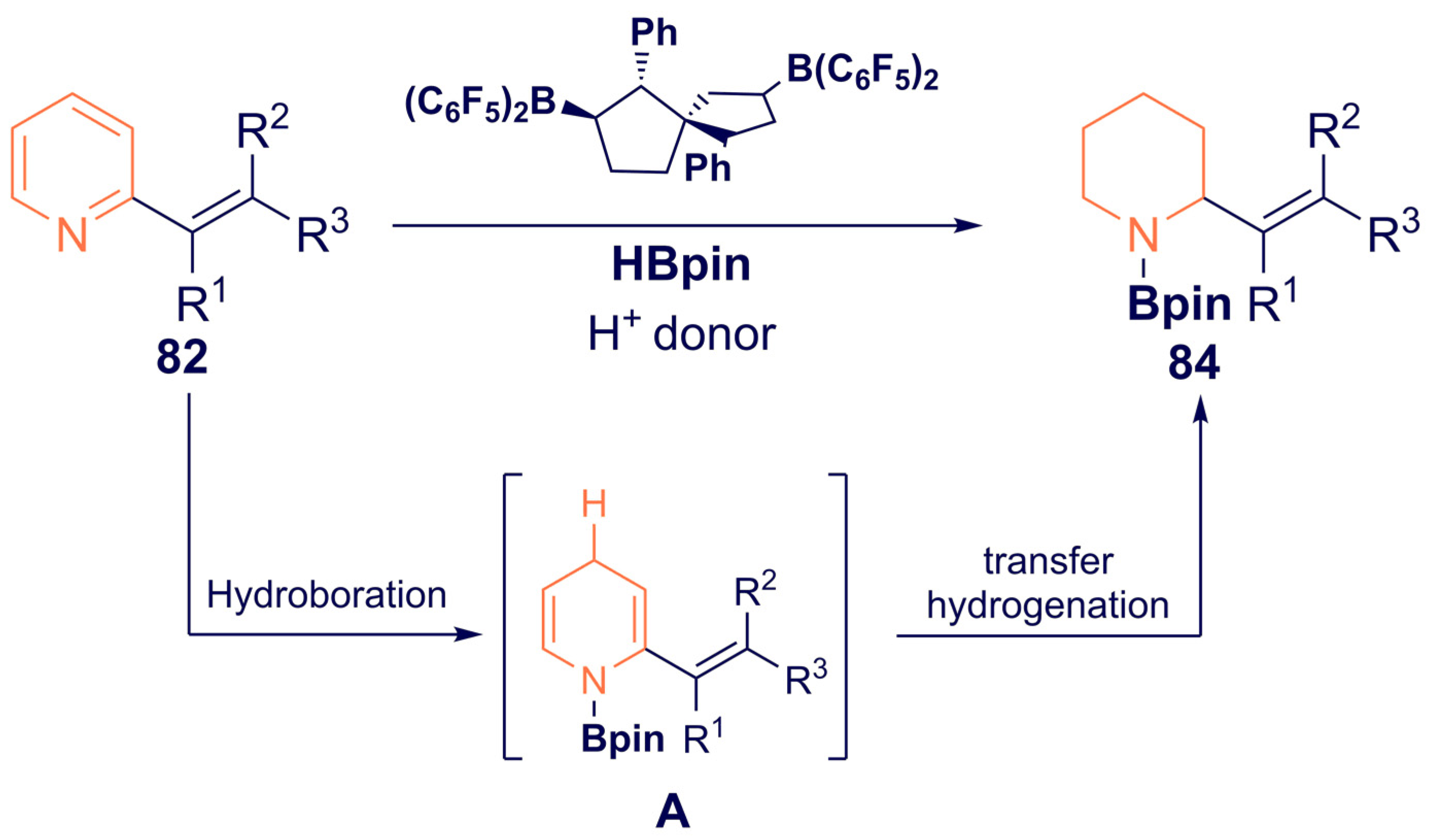{kind=link}
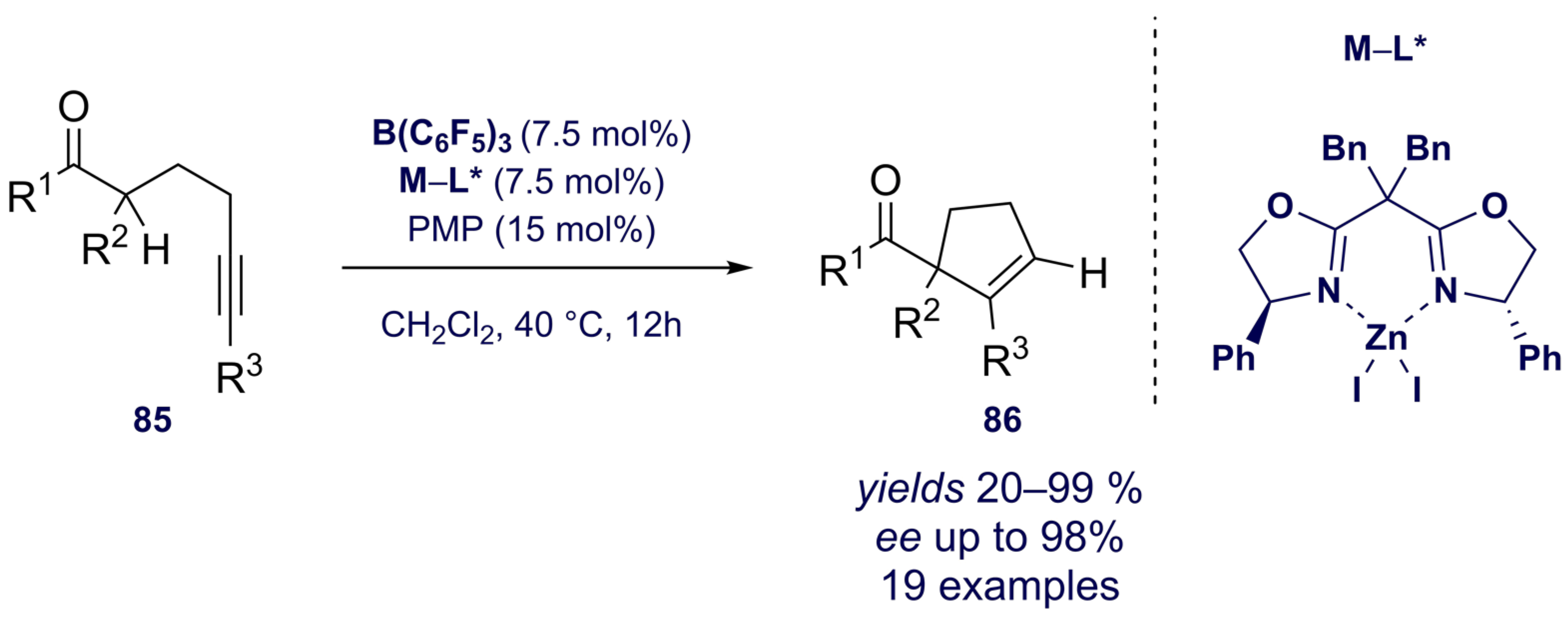{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
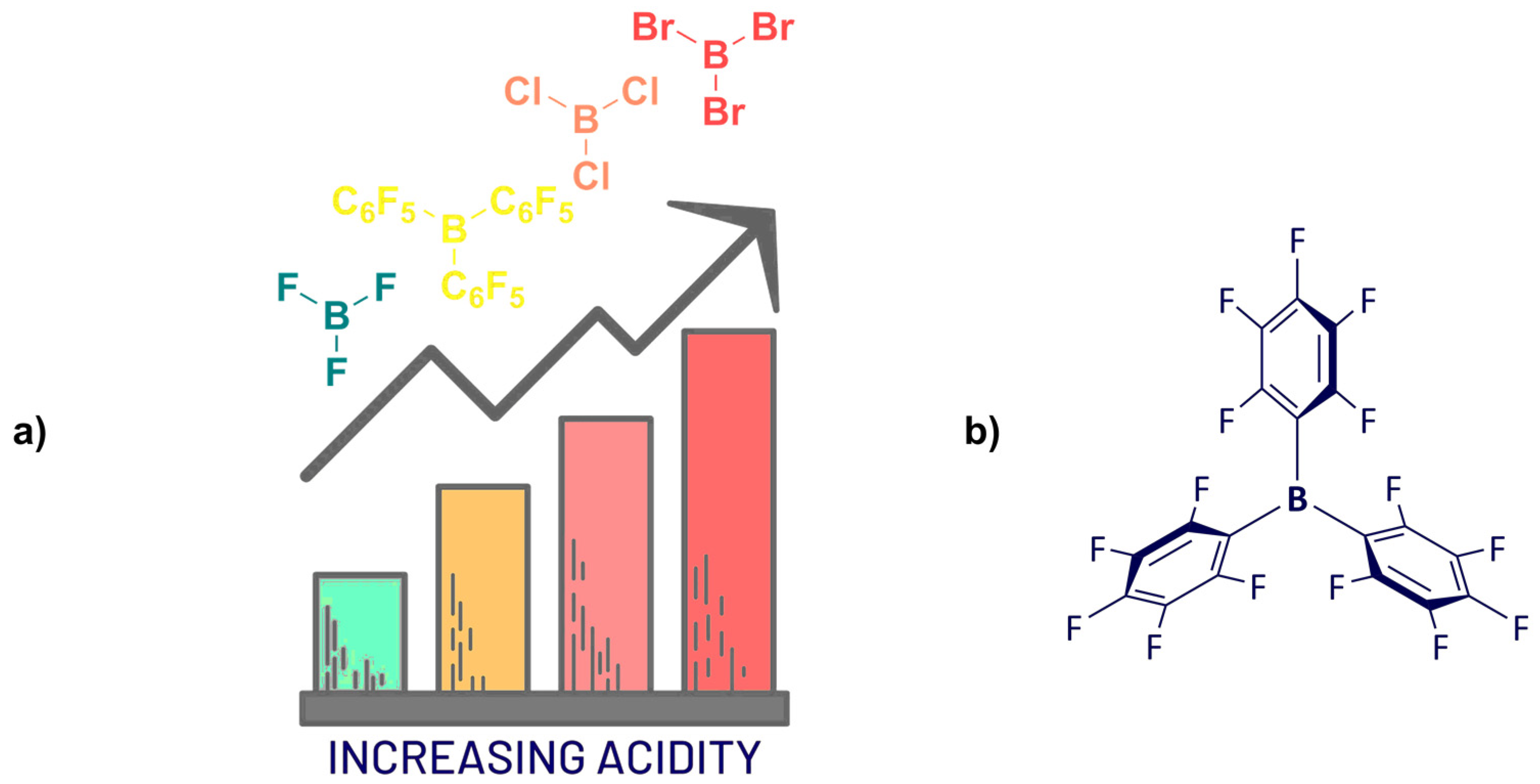
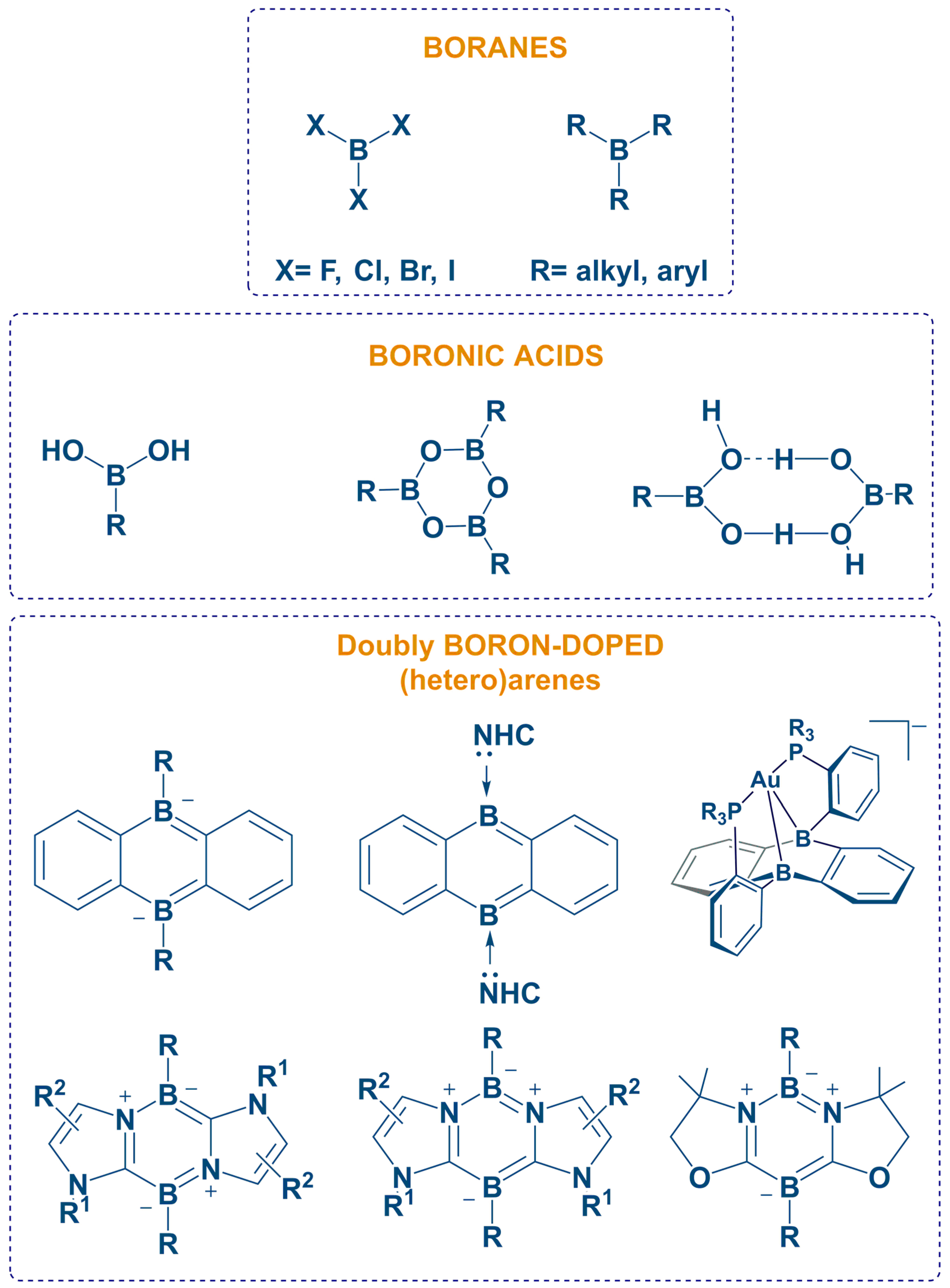
:1. Introduction
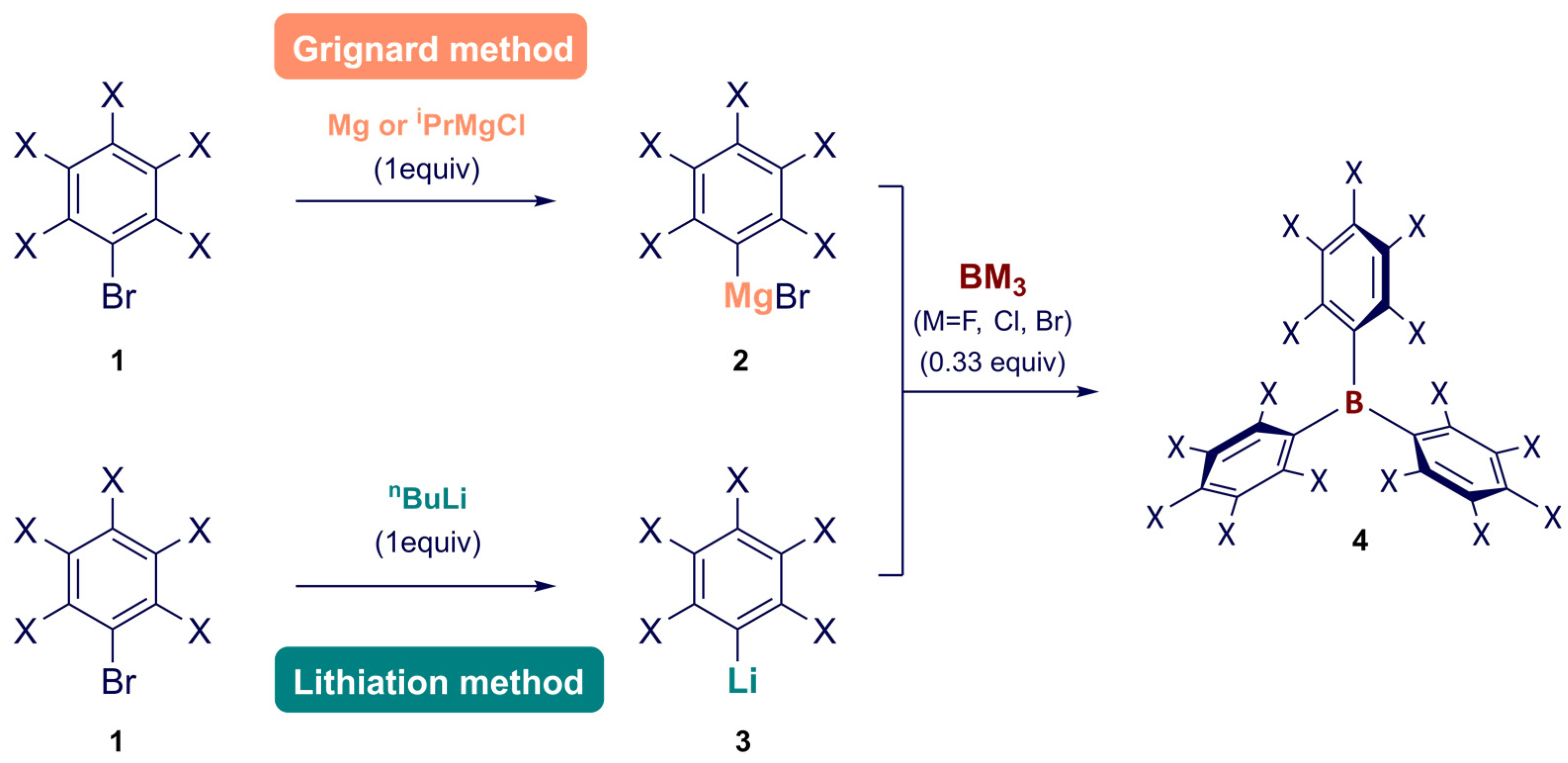
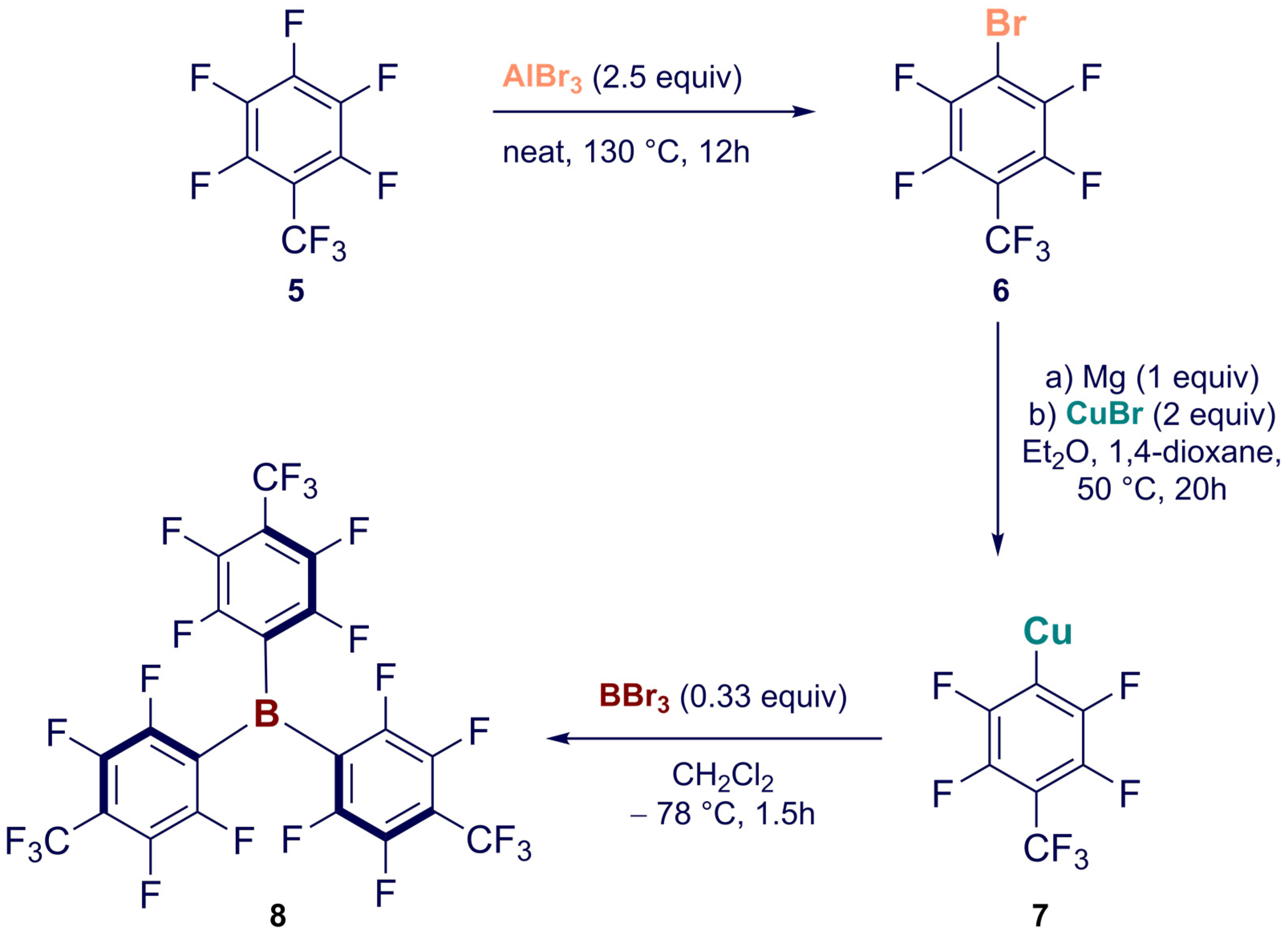
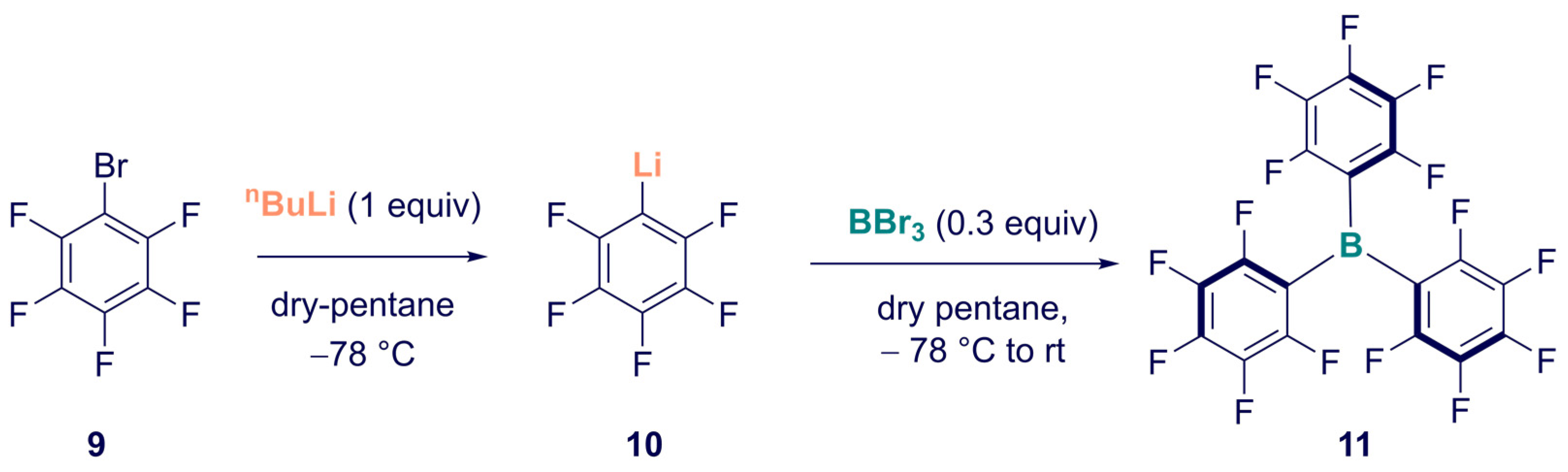
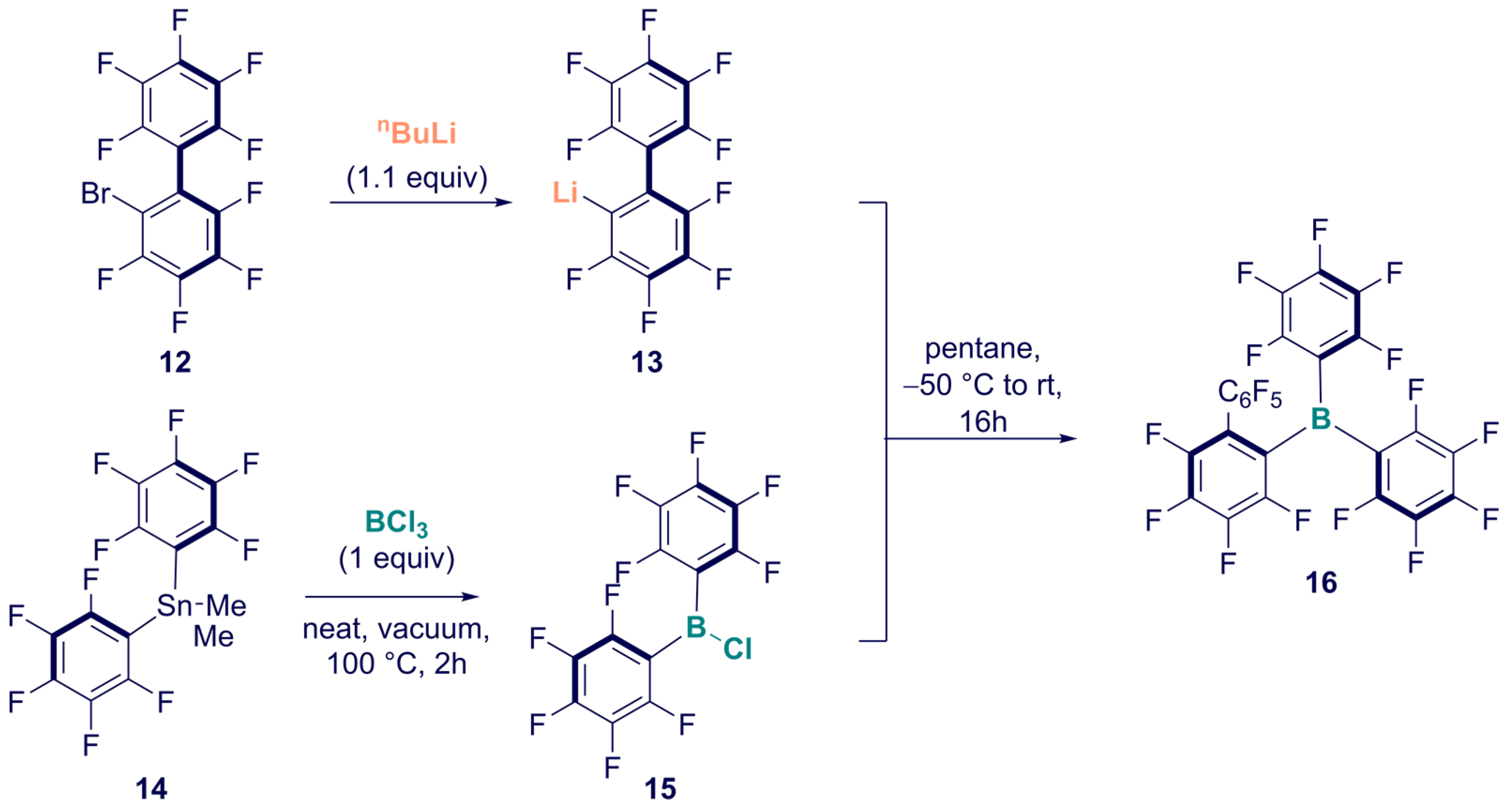
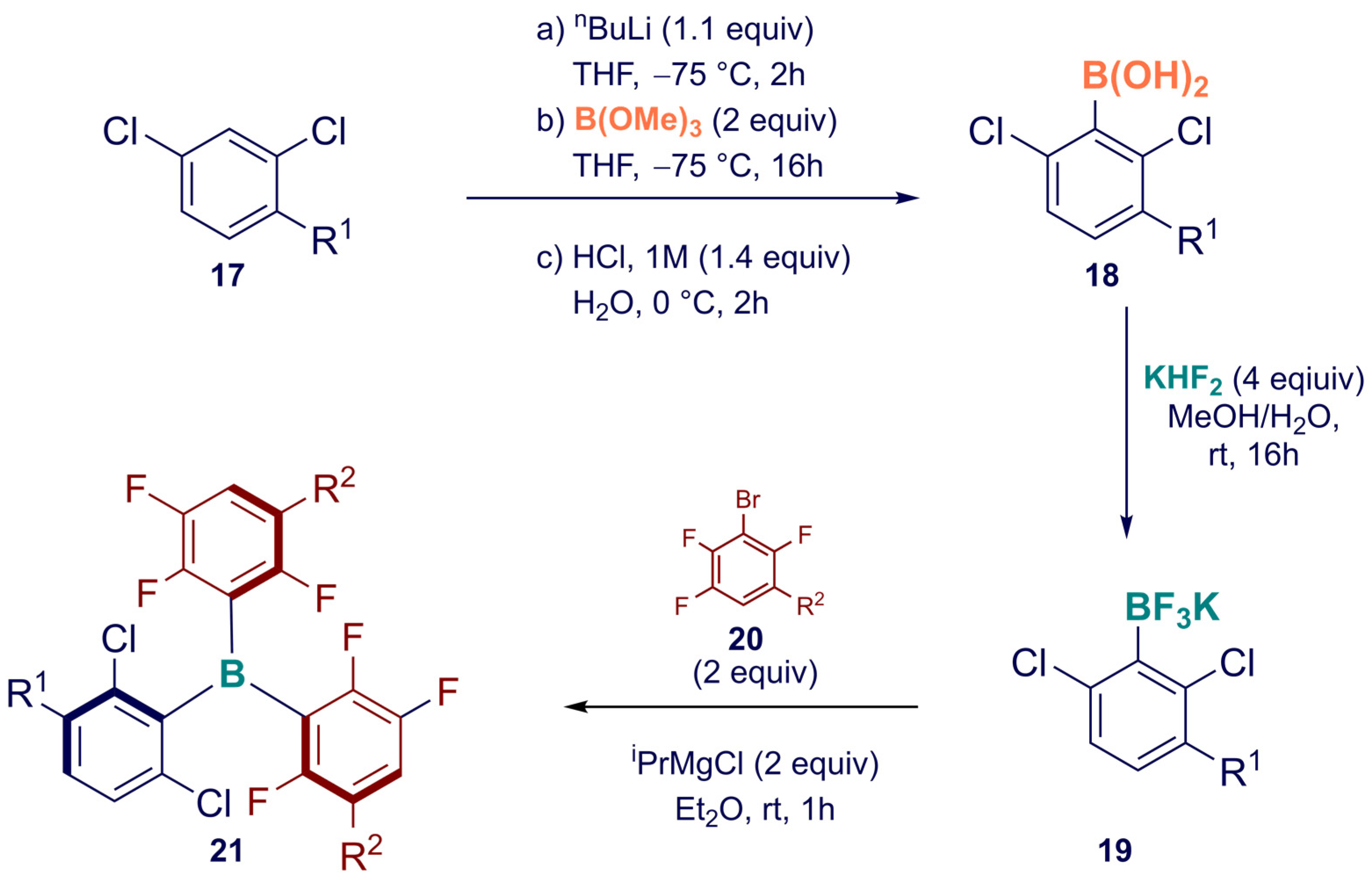
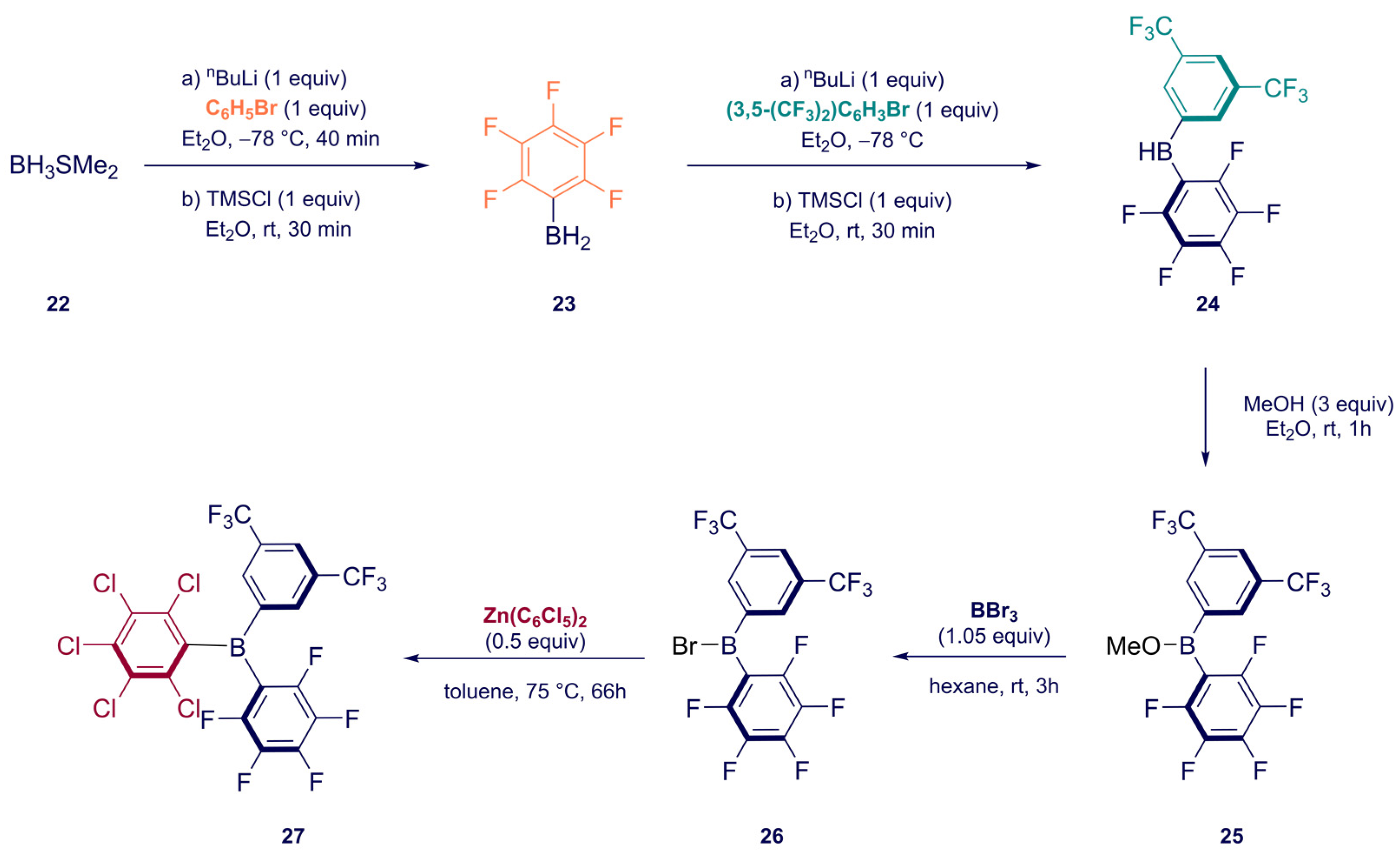
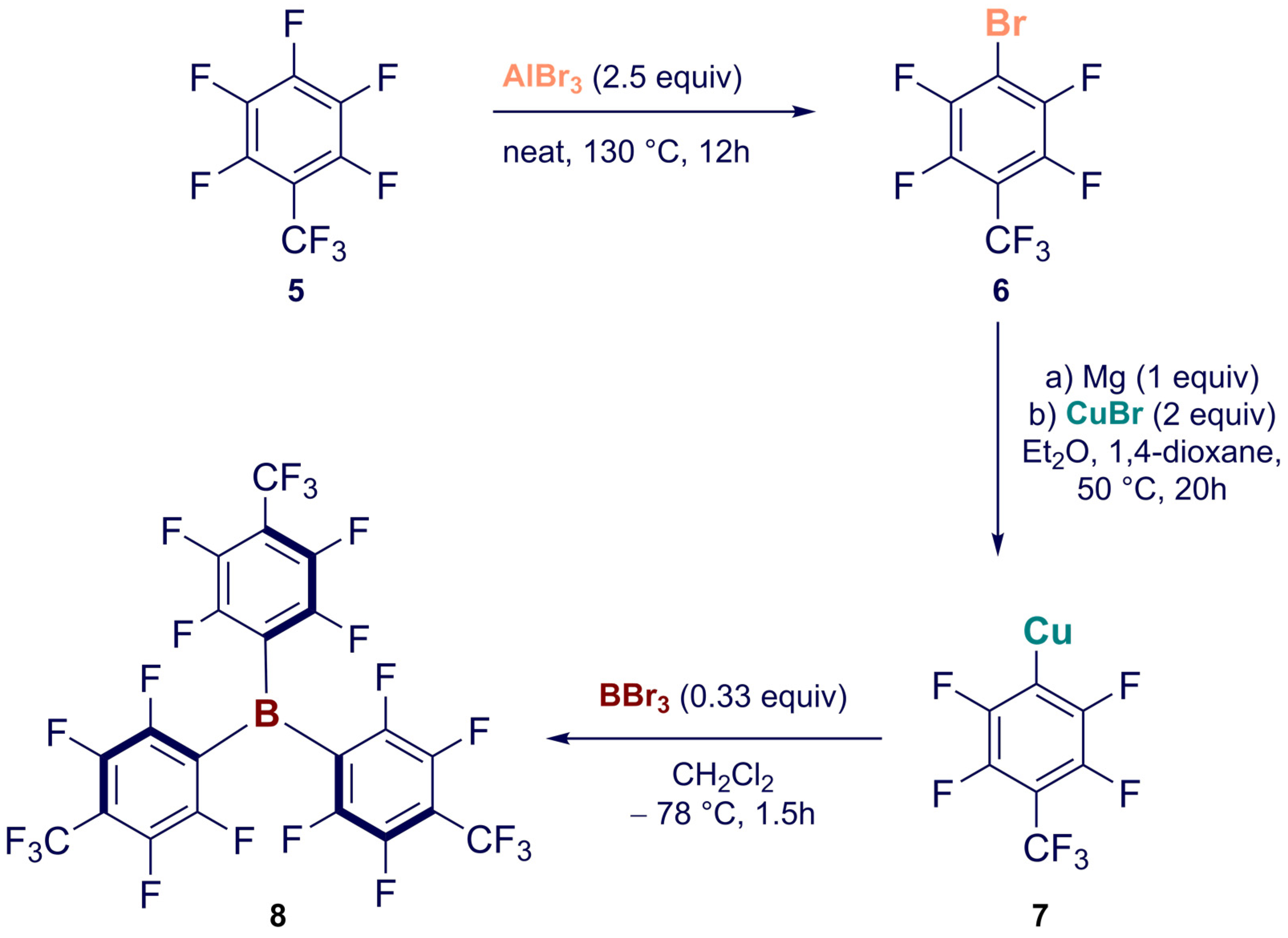
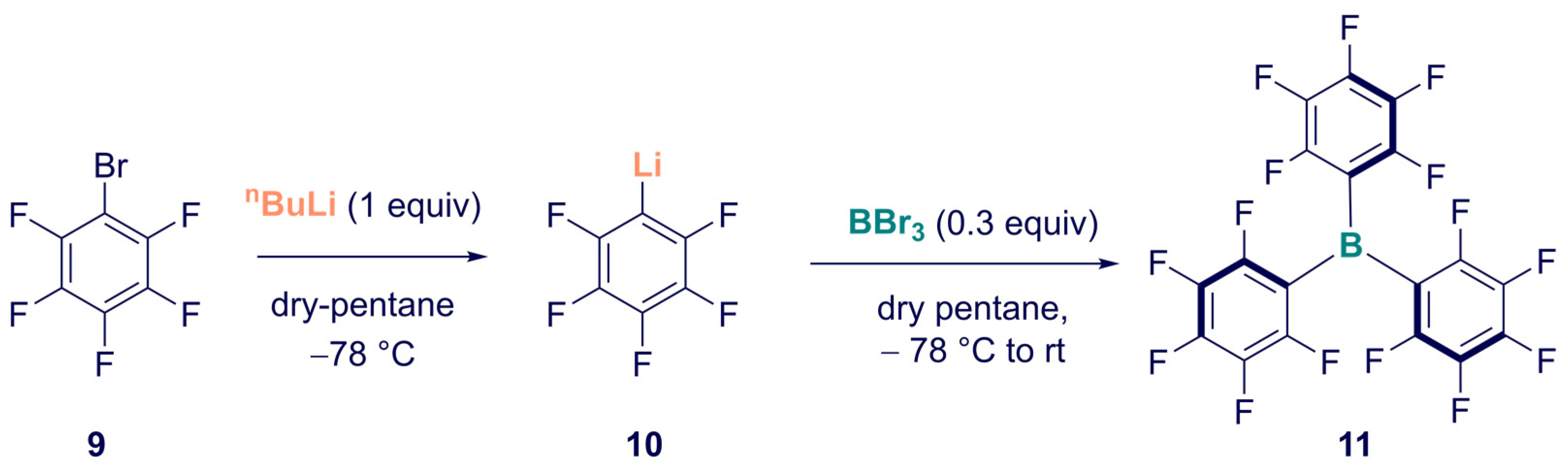
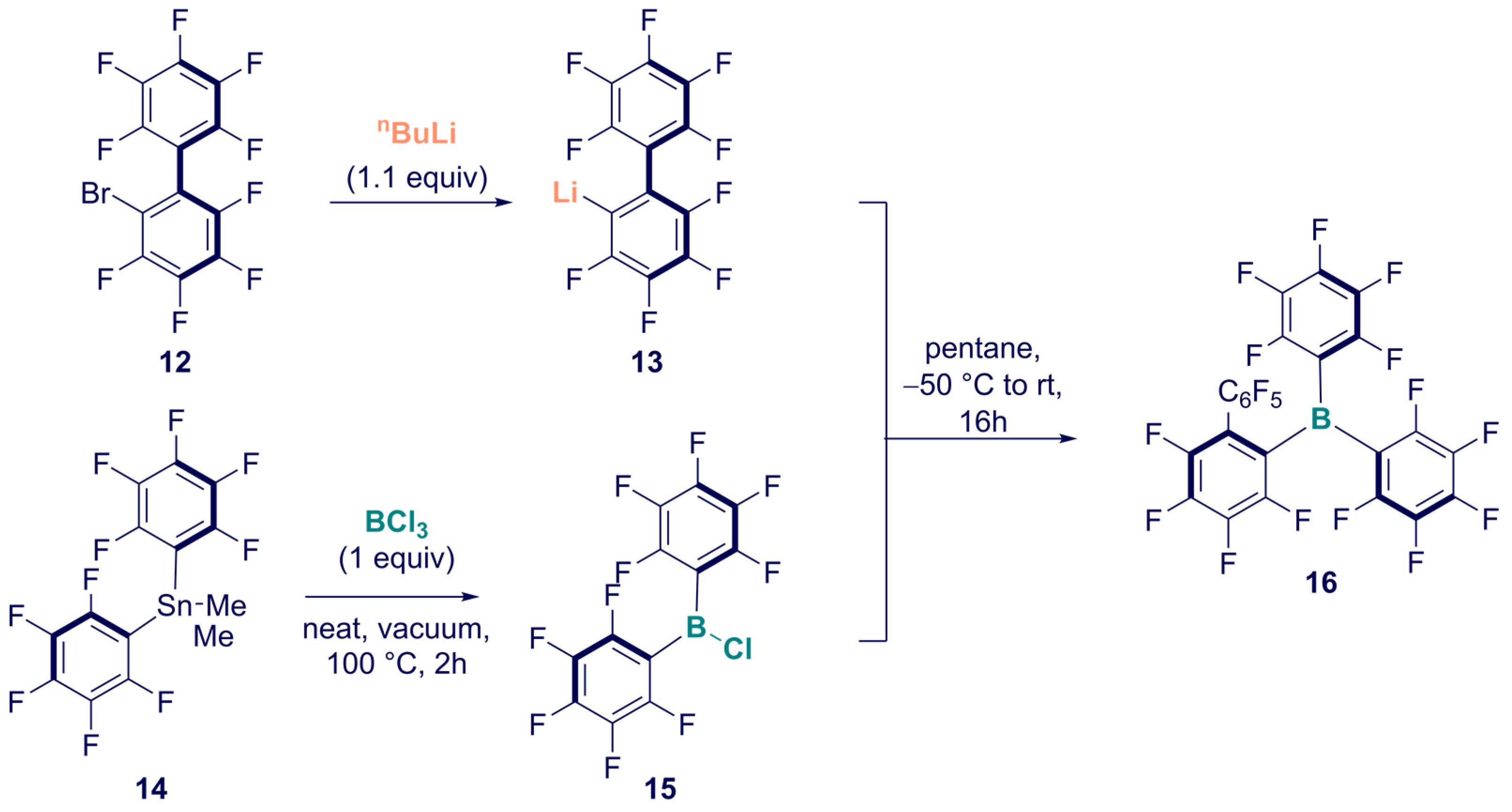
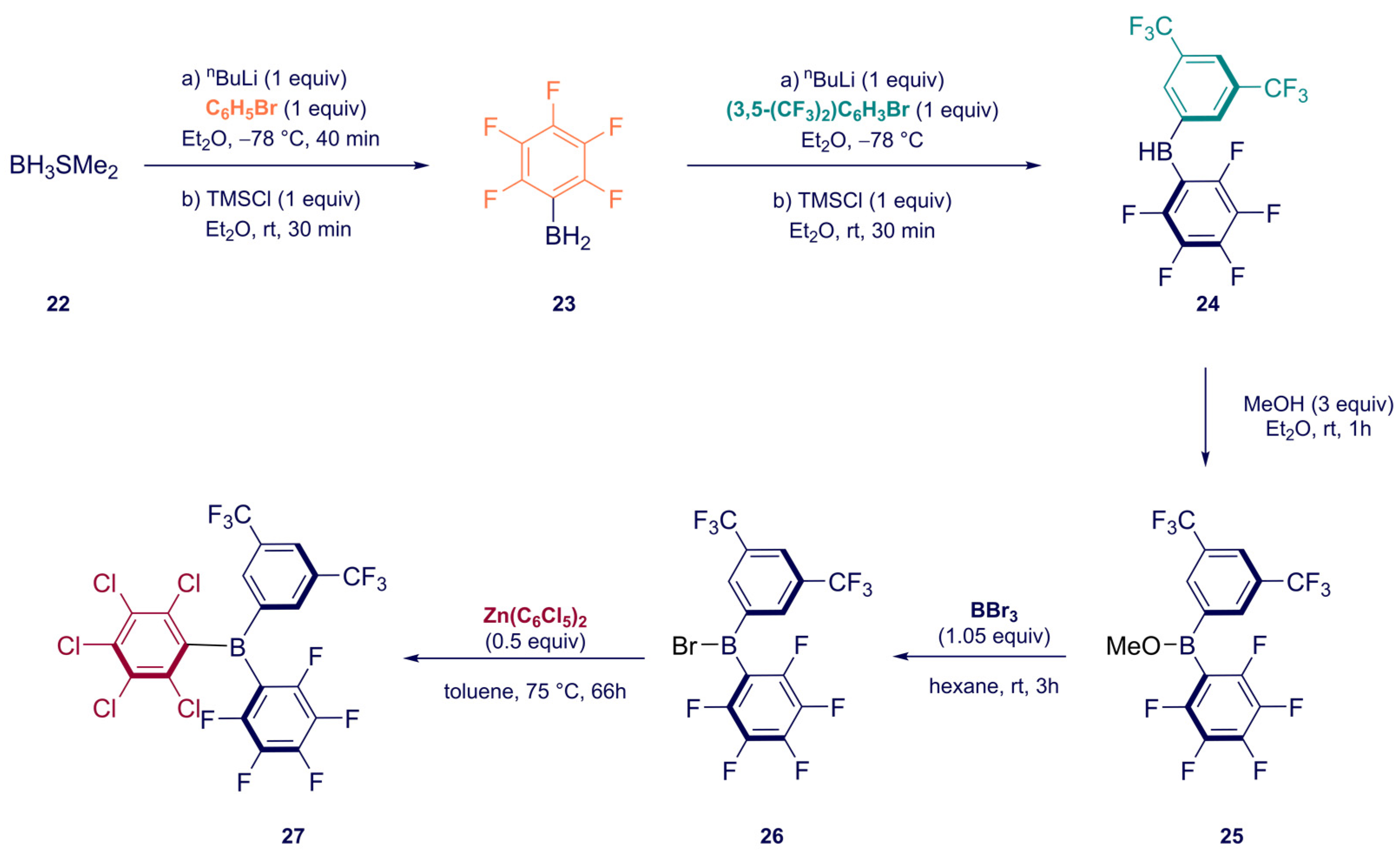
2. Synthesis
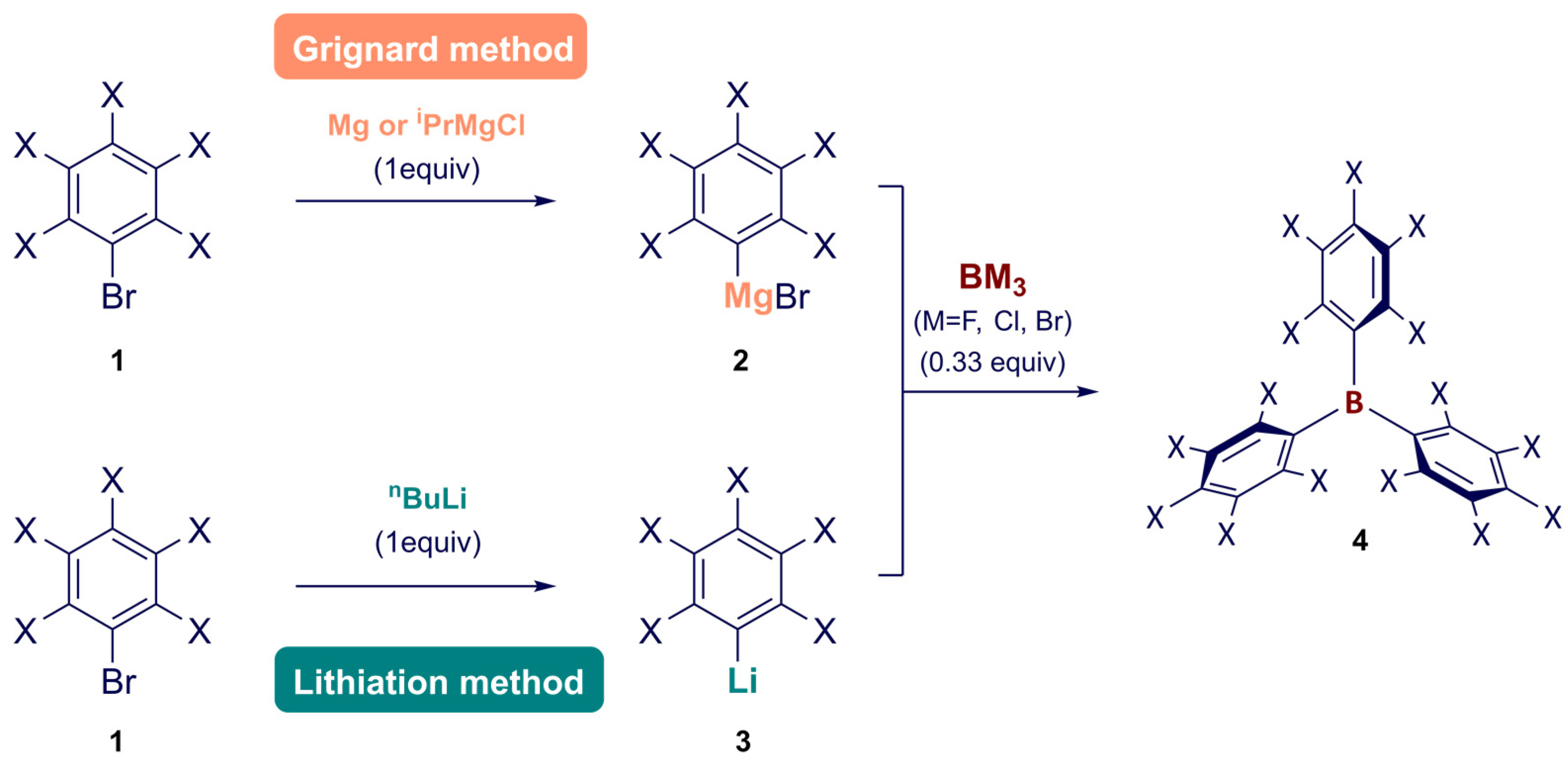
Synthesis of Homoleptic and Heteroleptic Halogenated Triarylboranes
3. Catalysis
3.1. Boron-Based Catalysis
3.2. Asymmetric Boron-Based Catalysis
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawson, J.R.; Melen, R.L. Recent developments and applications of Lewis acidic boron reagents. In Organometallic Chemistry; Fairlamb, I., Lynam, J.M., Patmore, N.J., Elliott, P., Eds.; The Royal Society of Chemistry: London, UK, 2017; Volume 41, pp. 1–27. [Google Scholar]
- Piers, W.E.; Chivers, T. Pentafluorophenylboranes: From obscurity to applications. Chem. Soc. Rev. 1997, 26, 345–354. [Google Scholar] [CrossRef]
- Massey, A.G.; Park, A.J.; Stone, F.G.A. Tris(pentafluoropheny)boron. Proc. Chem. Soc. 1963, 127, 212. [Google Scholar]
- Massey, A.G.; Park, A.J. Perfluorophenyl derivatives of the elements: I. Tris(pentafluorophenyl)boron. J. Organomet. Chem. 1964, 2, 245–250. [Google Scholar] [CrossRef]
- Klosin, J.; Fontaine, P.P.; Figueroa, R. Development of Group IV Molecular Catalysts for High Temperature Ethylene-α-Olefin Copolymerization Reactions. Acc. Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef]
- Janiak, C.; Lassahn, P.-G.; Lozan, V. Metal Complexes for the Vinyl Addition Polymerization of Norbornene: New Compound Classes and Activation Mechanism with B(C6F5)3/AlEt3. Macromol. Symp. 2006, 236, 88–99. [Google Scholar] [CrossRef]
- Focante, F.; Mercandelli, P.; Sironi, A.; Resconi, L. Complexes of tris(pentafluorophenyl)boron with nitrogen-containing compounds: Synthesis, reactivity and metallocene activation. Coord. Chem. Rev. 2006, 250, 170–188. [Google Scholar] [CrossRef]
- Erker, G.; Kehr, G.; Fröhlich, R. The (butadiene)zirconocene route to active homogeneous olefin polymerization catalysts. J. Organomet. Chem. 2005, 690, 6254–6262. [Google Scholar] [CrossRef]
- Ishihara, K.; Yamamoto, H. Arylboron Compounds as Acid Catalysts in Organic Synthetic Transformations. Eur. J. Org. Chem. 1999, 1999, 527–538. [Google Scholar] [CrossRef]
- Piers, W.E.; Sun, Y.; Lee, L.W.M. Zwitterionic metallocenes via reactions of organozirconocenes with highly electrophilic perfluorophenyl substituted boranes. Top. Catal. 1999, 7, 133–143. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E. Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters. J. Am. Chem. Soc. 1996, 118, 9440–9441. [Google Scholar] [CrossRef]
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCahill, J.S.J.; Welch, G.C.; Stephan, D.W. Reactivity of “Frustrated Lewis Pairs”: Three-Component Reactions of Phosphines, a Borane, and Olefins. Angew. Chem. 2007, 119, 5056–5059. [Google Scholar] [CrossRef]
- Yoshihiko, I.; Takeo, Y.; Eiichi, K.; Kenji, I. Method of Producing Tris(Pentafluorophenyl)Borane Using Pentafluorophenyl Alkyli Metal Salt Prepared From Pentafluorobenzene. European Patent EP0604962A1, 6 July 1994. [Google Scholar]
- Keess, S.; Simonneau, A.; Oestreich, M. Direct and Transfer Hydrosilylation Reactions Catalyzed by Fully or Partially Fluorinated Triarylboranes: A Systematic Study. Organometallics 2015, 34, 790–799. [Google Scholar] [CrossRef]
- Yin, Q.; Soltani, Y.; Melen, R.L.; Oestreich, M. BArF3-Catalyzed Imine Hydroboration with Pinacolborane Not Requiring the Assistance of an Additional Lewis Base. Organometallics 2017, 36, 2381–2384. [Google Scholar] [CrossRef] [Green Version]
- Carden, J.L.; Gierlichs, L.J.; Wass, D.F.; Browne, D.L.; Melen, R.L. Unlocking the catalytic potential of tris(3,4,5-trifluorophenyl)borane with microwave irradiation. Chem. Commun. 2019, 55, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Herrington, T.J.; Thom, A.J.W.; White, A.J.P.; Ashley, A.E. Novel H2 activation by a trisfig3,5-bis(trifluoromethyl)phenyl]borane frustrated Lewis pair. Dalton Trans. 2012, 41, 9019–9022. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Kemper, S.; Klare, H.F.T.; Oestreich, M. Boron Lewis Acid-Catalyzed Hydroboration of Alkenes with Pinacolborane: BArF3 Does What B(C6F5)3 Cannot Do! Chem. Eur. J. 2016, 22, 13840–13844. [Google Scholar] [CrossRef]
- Cornet, S.M.; Dillon, K.B.; Entwistle, C.D.; Fox, M.A.; Goeta, A.E.; Goodwin, H.P.; Marder, T.B.; Thompson, A.L. Synthesis and characterisation of some new boron compounds containing the 2,4,6-(CF3)3C6H2(fluoromes = Ar), 2,6-(CF3)2C6H3(fluoroxyl = Ar′), or 2,4-(CF3)2C6H3 (Ar″) ligands. Dalton Trans. 2003, 4395–4405. [Google Scholar] [CrossRef]
- Blagg, R.J.; Lawrence, E.J.; Resner, K.; Oganesyan, V.S.; Herrington, T.J.; Ashley, A.E.; Wildgoose, G.G. Exploring structural and electronic effects in three isomers of tris{bis(trifluoromethyl)phenyl}borane: Towards the combined electrochemical-frustrated Lewis pair activation of H2. Dalton Trans. 2016, 45, 6023–6031. [Google Scholar] [CrossRef] [Green Version]
- Shinji, T.; Mitsuhiro, A.; Michinori, O.; Fumio, T. Structural and Stereochemical Studies of Tris[2-(trifluoromethyl)phenyl]borane: Spontaneous Resolution, Stereodynamics, and Intramolecular C-F·B Interactions. Bull. Chem. Soc. Jpn. 2001, 73, 2357–2362. [Google Scholar]
- Li, L.; Marks, T.J. New Organo-Lewis Acids. Tris(β-perfluoronaphthyl)borane (PNB) as a Highly Active Cocatalyst for Metallocene-Mediated Ziegler−Natta α-Olefin Polymerization. Organometallics 1998, 17, 3996–4003. [Google Scholar] [CrossRef]
- Chen, Y.X.E.; Metz, M.V.; Li, L.; Stern, C.L.; Marks, T.J. Sterically Encumbered (Perfluoroaryl)Borane and Aluminate Cocatalysts for Tuning Cation−Anion Ion Pair Structure and Reactivity in Metallocene Polymerization Processes. A Synthetic, Structural, and Polymerization Study. J. Am. Chem. Soc. 1998, 120, 6287–6305. [Google Scholar] [CrossRef]
- Körte, L.A.; Schwabedissen, J.; Soffner, M.; Blomeyer, S.; Reuter, C.G.; Vishnevskiy, Y.V.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Tris(perfluorotolyl)borane—A Boron Lewis Superacid. Angew. Chem. Int. Ed. 2017, 56, 8578–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stern, C.L.; Marks, T.J. Bis(Pentafluorophenyl)(2-perfluorobiphenylyl)borane. A New Perfluoroarylborane Cocatalyst for Single-Site Olefin Polymerization. Organometallics 2000, 19, 3332–3337. [Google Scholar] [CrossRef]
- Gyömöre, Á.; Bakos, M.; Földes, T.; Pápai, I.; Domján, A.; Soós, T. Moisture-Tolerant Frustrated Lewis Pair Catalyst for Hydrogenation of Aldehydes and Ketones. ACS Catal. 2015, 5, 5366–5372. [Google Scholar] [CrossRef] [Green Version]
- Blagg, R.J.; Wildgoose, G.G. H2 activation using the first 1:1:1 hetero-tri(aryl)borane. RSC Adv. 2016, 6, 42421–42427. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Roy, S.; Chatterjee, I. Tris(pentafluorophenyl)borane catalyzed C–C and C–heteroatom bond formation. Org. Biomol. Chem. 2021, 19, 1230–1267. [Google Scholar] [CrossRef] [PubMed]
- Harder, S. Introduction to Early Main Group Organometallic Chemistry and Catalysis. In Early Main Group Metal Catalysis: Concepts and Reactions; Wiley—VCH: Weinheim, Germany, 2019; pp. 1–29. [Google Scholar]
- Stephan, D.W. Catalysis, FLPs, and Beyond. Chem 2020, 6, 1520–1526. [Google Scholar] [CrossRef]
- Hall, D.G. Boronic acid catalysis. Chem. Soc. Rev. 2019, 48, 3475–3496. [Google Scholar] [CrossRef]
- Prey, S.E.; Wagner, M. Threat to the Throne: Can Two Cooperating Boron Atoms Rival Transition Metals in Chemical Bond Activation and Catalysis? Adv. Synth. Catal. 2021, 363, 2290–2309. [Google Scholar] [CrossRef]
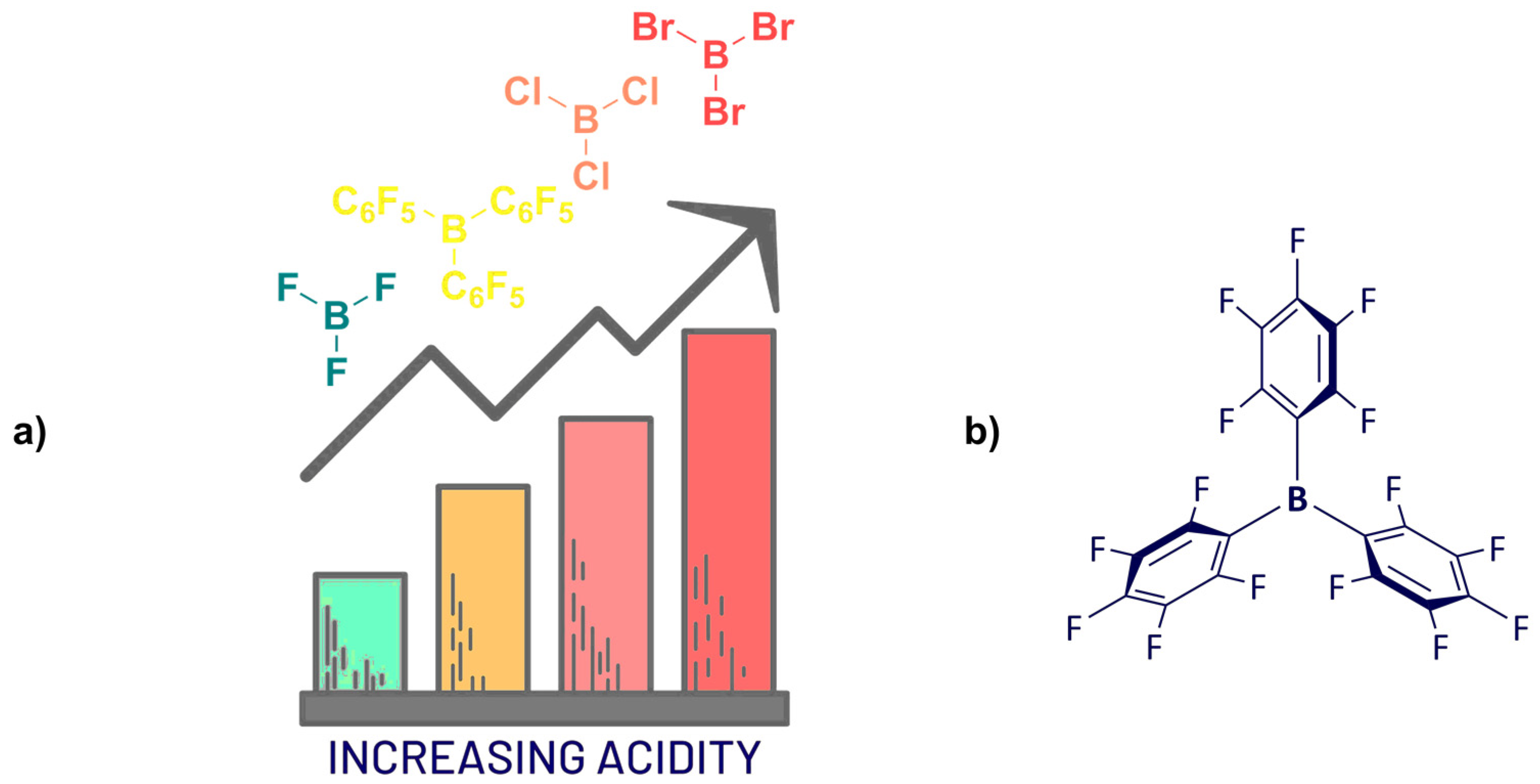
- Piers, W.E. The chemistry of perfluoroaryl boranes. In Advances in Organometallic Chemistry; West, R., Hill, A., Eds.; Academic Press: Cambridge, MA, USA, 2005; Volume 52, pp. 1–74. [Google Scholar]
- Laszlo, P.; Teston, M. Determination of the acidity of Lewis acids. J. Am. Chem. Soc. 1990, 112, 8750–8754. [Google Scholar] [CrossRef]
- Lawson, J.R.; Melen, R.L. Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, B.; Kinjo, R. Boron-Based Catalysts for C−C Bond-Formation Reactions. Chem. Asian J. 2018, 13, 1279–1292. [Google Scholar] [CrossRef]
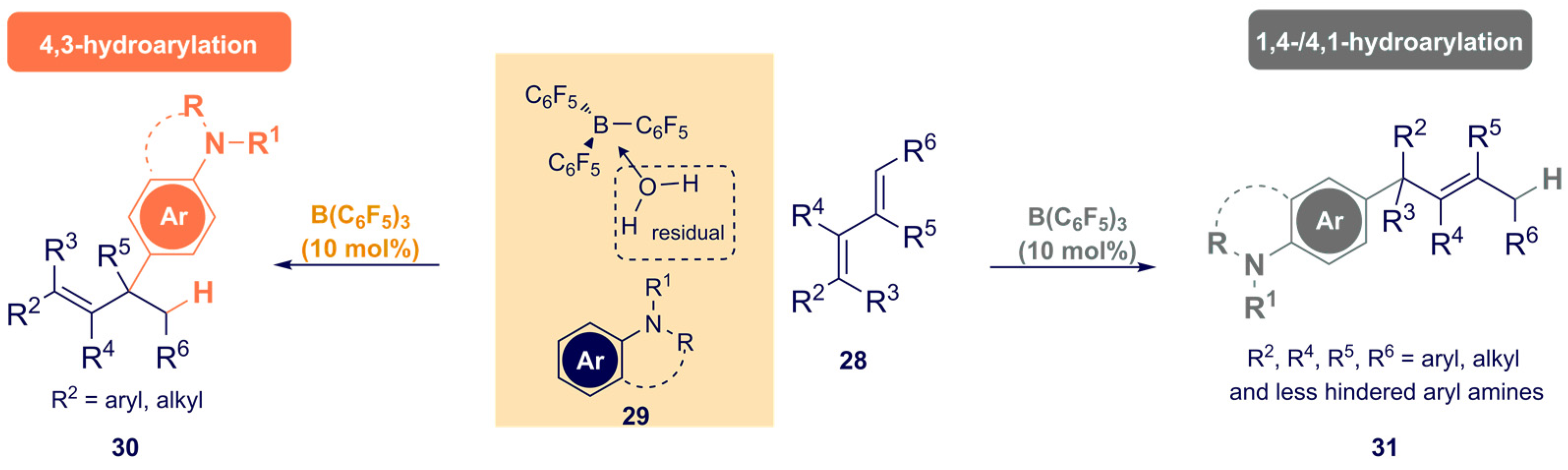
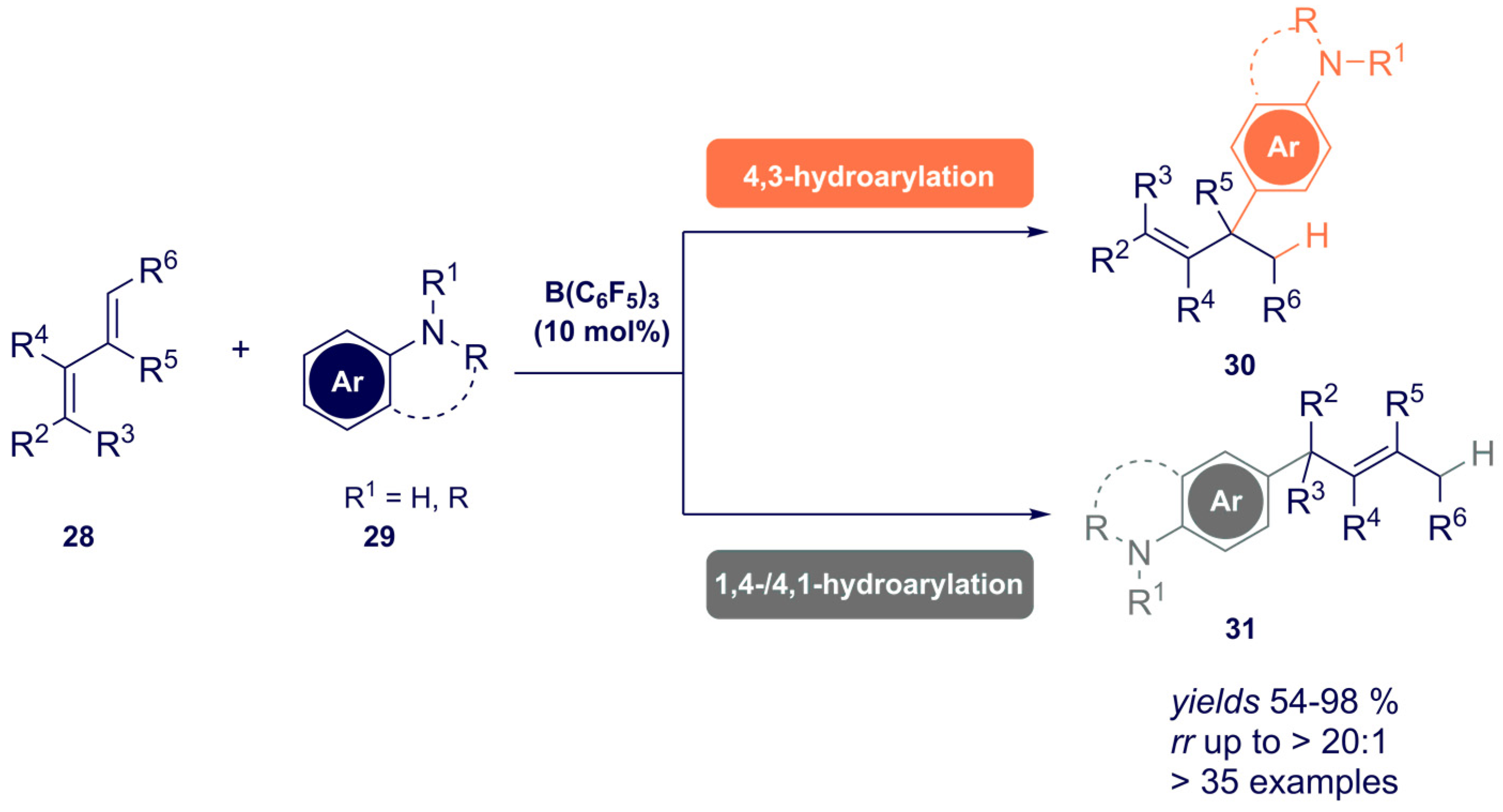
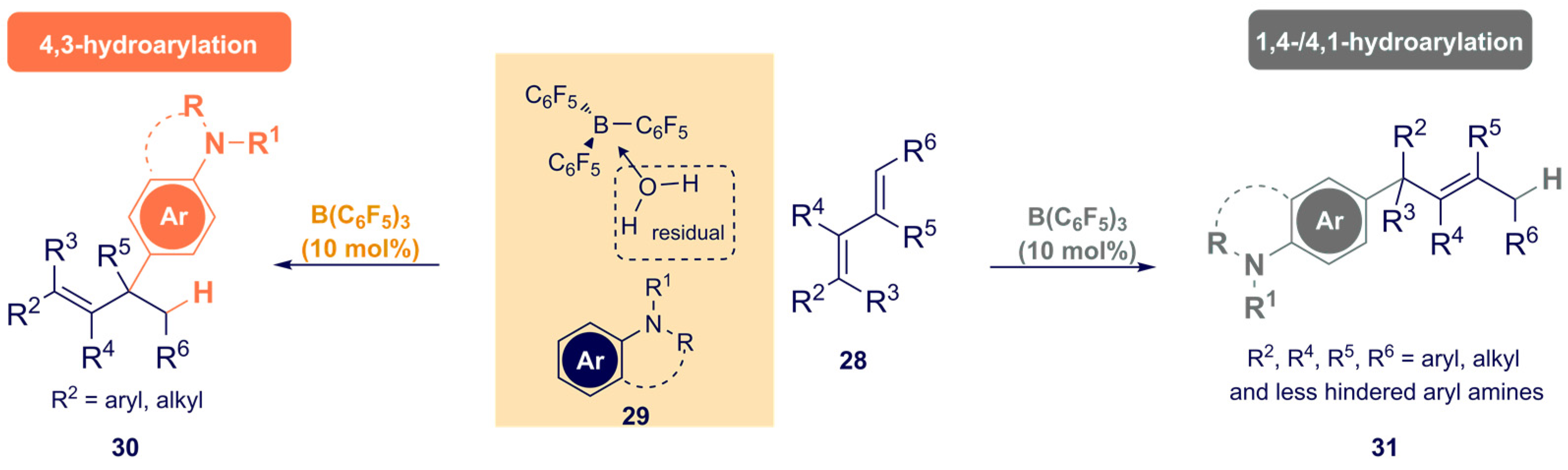
- Kumar, G.; Qu, Z.-W.; Grimme, S.; Chatterjee, I. Boron-Catalyzed Hydroarylation of 1,3-Dienes with Arylamines. Org. Lett. 2021, 23, 8952–8957. [Google Scholar] [CrossRef] [PubMed]
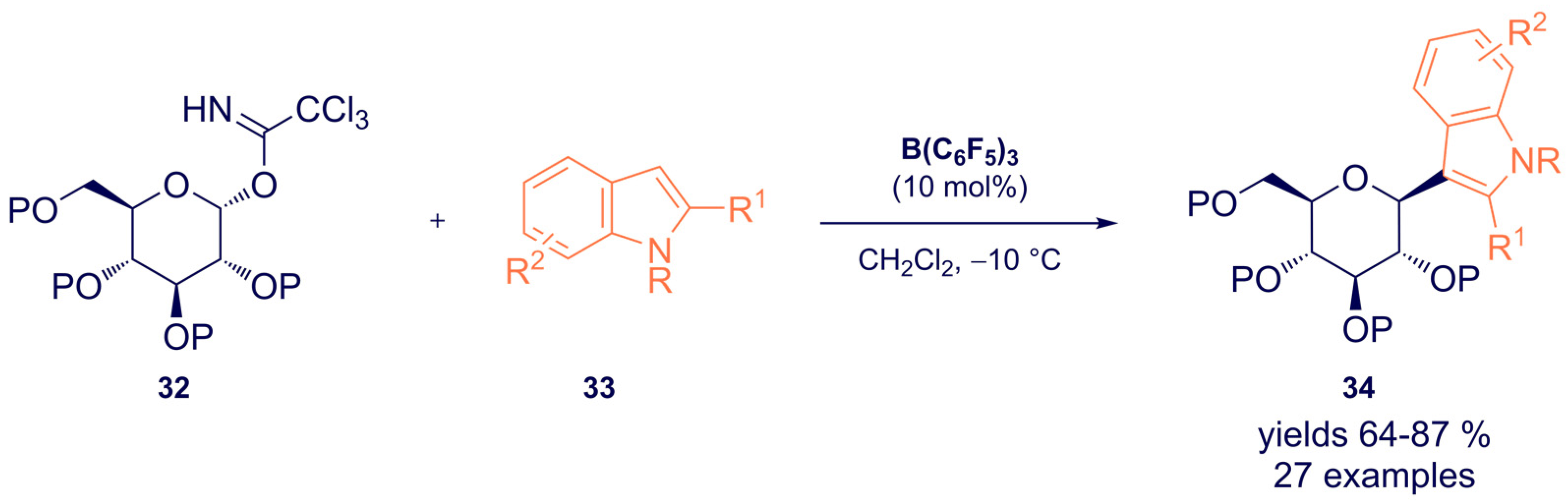
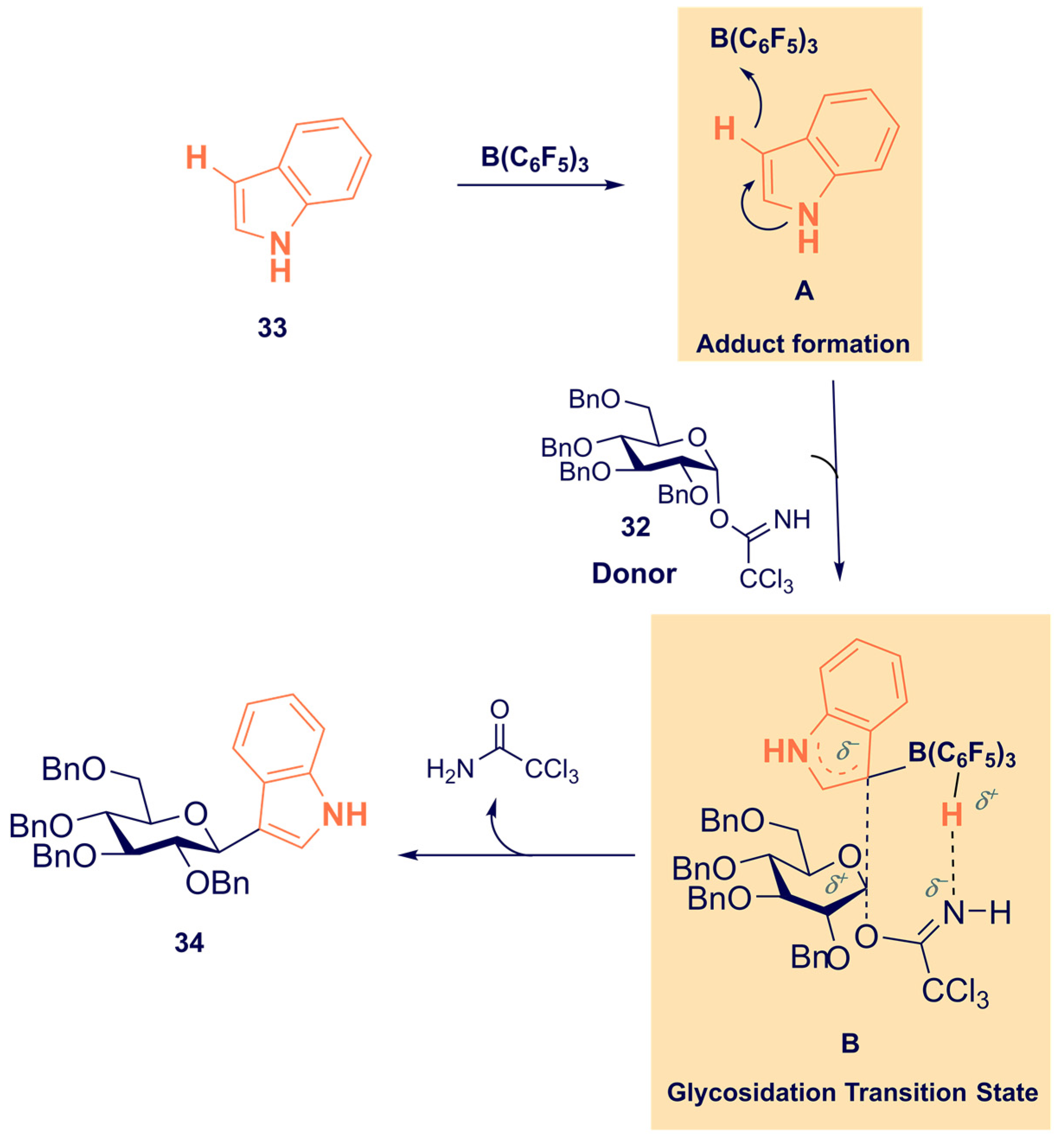
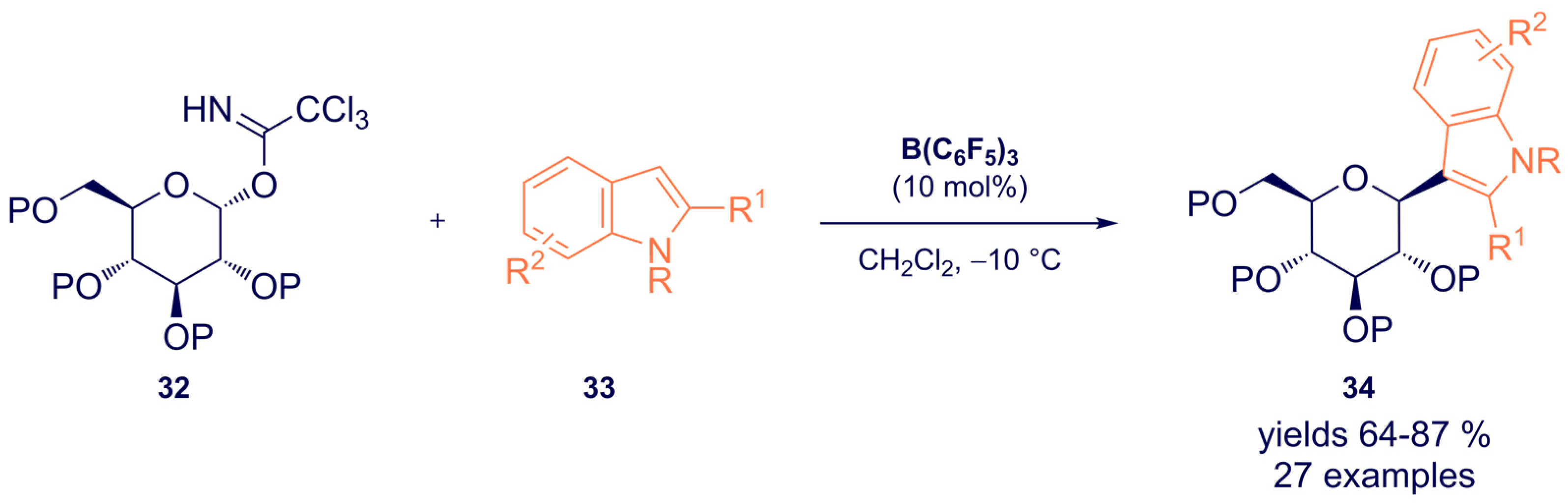
- Dubey, A.; Tiwari, A.; Mandal, P.K. Tris(pentafluorophenyl)borane-Catalyzed Stereoselective C-Glycosylation of Indoles with Glycosyl Trichloroacetimidates: Access to 3-Indolyl-C-glycosides. J. Org. Chem. 2021, 86, 8516–8526. [Google Scholar] [CrossRef] [PubMed]
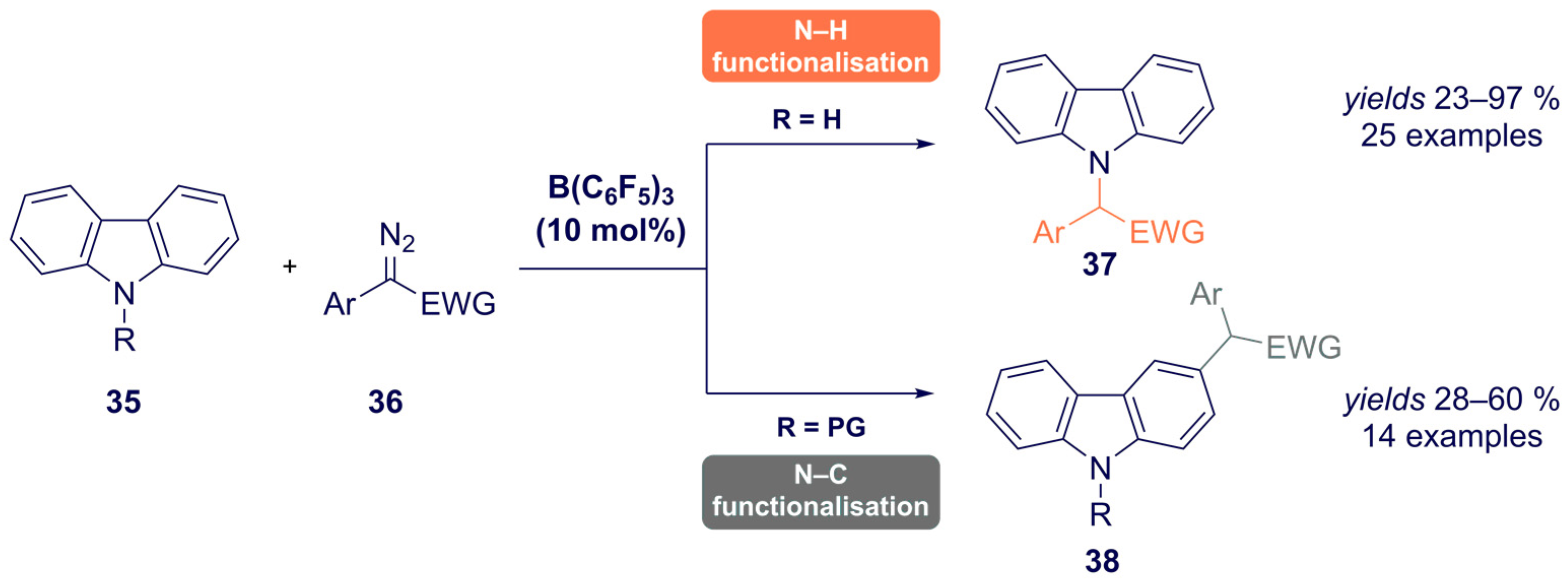
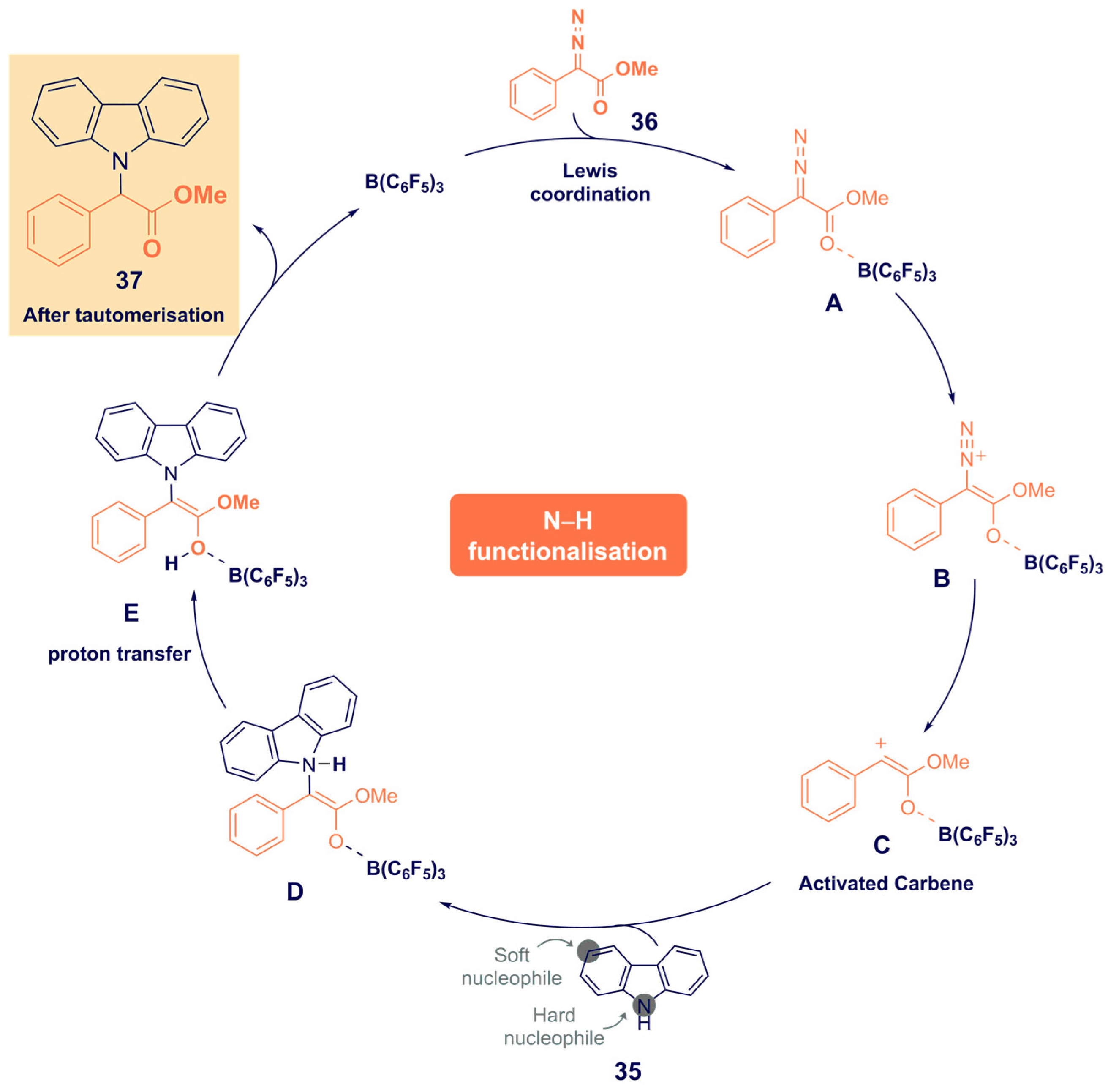
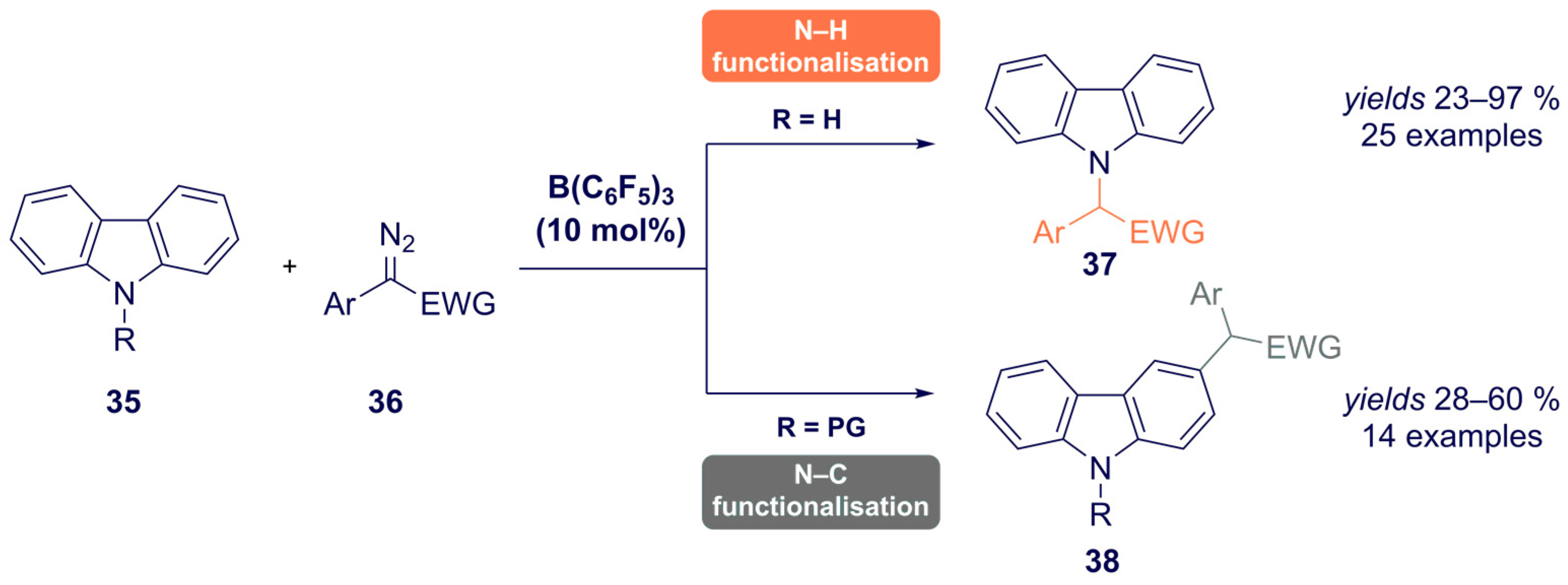
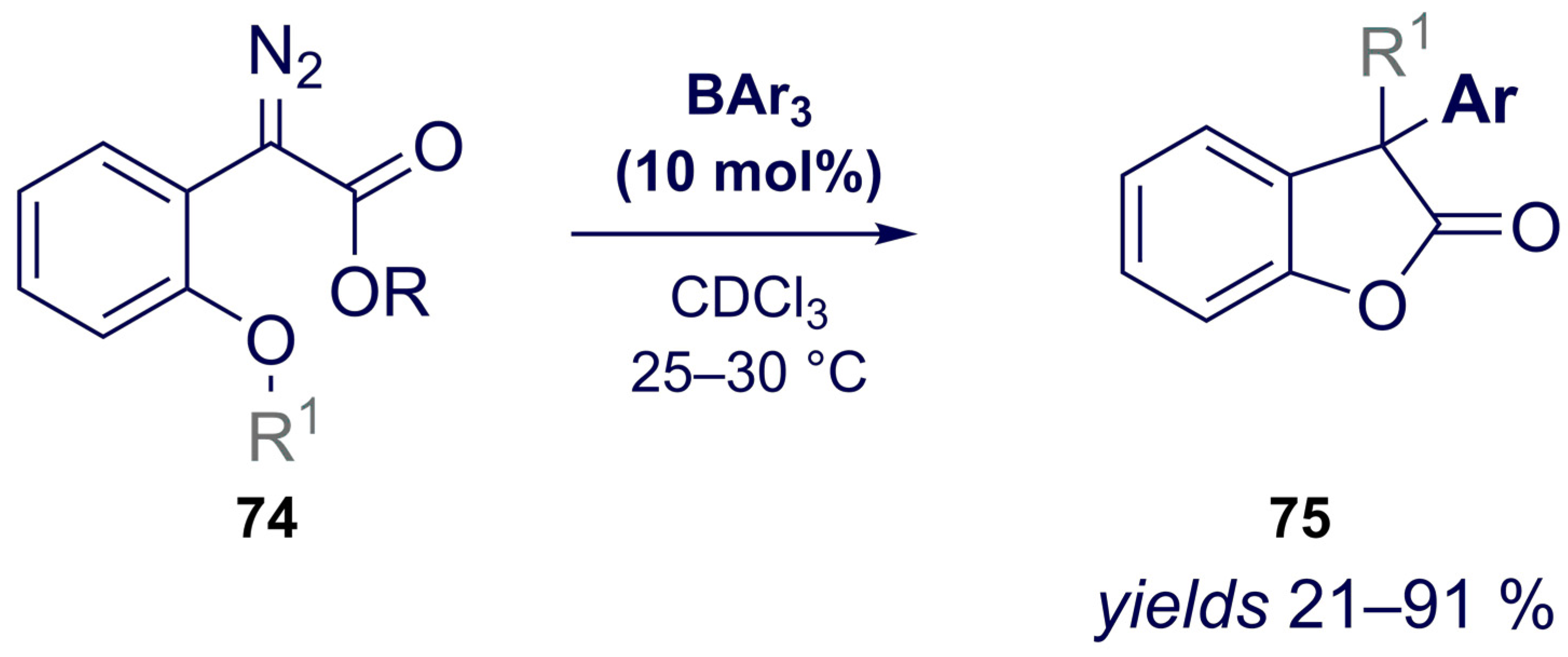
- He, F.; Koenigs, R.M. Borane-Catalyzed Carbazolation Reactions of Aryldiazoacetates. Org. Lett. 2021, 23, 5831–5835. [Google Scholar] [CrossRef]
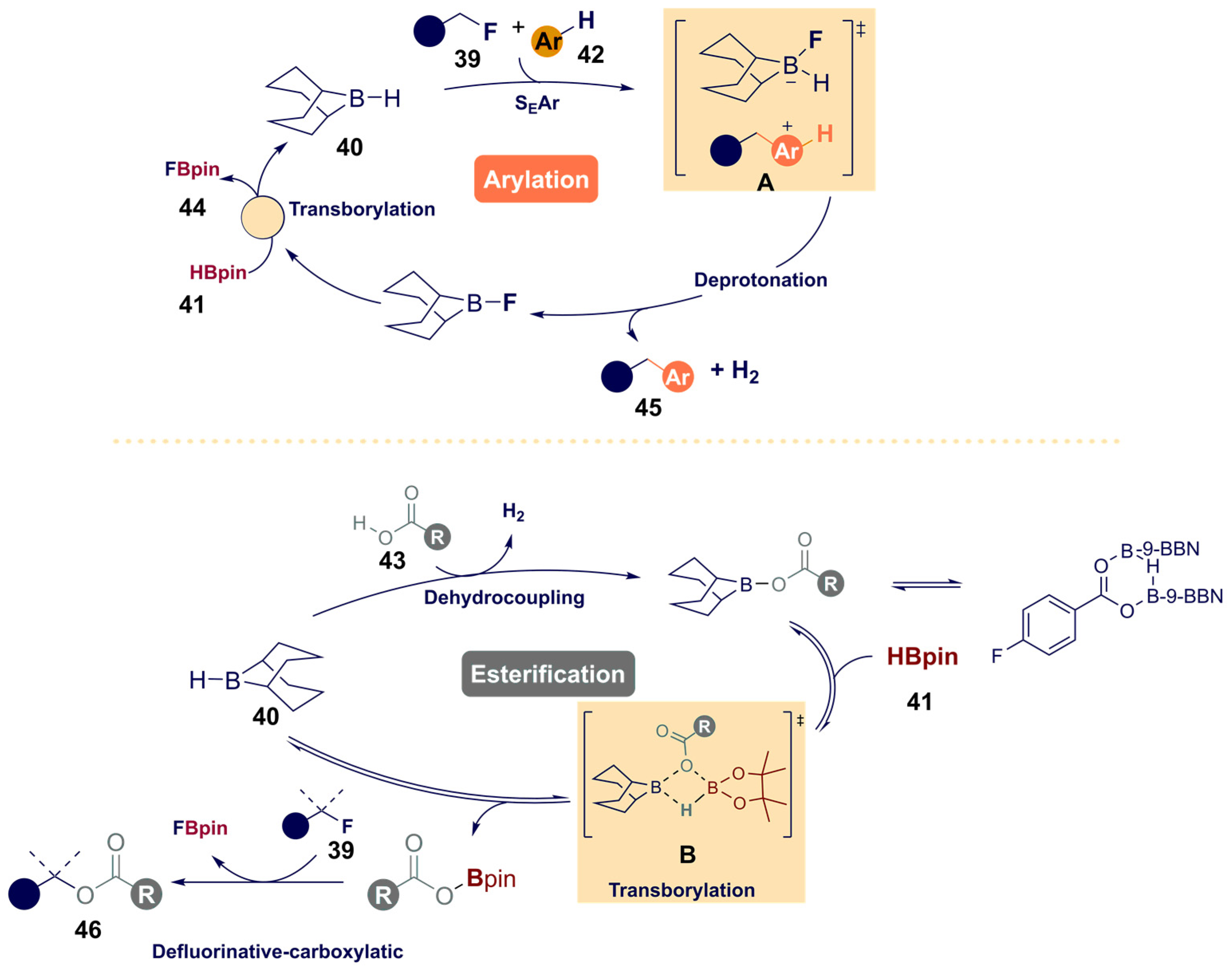
- Willcox, D.R.; Nichol, G.S.; Thomas, S.P. Borane-catalyzed C(sp3)-F bond arylation and esterification enabled by transborylation. ACS Catal. 2021, 11, 3190–3197. [Google Scholar] [CrossRef]
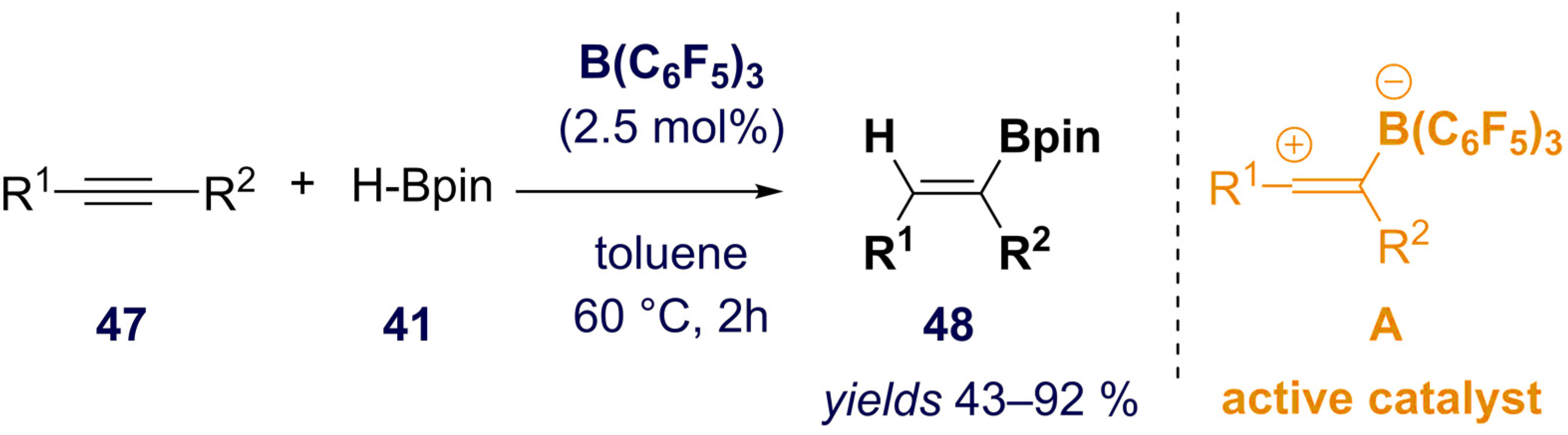
- Bismuto, A.; Cowley, M.J.; Thomas, S.P. Zwitterion-Initiated Hydroboration of Alkynes and Styrene. Adv. Synth. Catal. 2021, 363, 2382–2385. [Google Scholar] [CrossRef]
- Kiyokawa, K.; Urashima, N.; Minakata, S. Tris(pentafluorophenyl)borane-Catalyzed Formal Cyanoalkylation of Indoles with Cyanohydrins. J. Org. Chem. 2021, 86, 8389–8401. [Google Scholar] [CrossRef]
- Richter, S.C.; Oestreich, M. Chemoselective Deoxygenation of 2° Benzylic Alcohols through a Sequence of Formylation and B(C6F5)3-Catalyzed Reduction. Eur. J. Org. Chem. 2021, 2021, 2103–2106. [Google Scholar] [CrossRef]
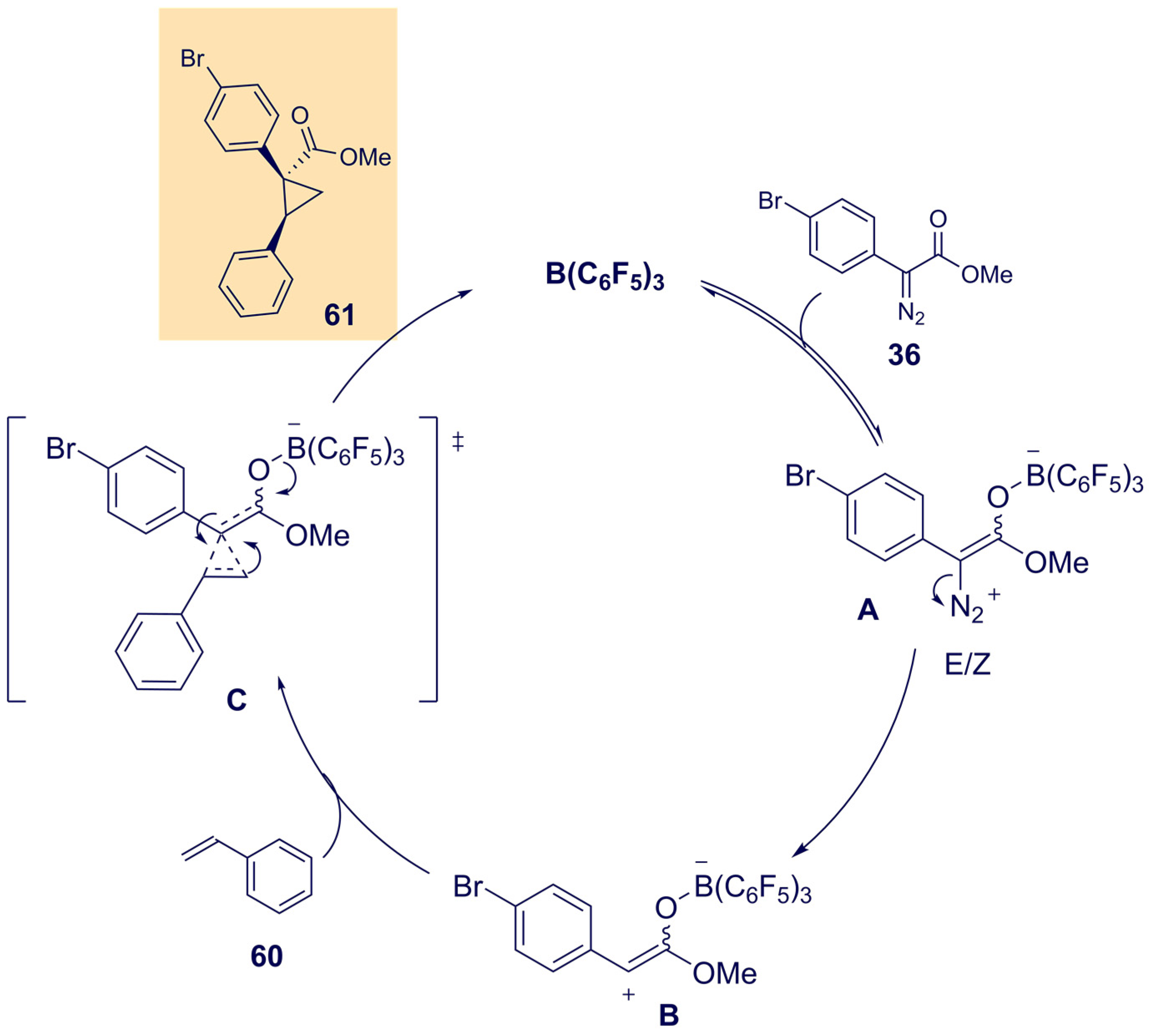
- Dasgupta, A.; Babaahmadi, R.; Slater, B.; Yates, B.F.; Ariafard, A.; Melen, R.L. Borane-Catalyzed Stereoselective C–H Insertion, Cyclopropanation, and Ring-Opening Reactions. Chem 2020, 6, 2364–2381. [Google Scholar] [CrossRef]
- Mancinelli, J.P.; Wilkerson-Hill, S.M. Tris(pentafluorophenyl)borane-Catalyzed Cyclopropanation of Styrenes with Aryldiazoacetates. ACS Catal. 2020, 10, 11171–11176. [Google Scholar] [CrossRef]
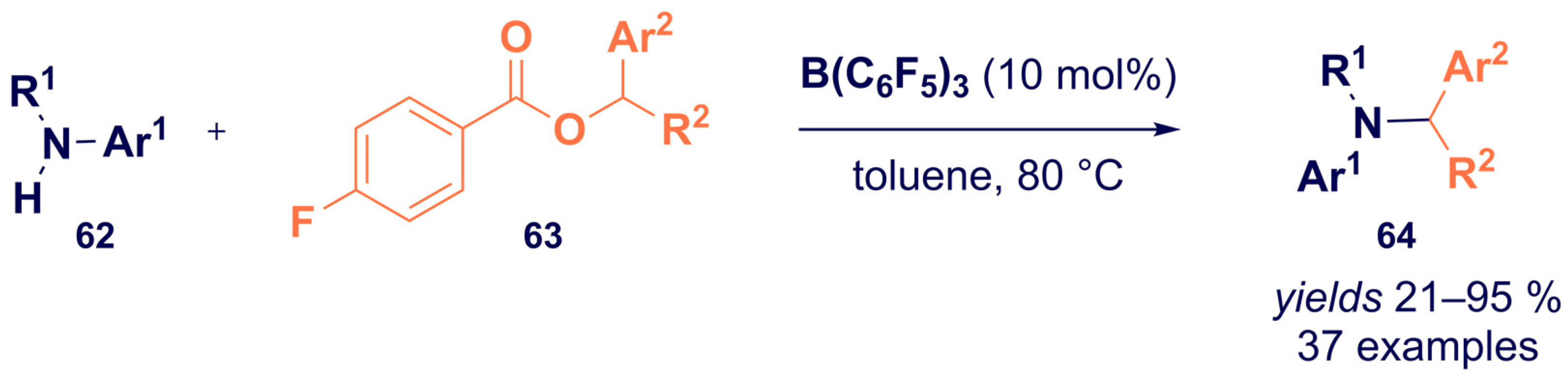
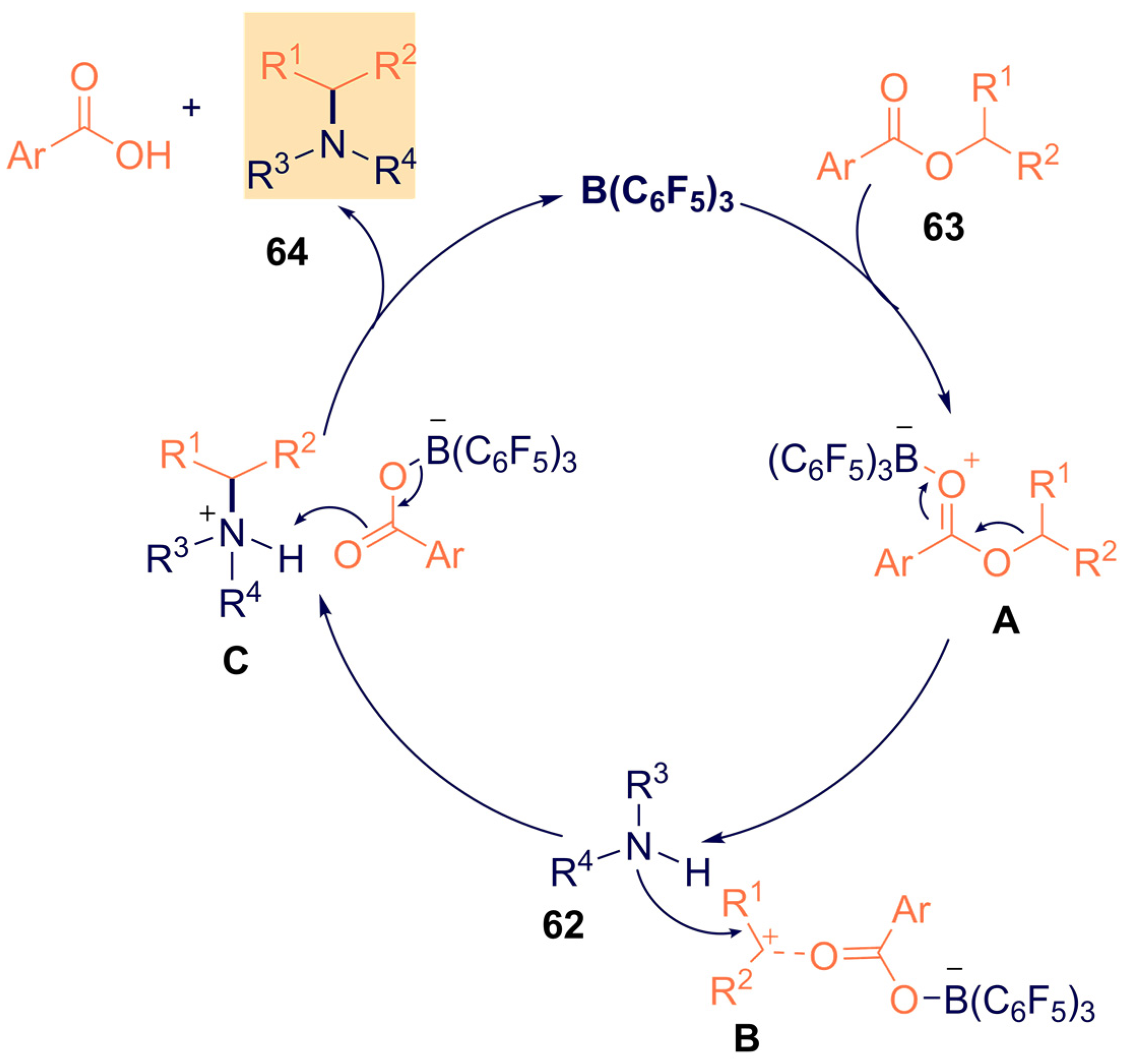
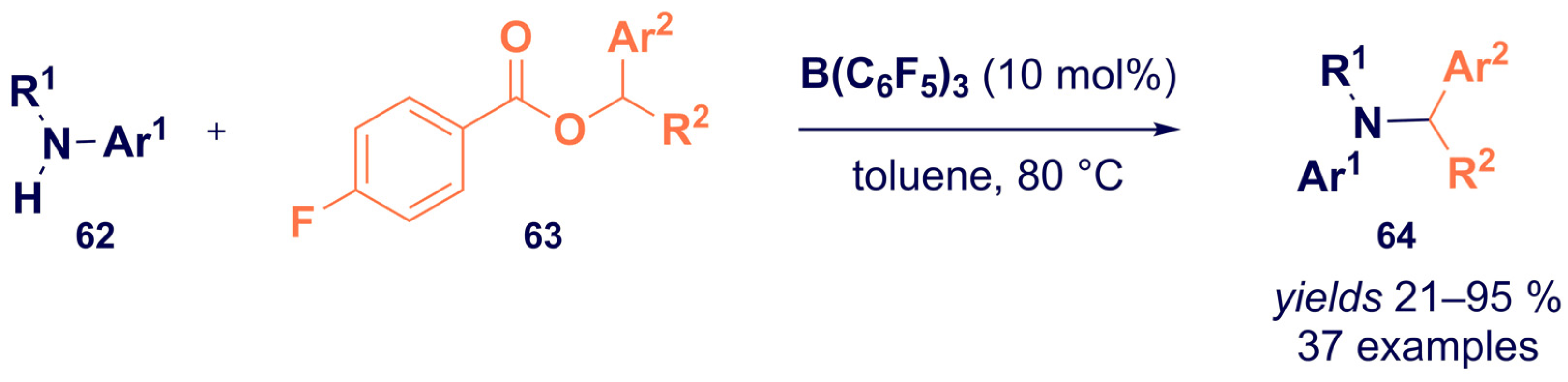
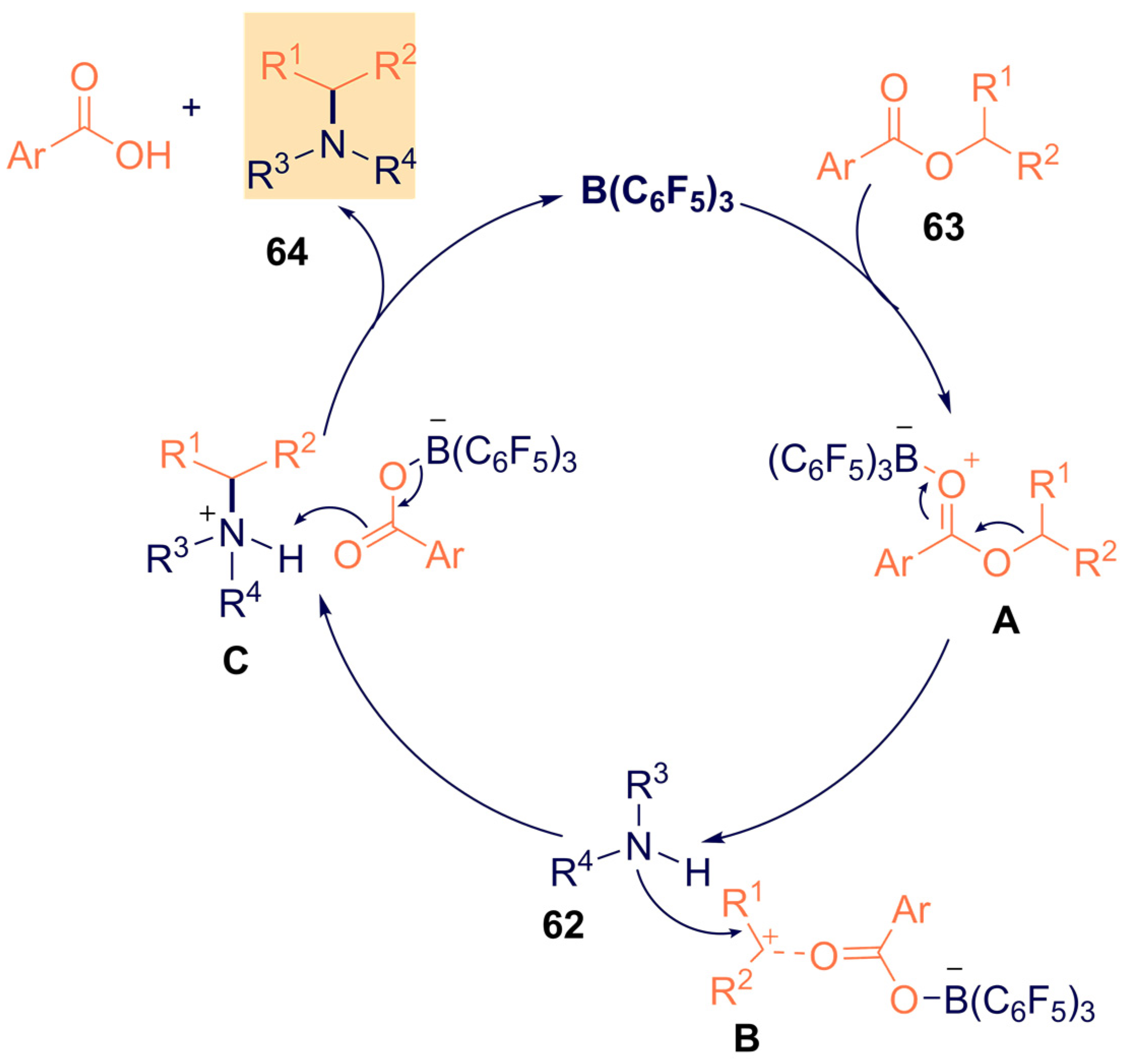
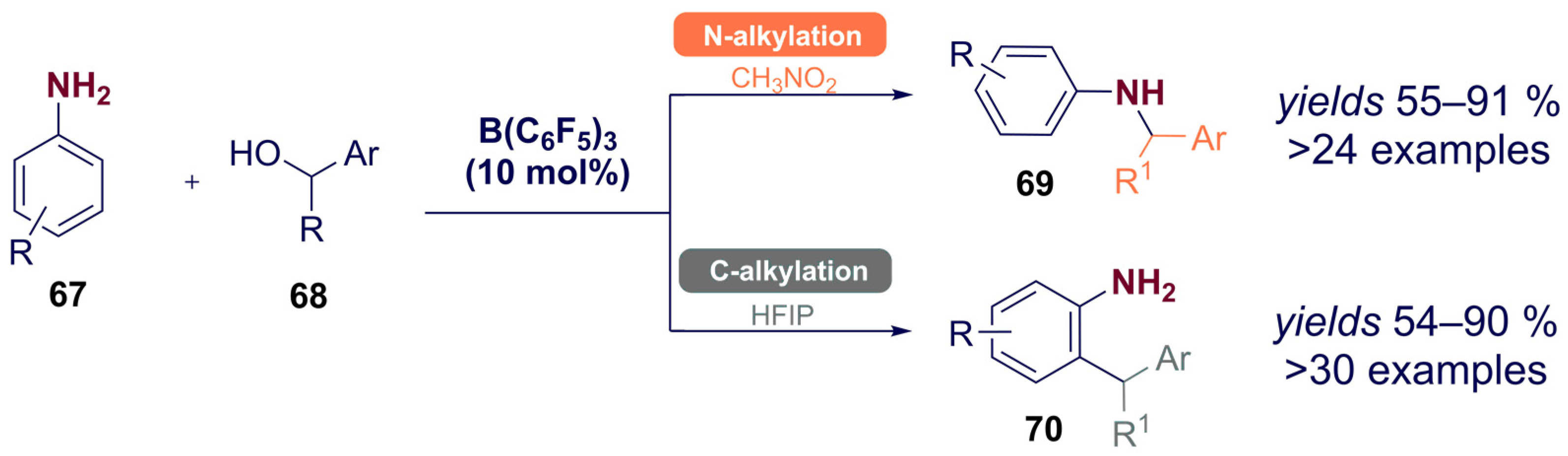
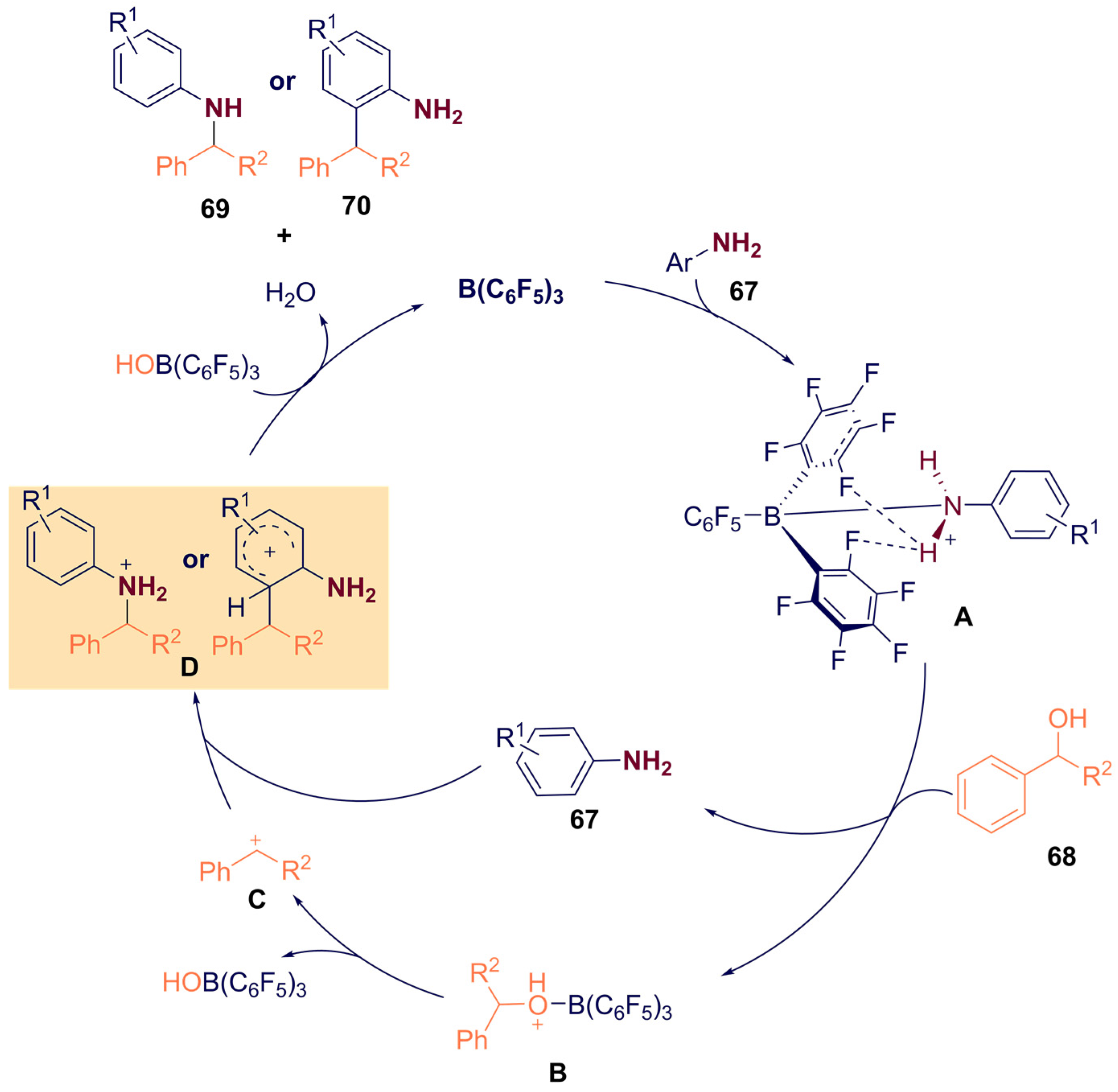
- Nori, V.; Dasgupta, A.; Babaahmadi, R.; Carlone, A.; Ariafard, A.; Melen, R.L. Triarylborane catalysed N -alkylation of amines with aryl esters. Catal. Sci. Technol. 2020, 10, 7523–7530. [Google Scholar] [CrossRef]
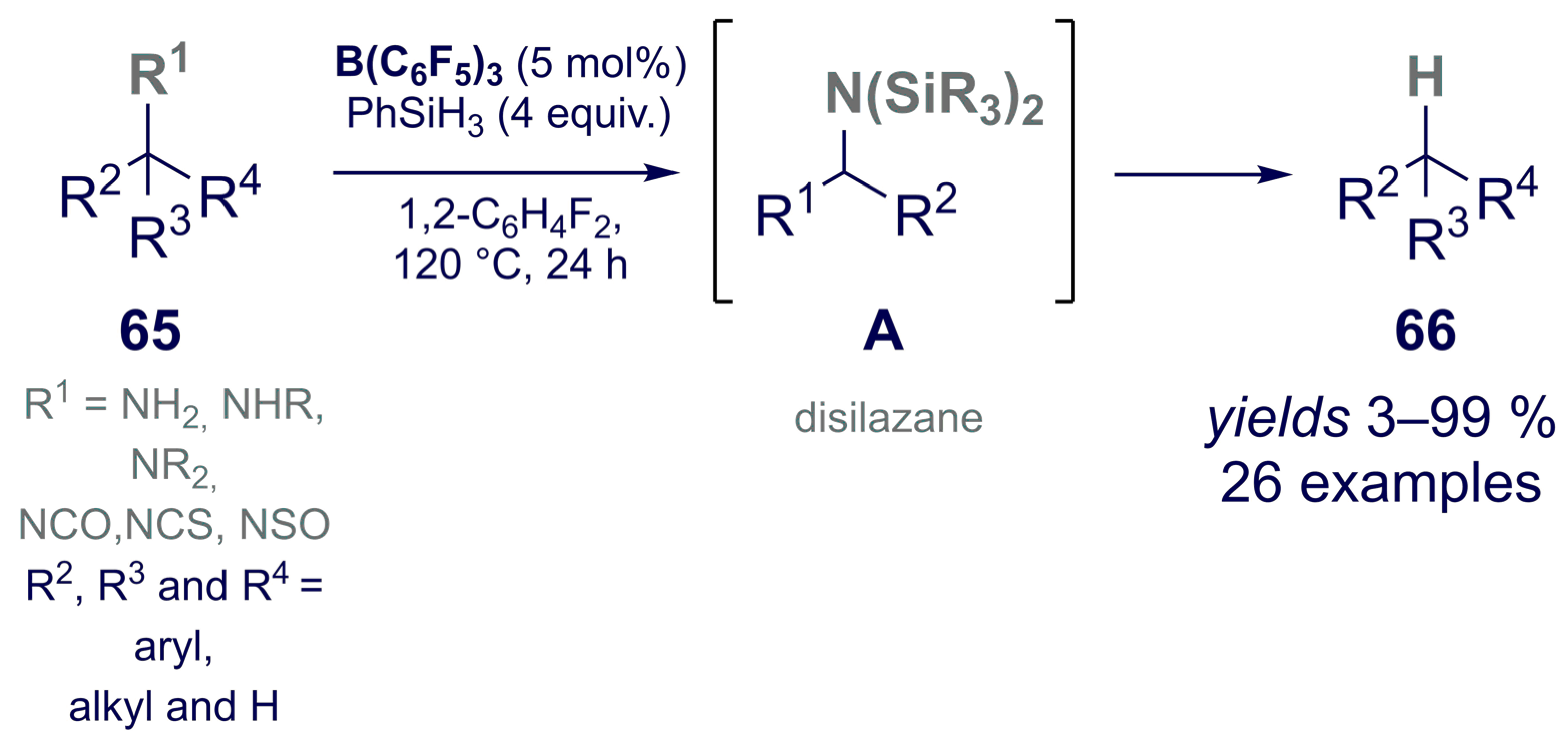
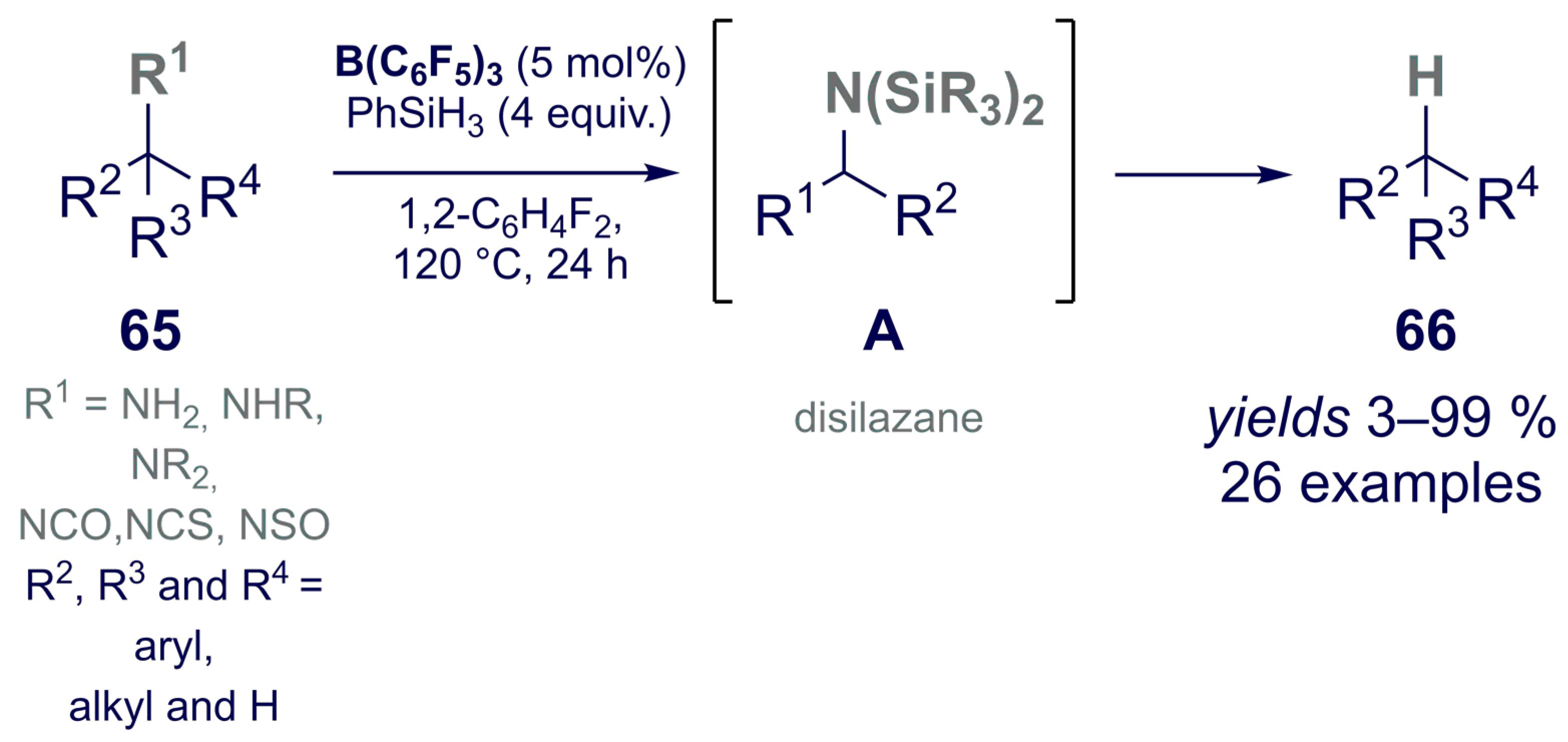
- Fang, H.; Oestreich, M. Reductive Deamination with Hydrosilanes Catalyzed by B(C6F5)3. Angew. Chem. Int. Ed. 2020, 59, 11394–11398. [Google Scholar] [CrossRef]
- Meng, S.-S.; Tang, X.; Luo, X.; Wu, R.; Zhao, J.-L.; Chan, A.S.C. Borane-Catalyzed Chemoselectivity-Controllable N-Alkylation and ortho C-Alkylation of Unprotected Arylamines Using Benzylic Alcohols. ACS Catal. 2019, 9, 8397–8403. [Google Scholar] [CrossRef]
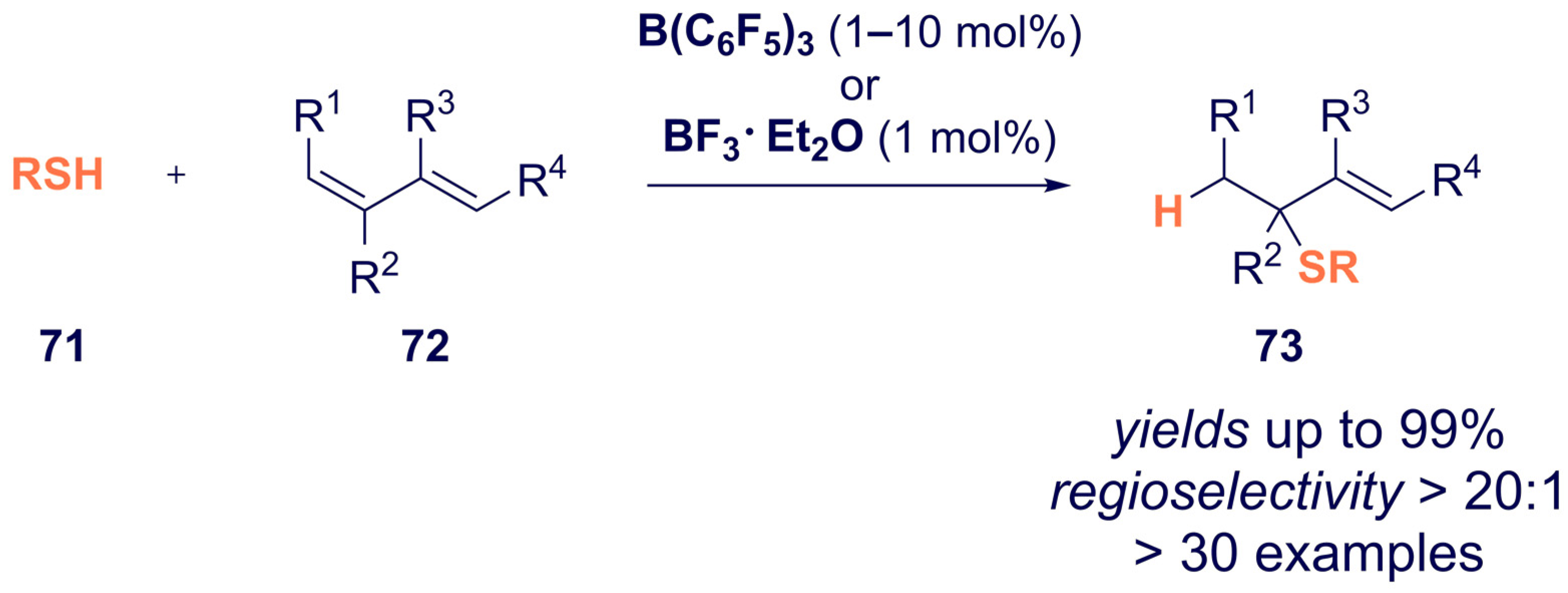
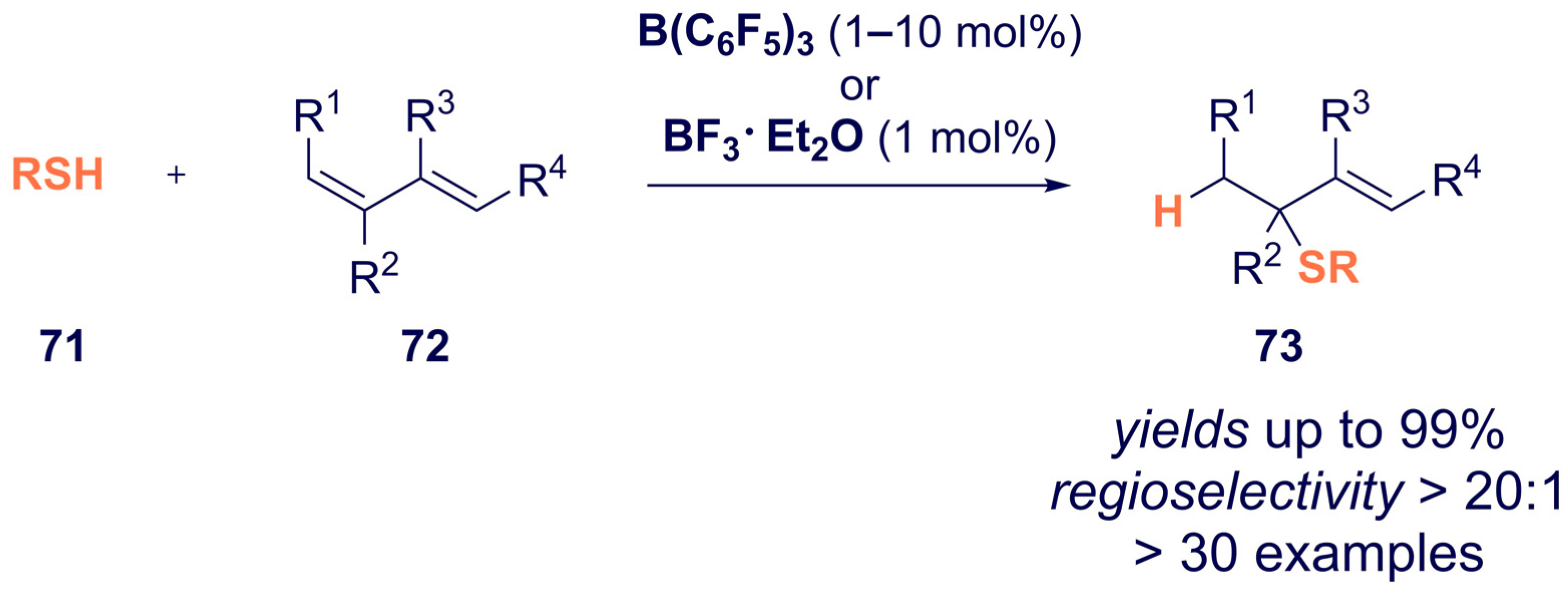
- Kumar, G.; Qu, Z.-W.; Ghosh, S.; Grimme, S.; Chatterjee, I. Boron Lewis Acid-Catalyzed Regioselective Hydrothiolation of Conjugated Dienes with Thiols. ACS Catal. 2019, 9, 11627–11633. [Google Scholar] [CrossRef]
- Santi, M.; Ould, D.M.C.; Wenz, J.; Soltani, Y.; Melen, R.L.; Wirth, T. Metal-Free Tandem Rearrangement/Lactonization: Access to 3,3-Disubstituted Benzofuran-2-(3H)-ones. Angew. Chem. Int. Ed. 2019, 58, 7861–7865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.-J.; Liu, N.; Liu, Q.-F.; Sun, W.; Wang, X.-C. Borane-Catalyzed Direct Asymmetric Vinylogous Mannich Reactions of Acyclic α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2021, 143, 3054–3059. [Google Scholar] [CrossRef]
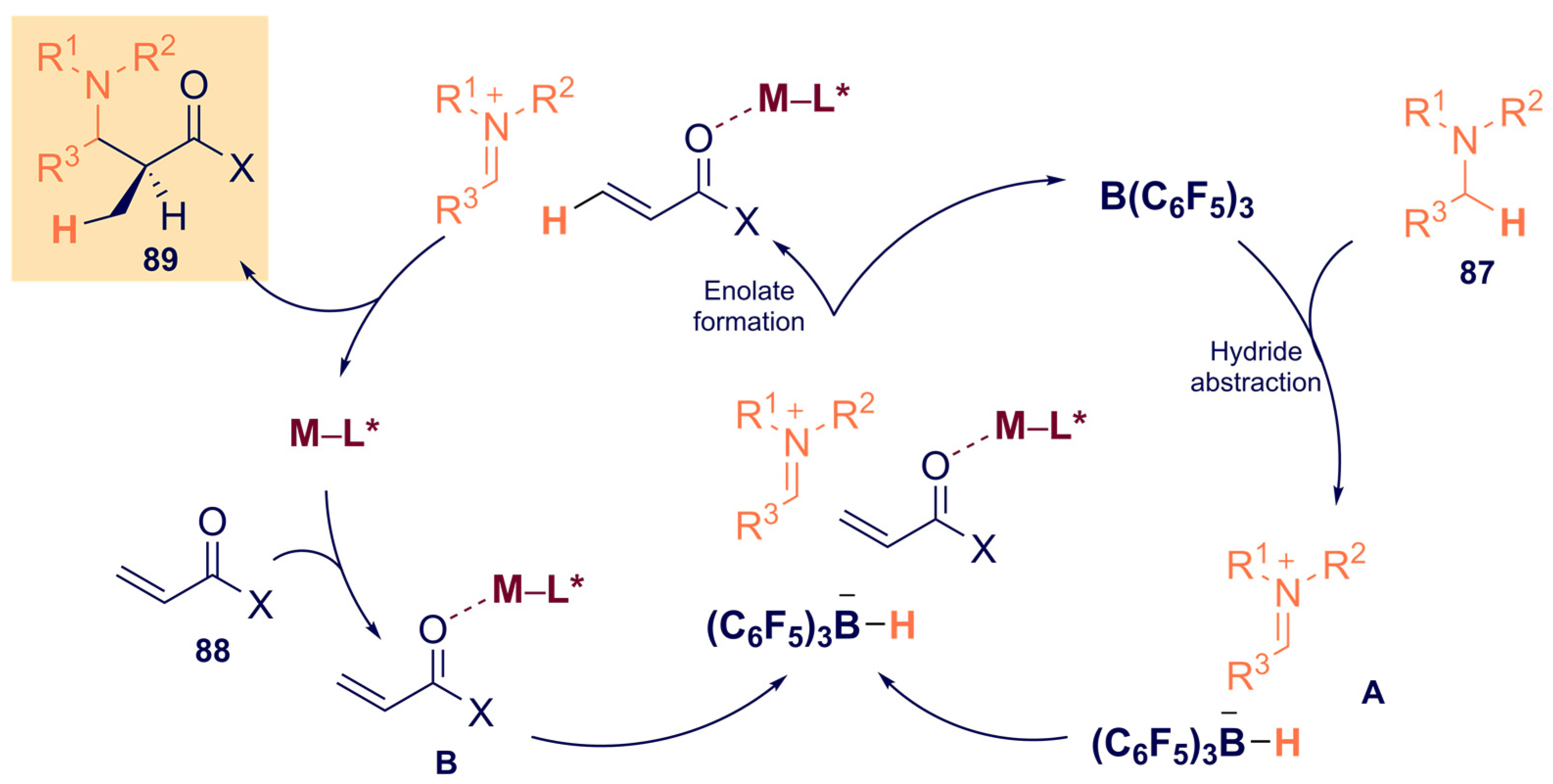
- Chang, Y.; Cao, M.; Chan, J.Z.; Zhao, C.; Wang, Y.; Yang, R.; Wasa, M. Enantioselective Synthesis of N-Alkylamines through β-Amino C-H Functionalization Promoted by Cooperative Actions of B(C6F5)3 and a Chiral Lewis Acid Co-Catalyst. J. Am. Chem. Soc. 2021, 143, 2441–2455. [Google Scholar] [CrossRef]
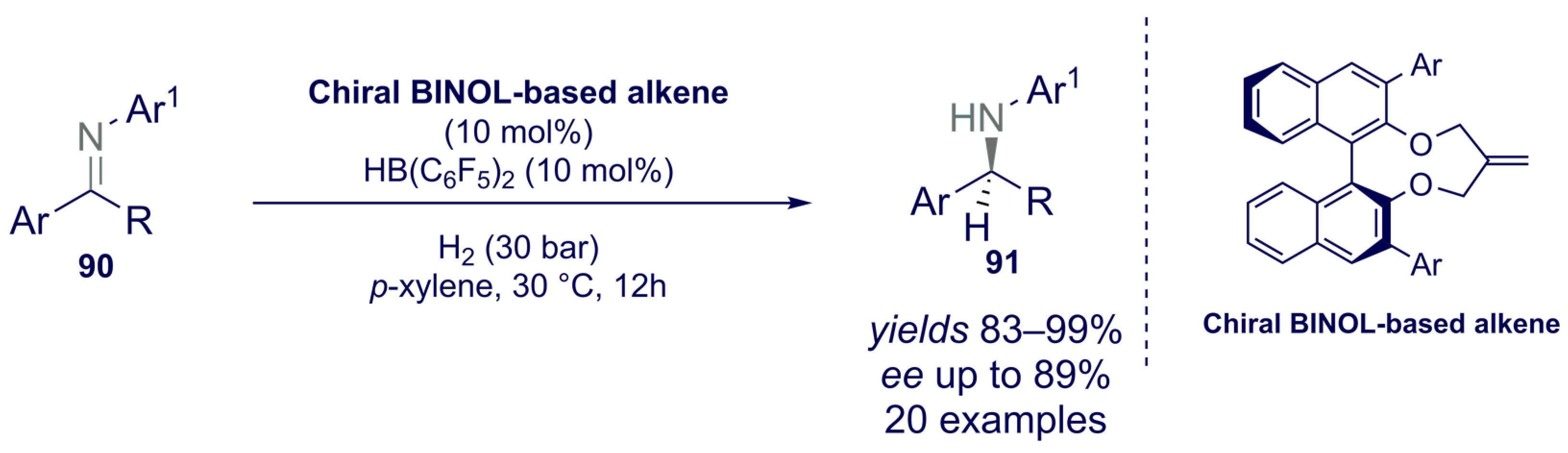
- Tian, J.-J.; Yang, Z.-Y.; Liang, X.-S.; Liu, N.; Hu, C.-Y.; Tu, X.-S.; Li, X.; Wang, X.-C. Borane-Catalyzed Chemoselective and Enantioselective Reduction of 2-Vinyl-Substituted Pyridines. Angew. Chem. 2020, 132, 18610–18614. [Google Scholar] [CrossRef]
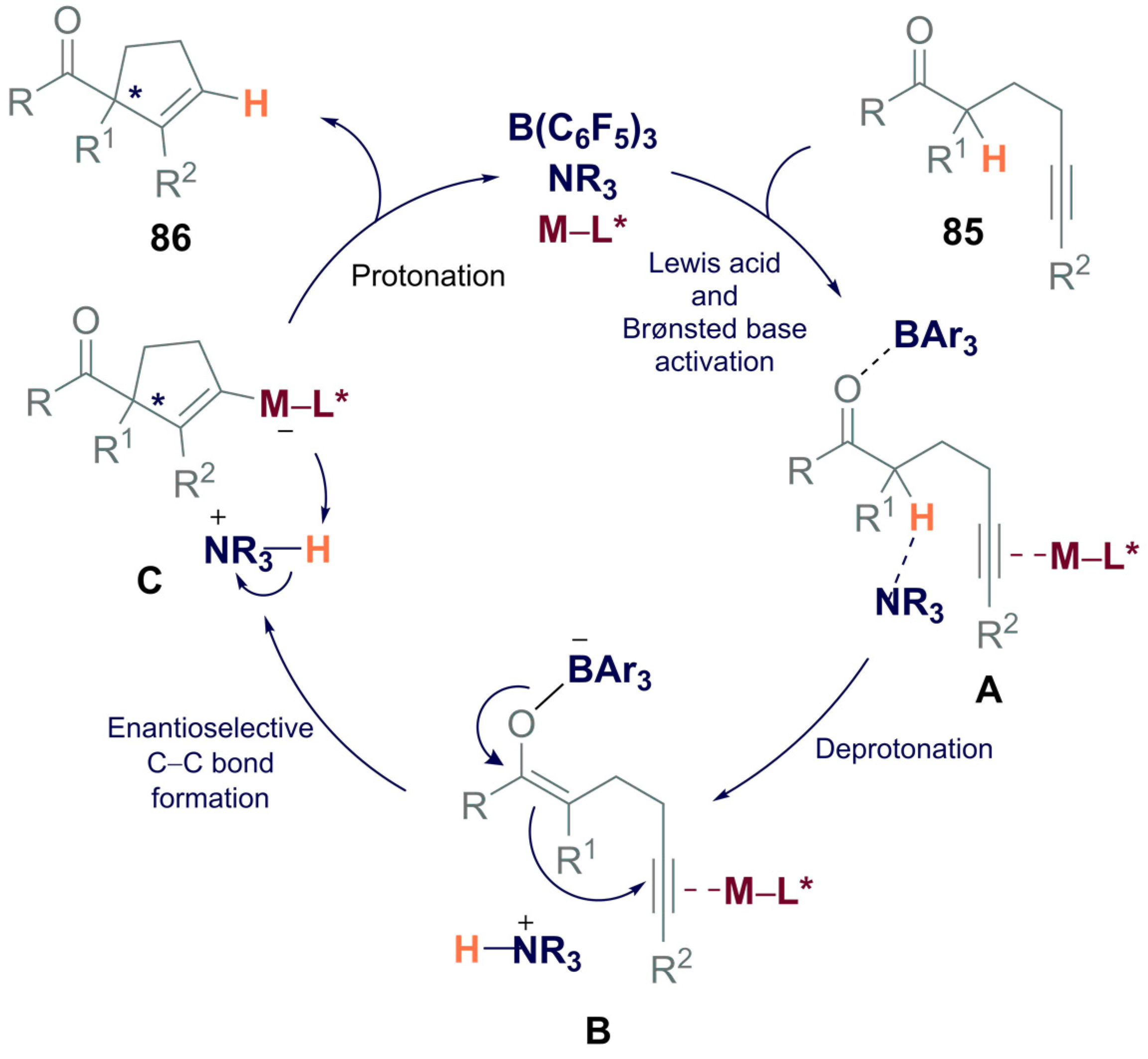
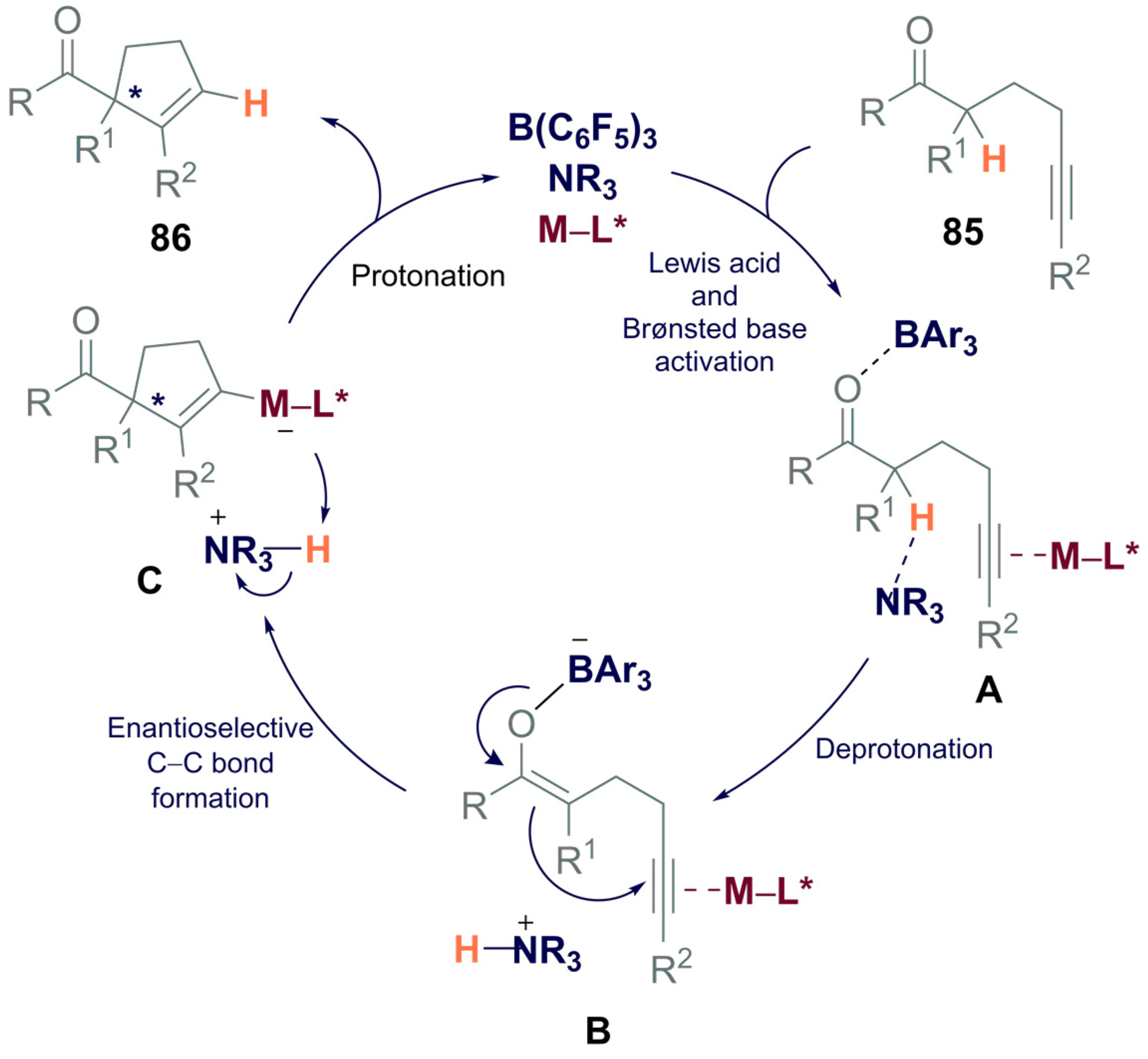
- Cao, M.; Yesilcimen, A.; Wasa, M. Enantioselective Conia-Ene-Type Cyclizations of Alkynyl Ketones through Cooperative Action of B(C6F5)3, N-Alkylamine and a Zn-Based Catalyst. J. Am. Chem. Soc. 2019, 141, 4199–4203. [Google Scholar] [CrossRef] [PubMed]
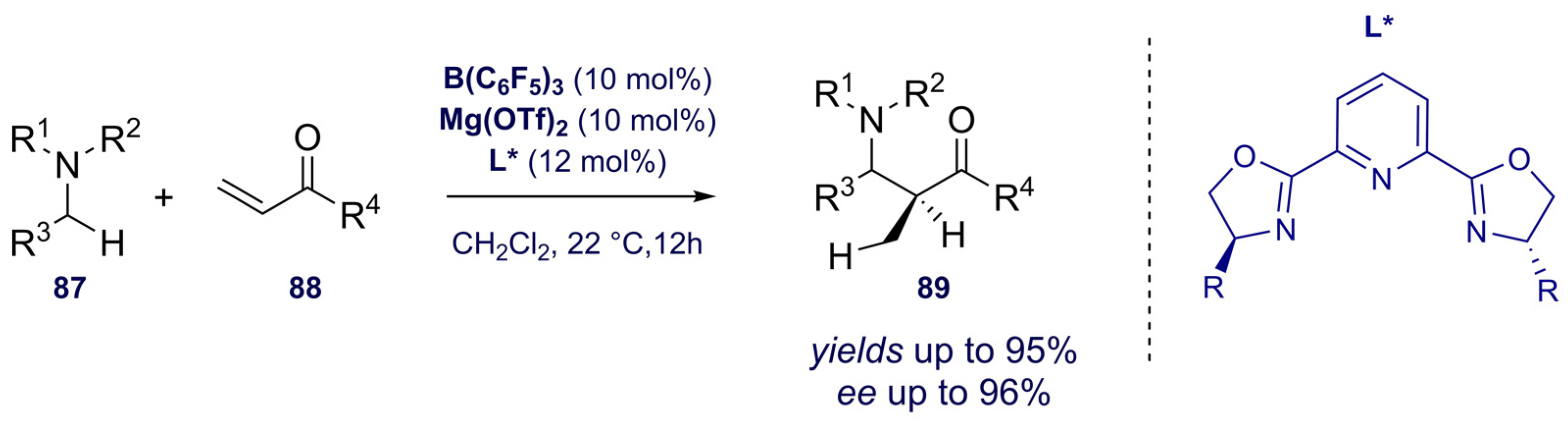
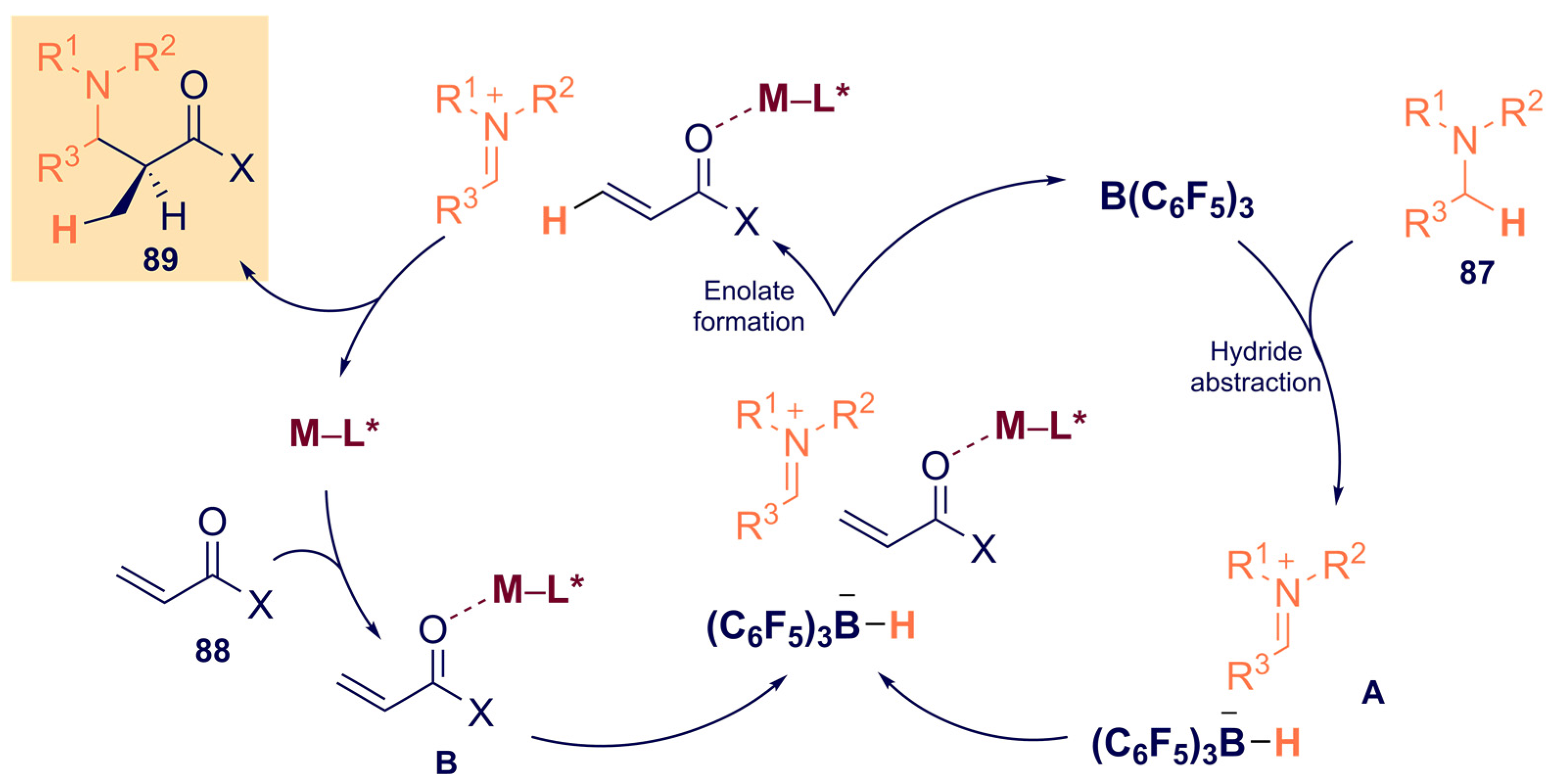
- Shang, M.; Chan, J.Z.; Cao, M.; Chang, Y.; Wang, Q.; Cook, B.; Torker, S.; Wasa, M. C-H Functionalization of Amines via Alkene-Derived Nucleophiles through Cooperative Action of Chiral and Achiral Lewis Acid Catalysts: Applications in Enantioselective Synthesis. J. Am. Chem. Soc. 2018, 140, 10593–10601. [Google Scholar] [CrossRef]
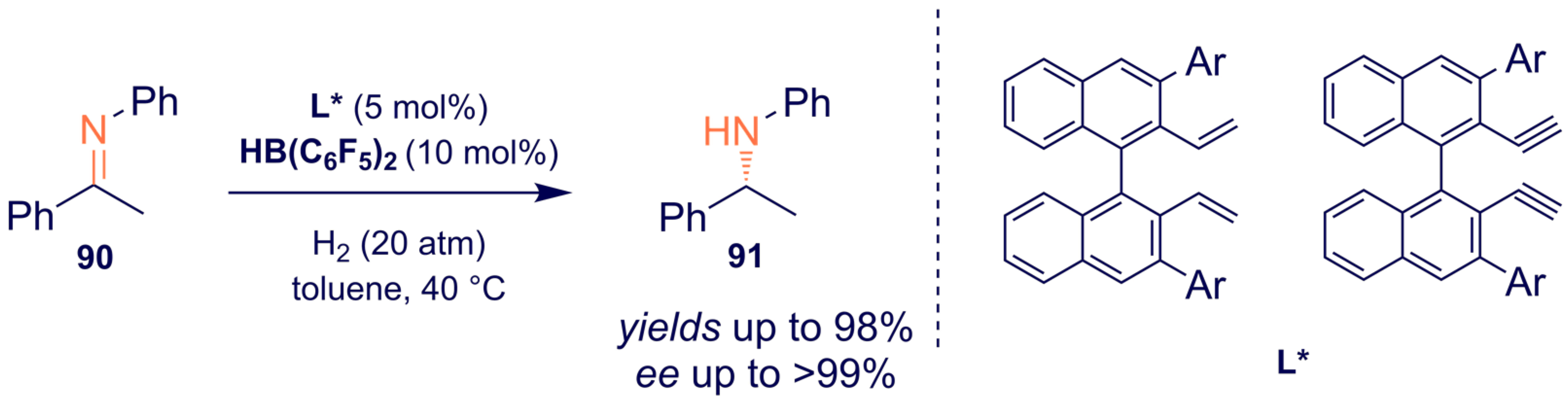
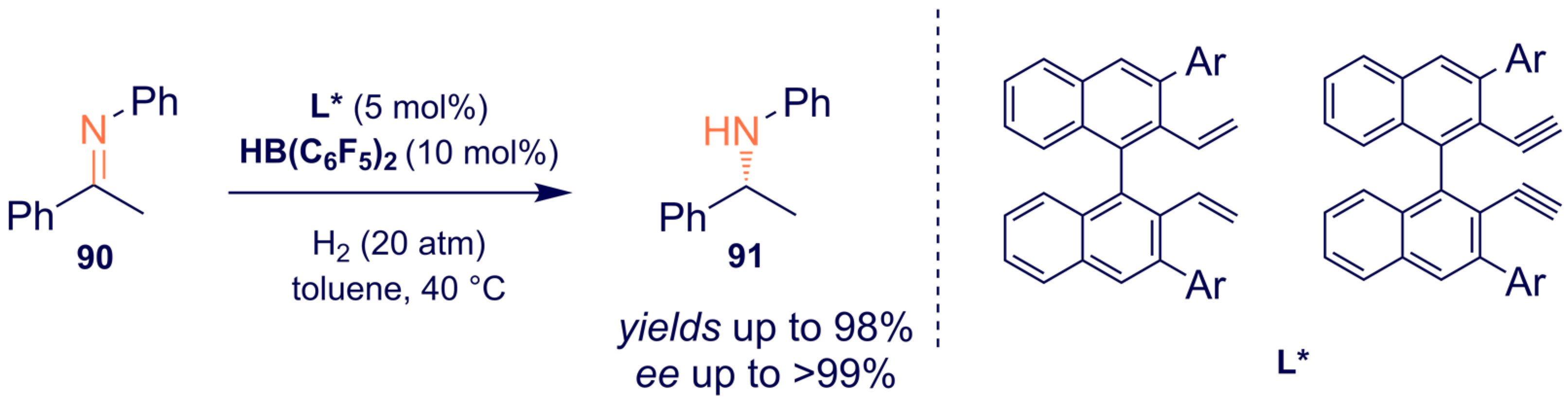
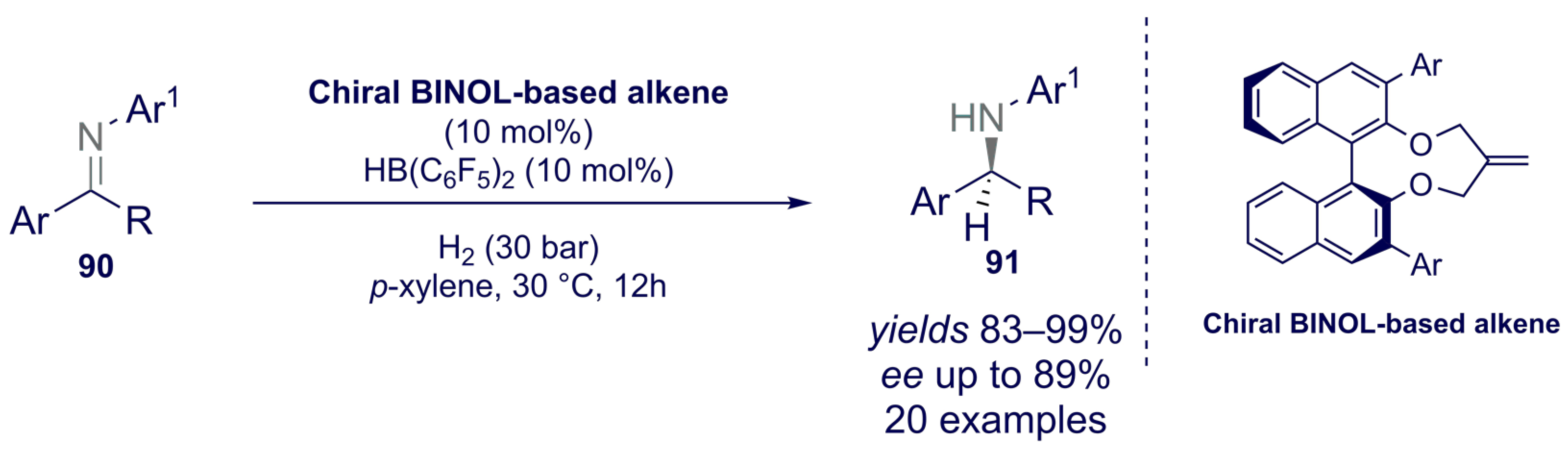
- Meng, W.; Feng, X.; Du, H. Frustrated Lewis Pairs Catalyzed Asymmetric Metal-Free Hydrogenations and Hydrosilylations. Acc. Chem. Res. 2017, 51, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, T.; Meng, W.; Du, H. Asymmetric hydrogenation of imines with chiral alkene-derived boron Lewis acids. Org. Biomol. Chem. 2018, 16, 8686–8689. [Google Scholar] [CrossRef] [PubMed]
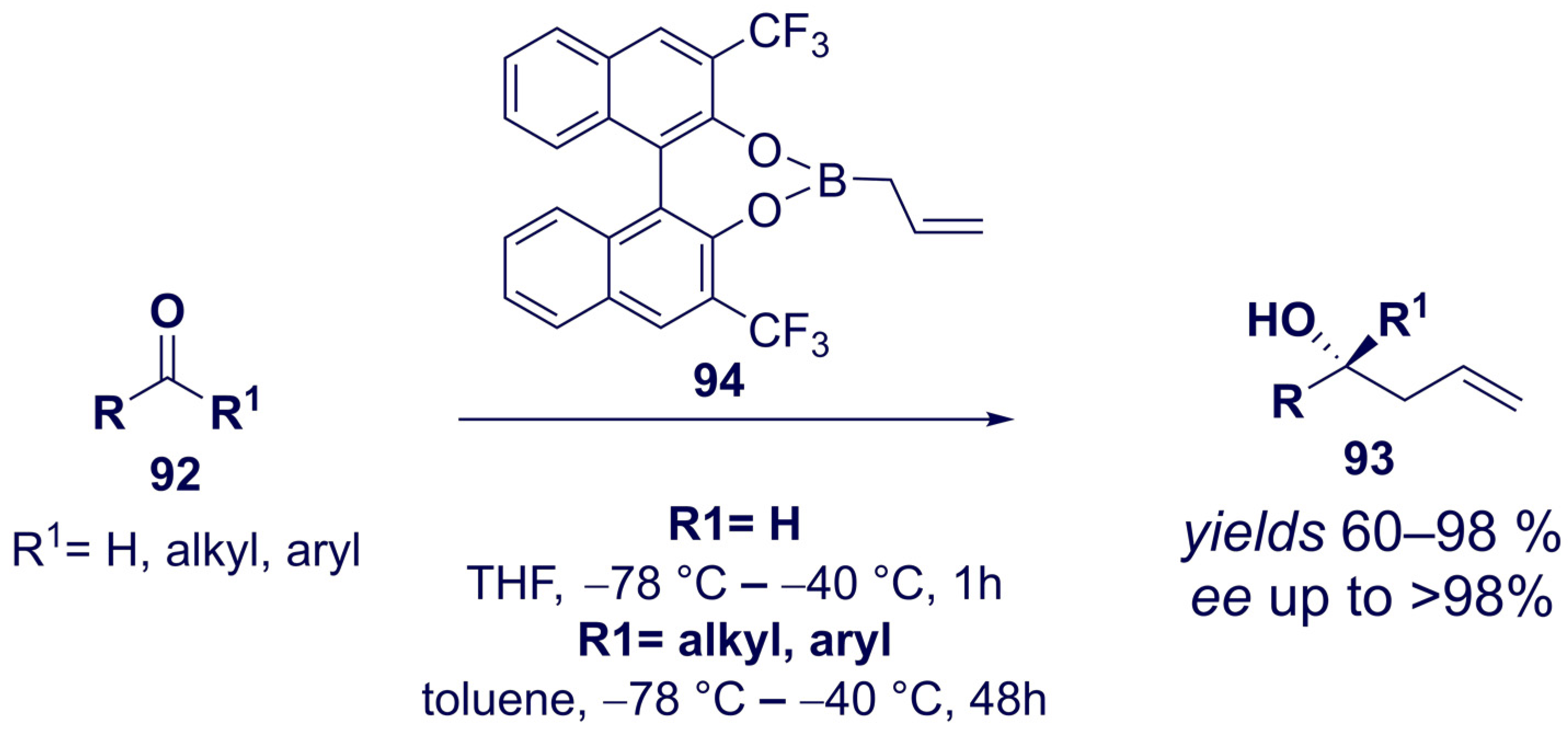
- Wu, T.R.; Shen, L.; Chong, J.M. Asymmetric Allylboration of Aldehydes and Ketones Using 3,3‘-Disubstitutedbinaphthol-Modified Boronates. Org. Lett. 2004, 6, 2701–2704. [Google Scholar] [CrossRef]




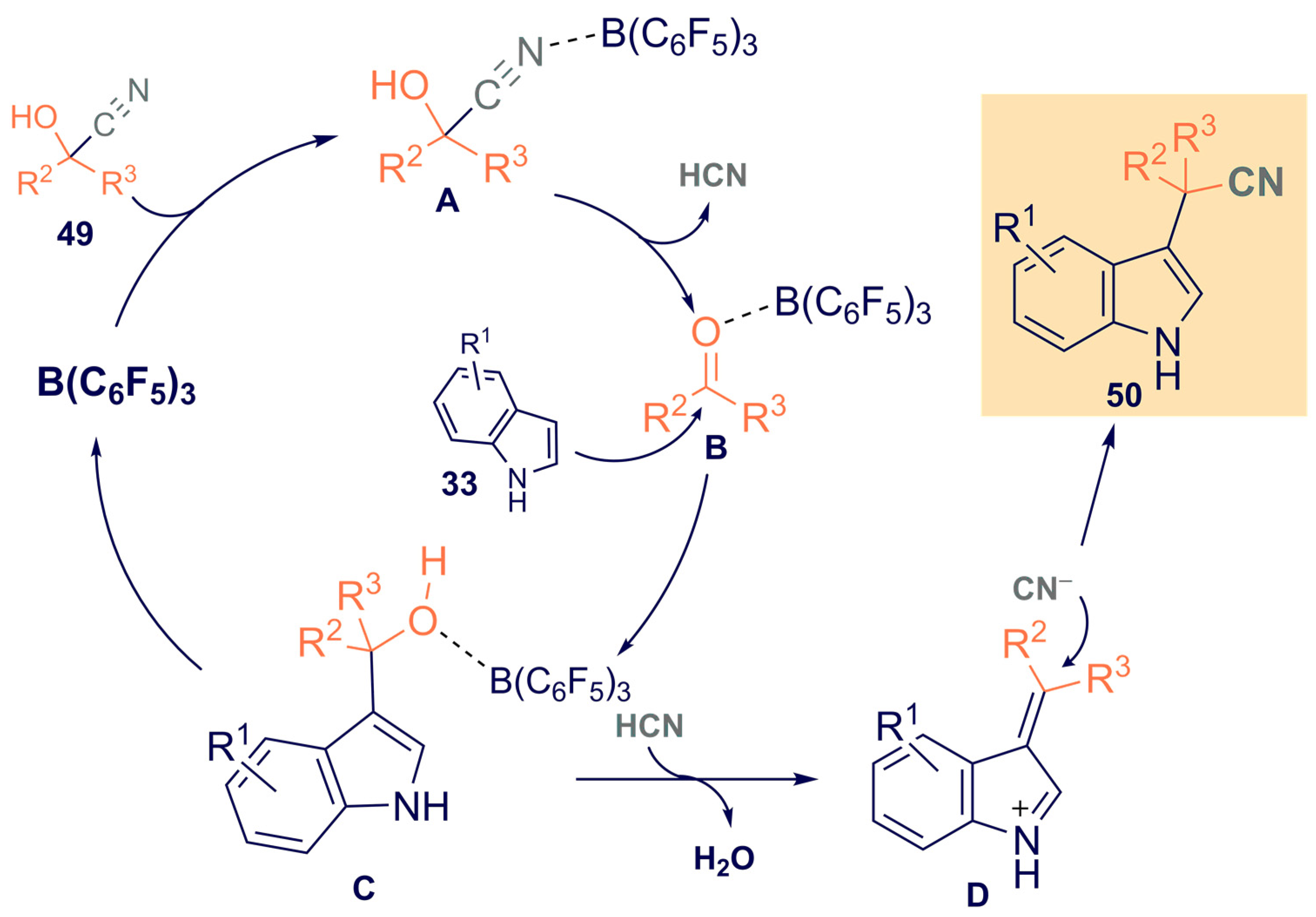

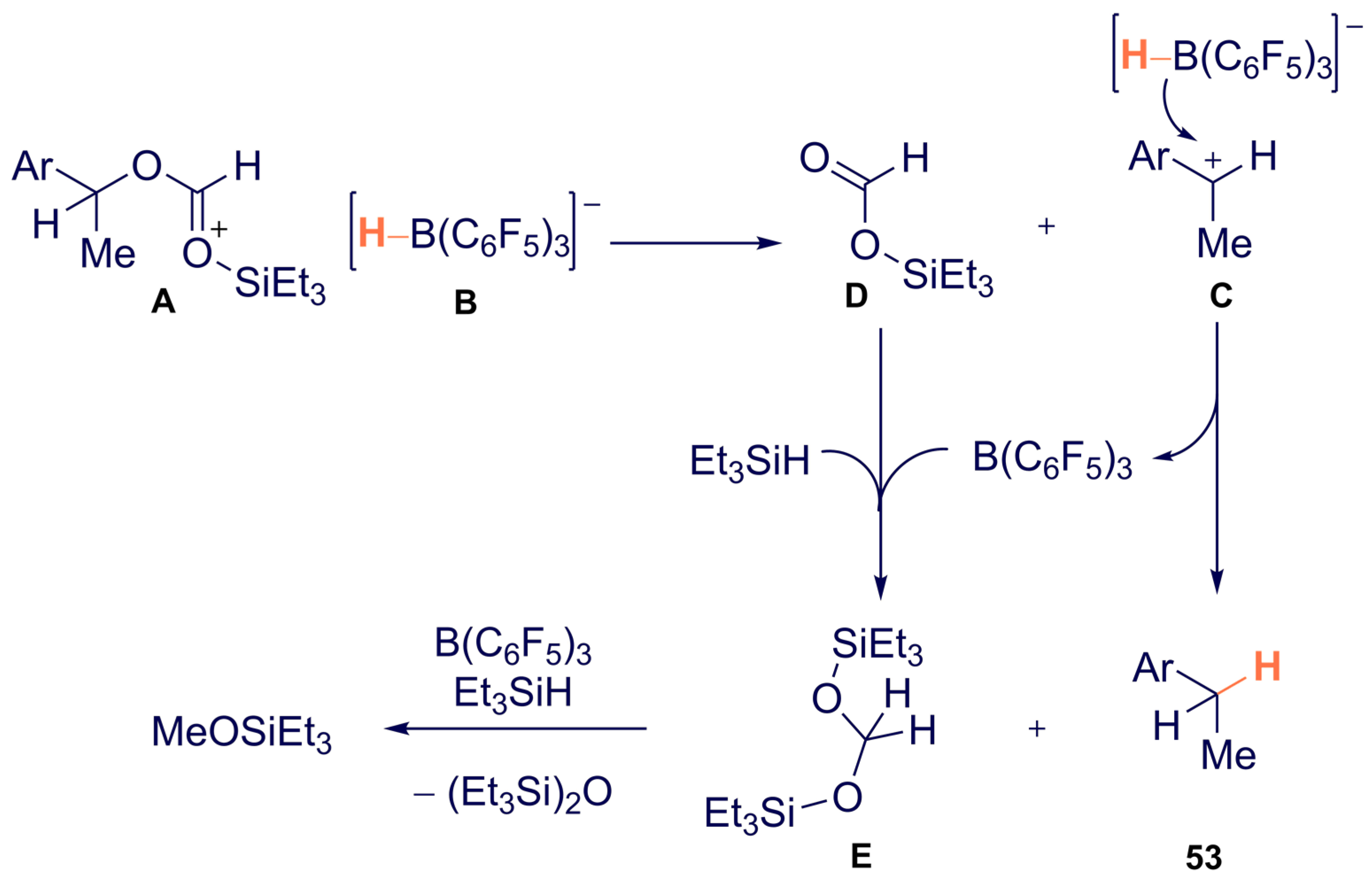
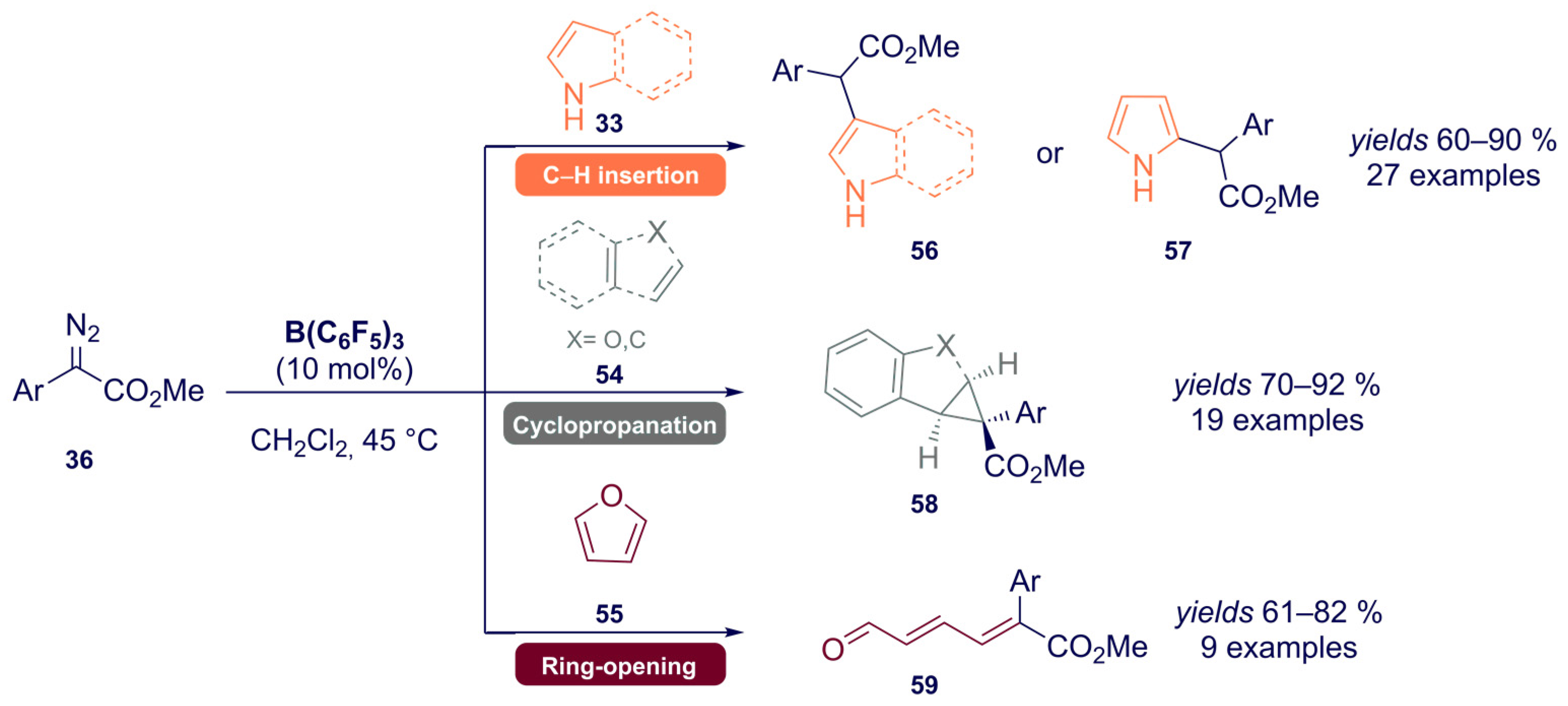
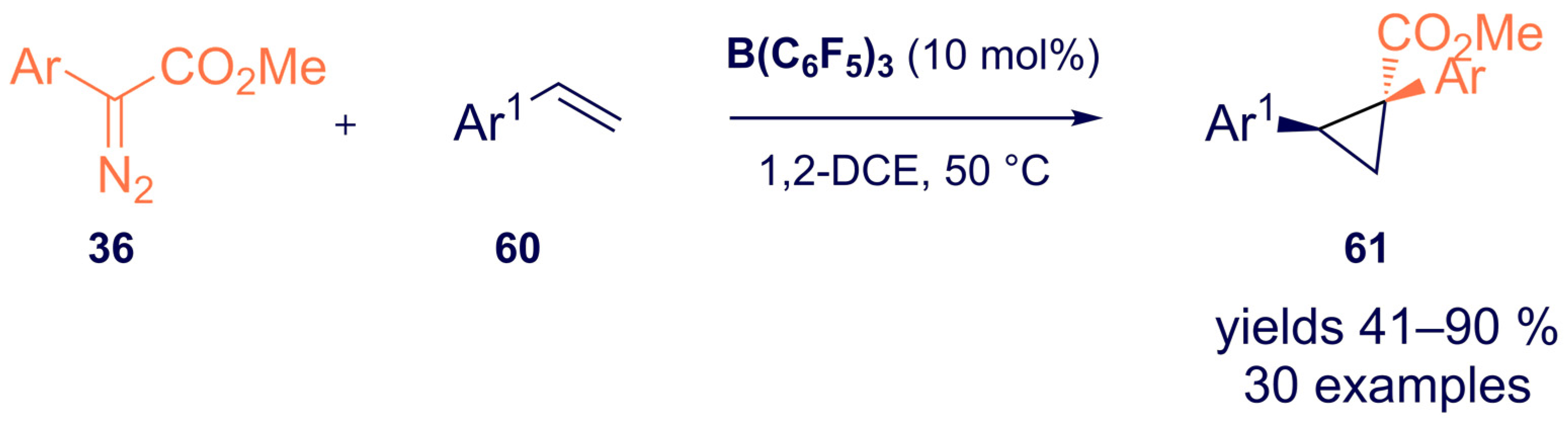





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nori, V.; Pesciaioli, F.; Sinibaldi, A.; Giorgianni, G.; Carlone, A. Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts 2022, 12, 5. https://doi.org/10.3390/catal12010005
Nori V, Pesciaioli F, Sinibaldi A, Giorgianni G, Carlone A. Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts. 2022; 12(1):5. https://doi.org/10.3390/catal12010005
Chicago/Turabian StyleNori, Valeria, Fabio Pesciaioli, Arianna Sinibaldi, Giuliana Giorgianni, and Armando Carlone. 2022. "Boron-Based Lewis Acid Catalysis: Challenges and Perspectives" Catalysts 12, no. 1: 5. https://doi.org/10.3390/catal12010005
APA StyleNori, V., Pesciaioli, F., Sinibaldi, A., Giorgianni, G., & Carlone, A. (2022). Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts, 12(1), 5. https://doi.org/10.3390/catal12010005