
PT-BI Co-Deposit Shell on AU Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
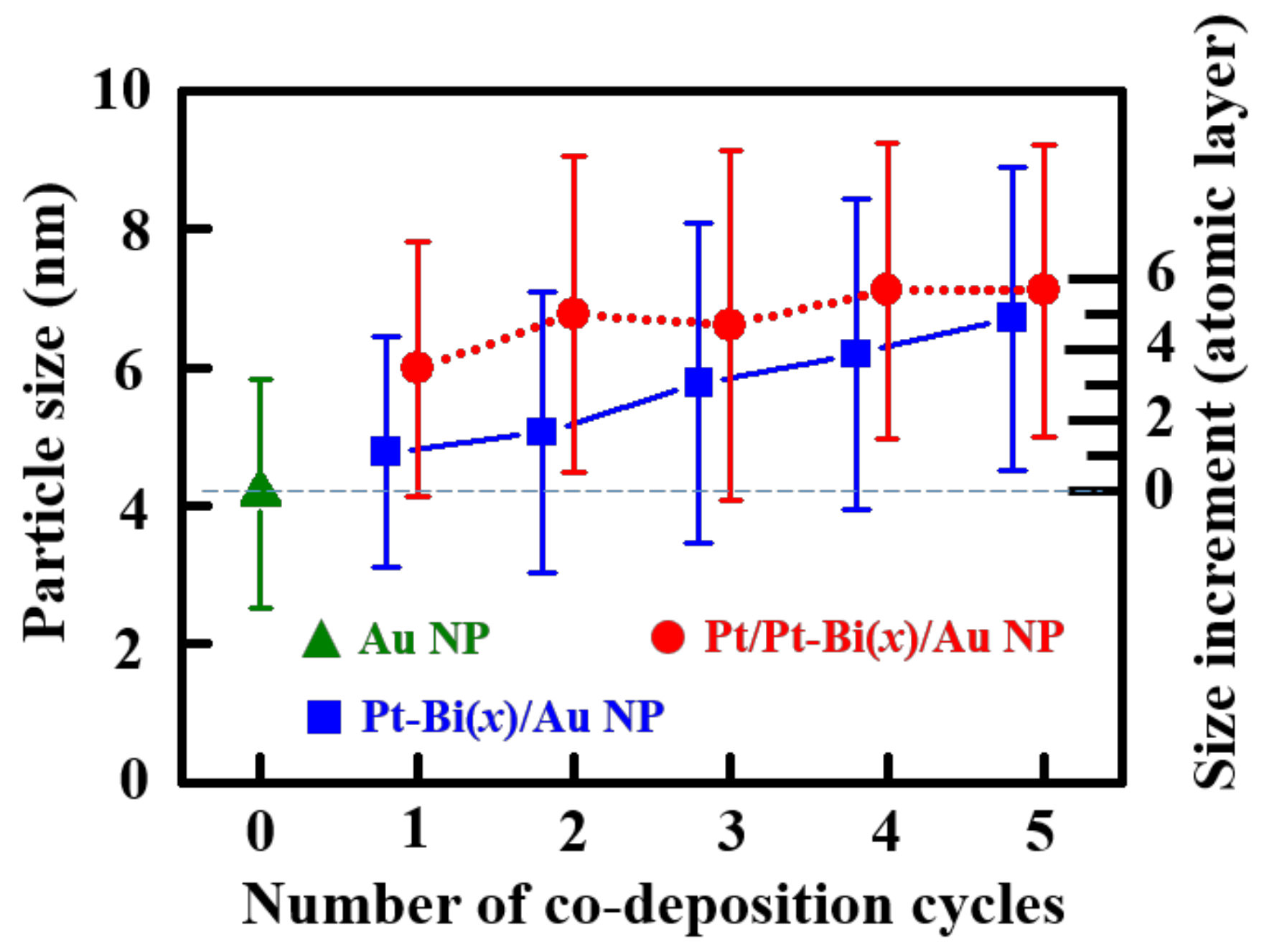
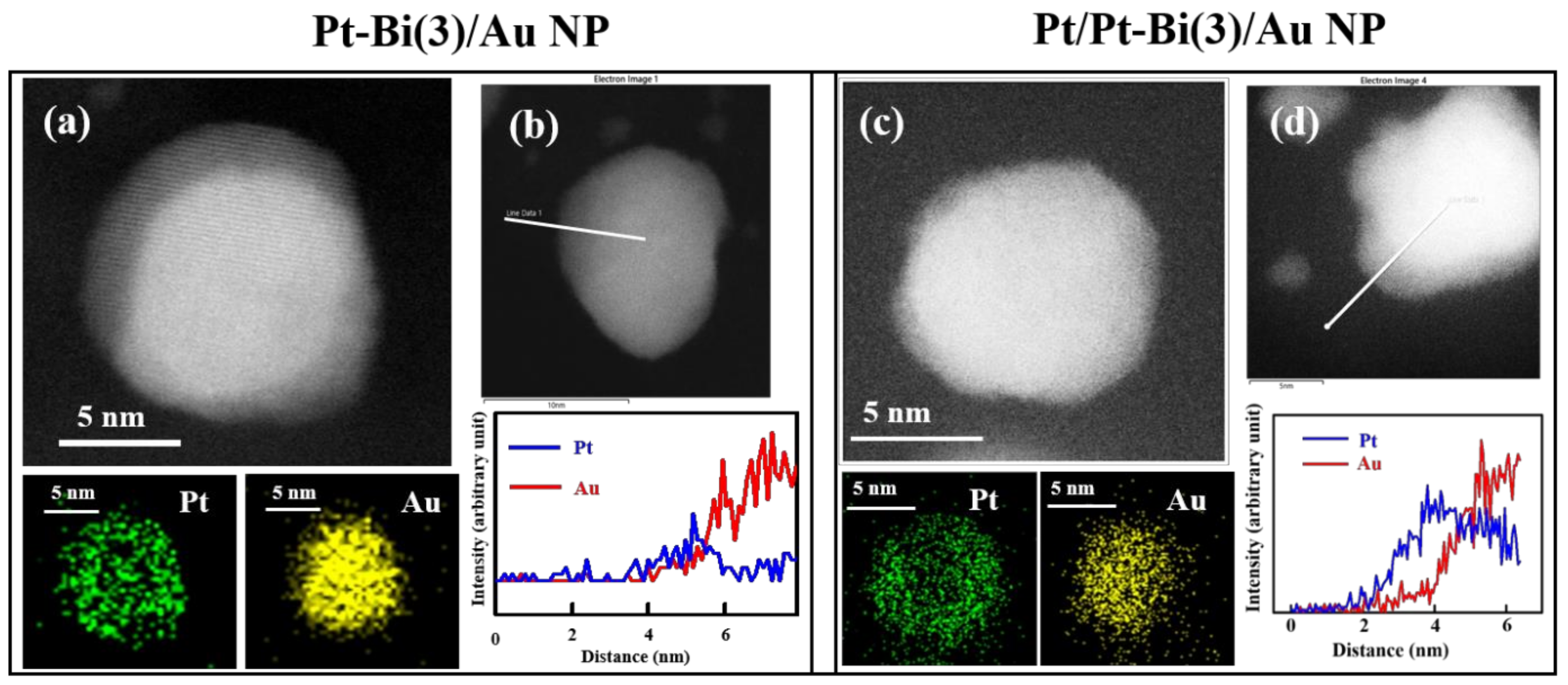
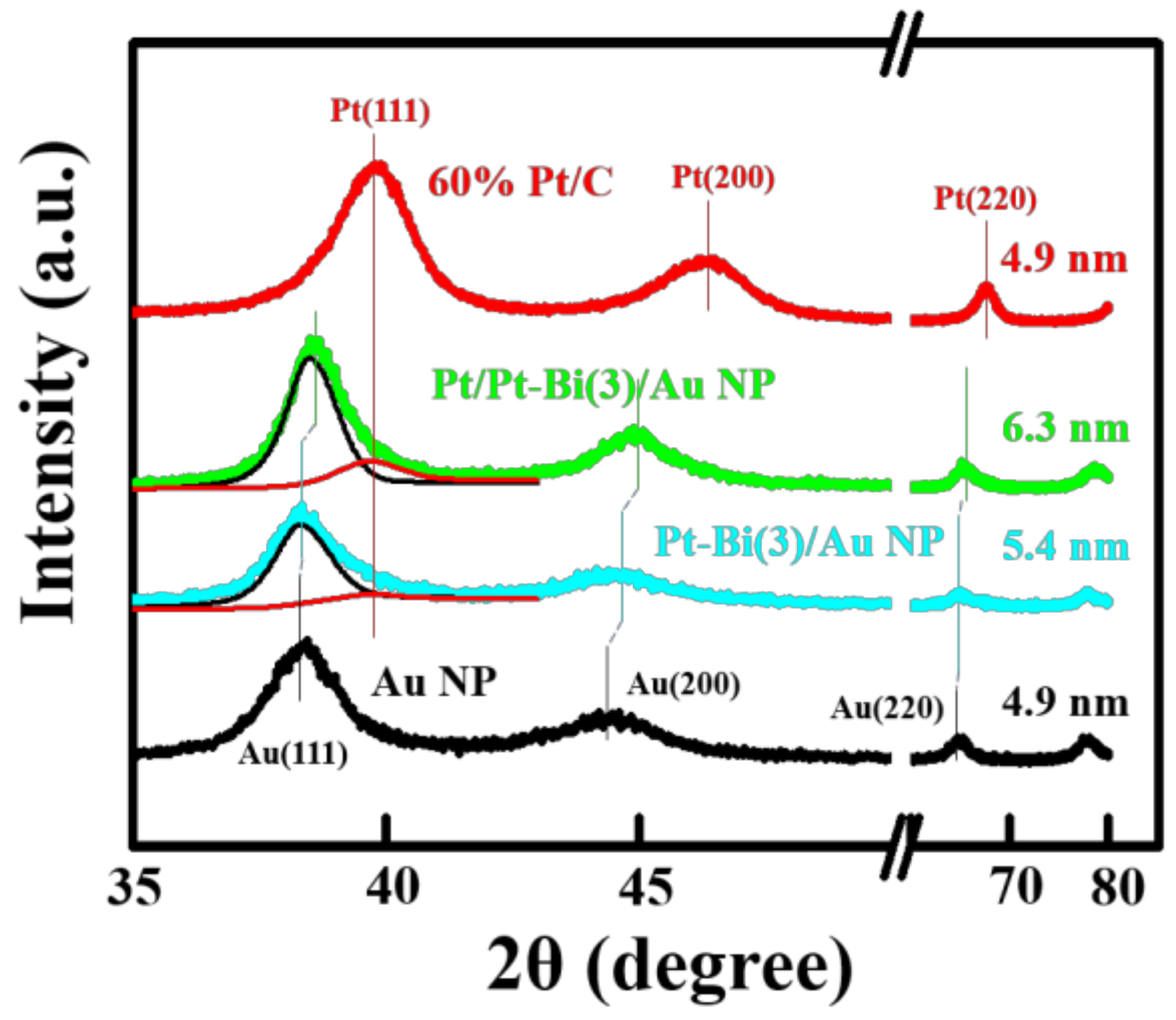
2.1. Structural Analysis
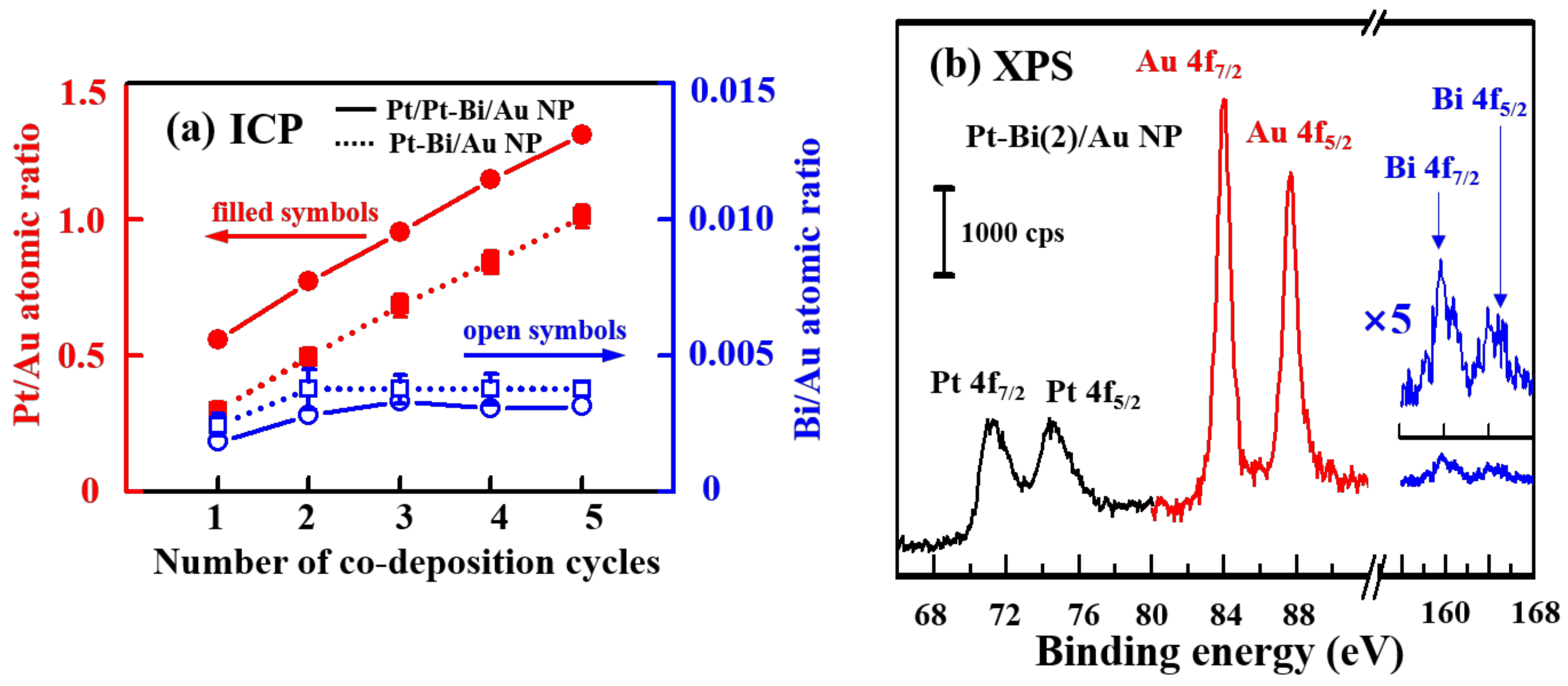
2.2. Compositional Analysis
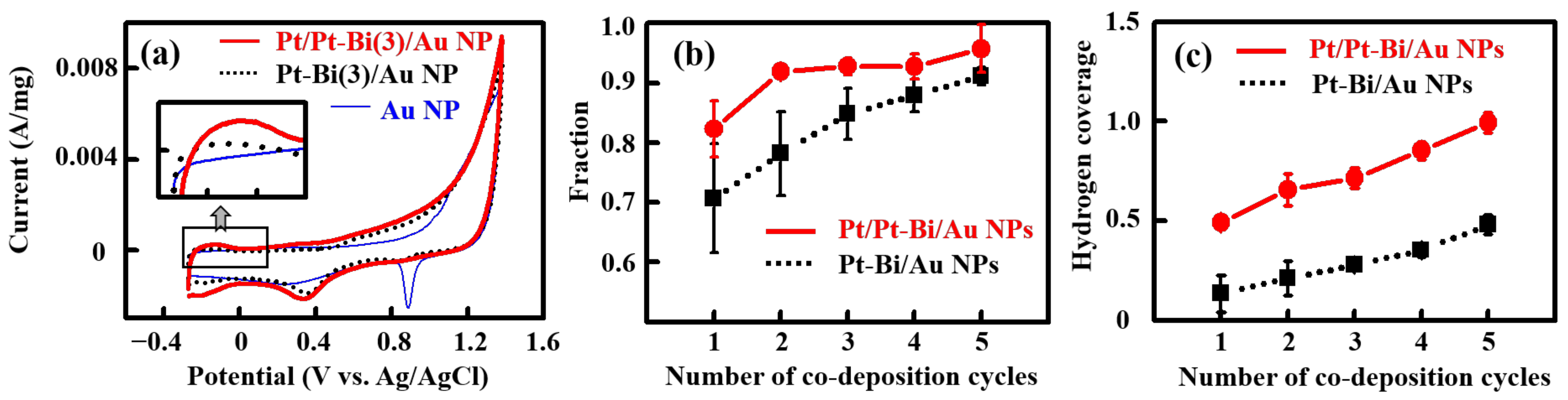
2.3. Electrochemical Behavior
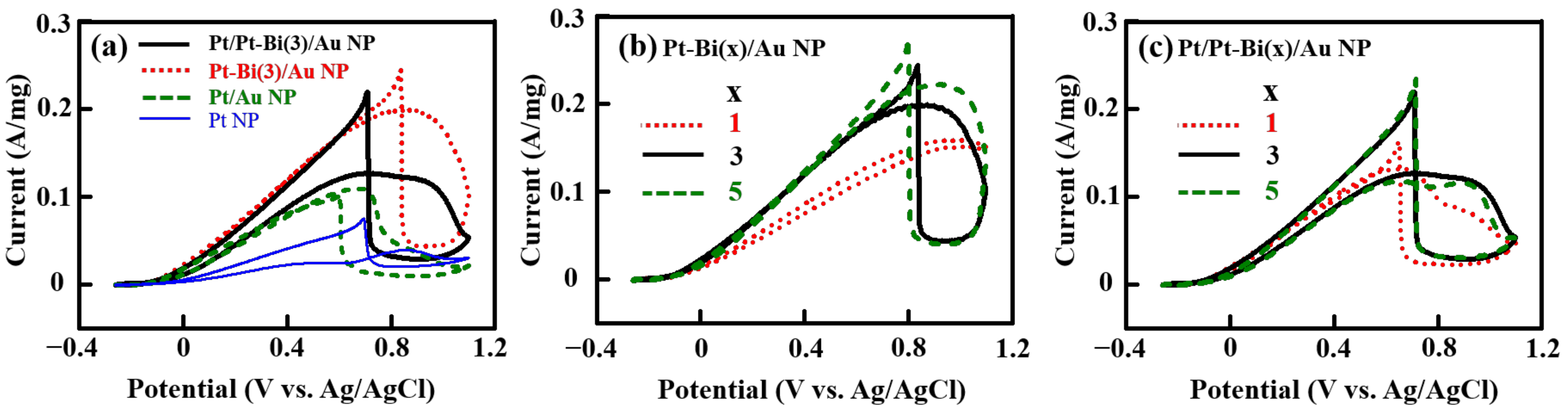
2.4. FAO Behavior
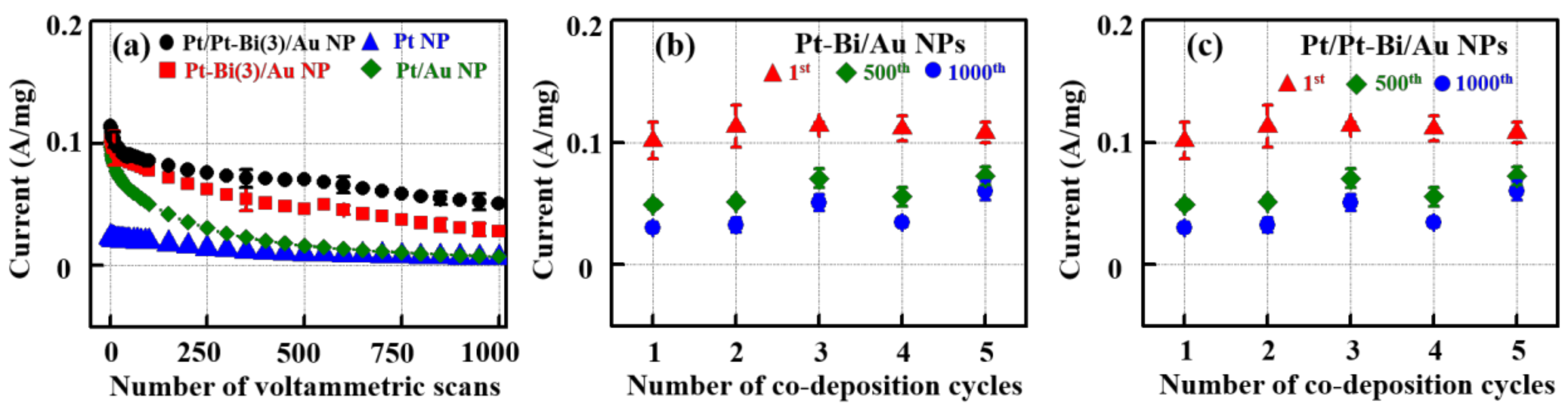
2.5. Current Decays in Various FAO Catalysts
3. Materials and Methods
3.1. Nanoparticles
3.2. Co-Deposition of Pt and Bi Using Irreversible Adsorption
3.3. Electrochemical Measurements
3.4. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Enthaler, S.; Langermann, J.V.; Schmidt, T. Carbon dioxide and formic acid—the couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010, 3, 1207–1217. [Google Scholar] [CrossRef]
- Peng, Y.P.; Yeh, Y.T.; Shah, S.I.; Huang, C.P. Concurrent photoelectrochemical reduction of CO2 and oxidation of methyl orange using nitrogen-doped TiO2. Appl. Catal. B 2012, 123–124, 414–423. [Google Scholar] [CrossRef]
- Jhong, H.R.M.; Ma, S.; Kenis, P.J.A. Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges and future opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar] [CrossRef]
- Qin, G.; Zhang, Y.; Ke, X.; Tong, X.; Sun, Z.; Liang, M.; Xue, S. Photocatalytic reduction of carbon dioxide to formic acid, formaldehyde, and methanol using dye-sensitized TiO2 film. Appl. Catal. B 2013, 129, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Lyu, L.; Zeng, X.; Yun, J.; Wei, F.; Jin, F. No catalyst addition and highly efficient dissociation of H2O for the reduction of CO2 to formic acid with Mn. Environ. Sci. Technol. 2014, 48, 6003–6009. [Google Scholar] [CrossRef]
- Won, D.H.; Choi, C.H.; Chung, J.; Woo, S.I. Photoelectrochemical production of formic acid and methanol from carbon dioxide on metal-decorated CuO/Cu2O-layered thin films under visible light irradiation. Appl. Catal. B 2014, 158–159, 217–223. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured Tin Catalysts for Selective Electrochemical Reduction of Carbon Dioxide to Formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef]
- Baran, T.; Wojtyła, S.; Dibenedetto, A.; Aresta, M.; Macyk, W. Zinc sulfide functionalized with ruthenium nanoparticles for photocatalytic reduction of CO2. Appl. Catal. B. 2015, 178, 170–176. [Google Scholar] [CrossRef]
- Kortlever, R.; Peters, I.; Koper, S.; Koper, M.T.M. Electrochemical CO2 reduction to formic acid at Low Overpotential and with high faradaic efficiency on carbonsupported bimetallic Pd–Pt nanoparticles. ACS Catal. 2015, 5, 3916–3923. [Google Scholar] [CrossRef]
- Yoshitomi, F.; Sekizawa, K.; Maeda, K.; Ishitani, O. Selective formic acid production via CO2 reduction with visible light using a hybrid of a perovskite tantalum oxynitride and a binuclear ruthenium(II) complex. ACS Appl. Mater. Interfaces 2015, 7, 13092–13097. [Google Scholar] [CrossRef]
- Bitar, Z.; Fecant, A.; Trela-Baudot, E.; Chardon-Noblat, S.; Pasquier, D. Electrocatalytic reduction of carbon dioxide on indium coated gas diffusion electrodes—comparison with indium foil. Appl. Catal. B 2016, 189, 172–180. [Google Scholar] [CrossRef]
- Kayan, D.B.; Köleli, F. Simultaneous electrocatalytic reduction of dinitrogen and carbon dioxide on conducting polymer electrodes. Appl. Catal. B 2016, 181, 88–93. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Su, T.; Tian, H.; Qin, Z.; Ji, H. Preparation and characterization of Cu modified BiYO3 for carbon dioxide reduction to formic acid. Appl. Catal. B 2017, 202, 364–373. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Zhang, D.; Li, G.; Li, H. Microwave irradiation induced UIO-66-NH2 anchored on graphene with high activity for photocatalytic reduction of CO2. Appl. Catal. B 2018, 228, 47–53. [Google Scholar] [CrossRef]
- Ha, S.; Larsen, R.; Zhu, Y.; Masel, R.I. Direct Formic Acid Fuel Cells with 600 mA cm–2 at 0.4 V and 22 °C. Fuel Cells 2004, 4, 337–343. [Google Scholar] [CrossRef]
- Ha, S.; Dunbar, Z.; Masel, R.I. Characterization of a high performing passive direct formic acid fuel cell. J. Power Sources 2006, 158, 129–136. [Google Scholar] [CrossRef]
- Chen, W.; Kim, J.; Sun, S.H.; Chen, S.W. Electro-oxidation of formic acid catalyzed by FePt nanoparticles. Phys. Chem. Chem. Phys. 2006, 8, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Tian, N.; Zhou, Z.Y.; Sun, S.G.; Ding, Y.; Wang, Z.L. Synthesis of Tetrahexahedral Platinum Nanocrystals with High-Index Facets and High Electro-Oxidation Activity. Science 2007, 316, 732–735. [Google Scholar] [CrossRef]
- Chen, W.; Kim, J.M.; Xu, L.P.; Sun, S.H.; Chen, S.W. Langmuir−Blodgett Thin Films of Fe20Pt80 Nanoparticles for the Electrocatalytic Oxidation of Formic Acid. J. Phys. Chem. C 2007, 111, 13452–13459. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.C.; Holt-Hindle, P. Platinum-Based Nanostructured Materials: Synthesis, Properties, and Applications. Chem. Rev. 2010, 110, 3767–3804. [Google Scholar] [CrossRef]
- Ren, M.; Kang, Y.; He, W.; Zou, Z.; Xue, X.; Akins, D.L.; Yang, H.; Feng, S. Origin of performance degradation of palladium-based direct formic acid fuel cells. Appl. Catal. B 2011, 104, 49–53. [Google Scholar] [CrossRef]
- Yuan, D.W.; Liu, Z.R. Atomic ensemble effects on formic acid oxidation on PdAu electrode studied by first-principles calculations. J. Power Sources 2013, 224, 241–249. [Google Scholar] [CrossRef]
- Adams, B.D.; Asmussen, R.M.; Ostrom, C.K.; Chen, A. Synthesis and Comparative Study of Nanoporous Palladium-Based Bimetallic Catalysts for Formic Acid Oxidation. J. Phys. Chem. C 2014, 118, 29903–29910. [Google Scholar] [CrossRef]
- Lu, Y.; Du, S.; Steinberger-Wilckens, R. One-dimensional nanostructured electrocatalysts for polymer electrolyte membrane fuel cells—A review. Appl. Catal. B 2016, 199, 292–314. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, M.; Li, H.; Li, Y.; Ye, J.; Cao, Z.; Fang, M.; Kuang, Q.; Zheng, J.; Xie, Z. Stable palladium hydride as a superior anode electrocatalyst for direct formic acid fuel cells. Nano Energy 2018, 44, 127–134. [Google Scholar] [CrossRef]
- Hu, S.; Che, F.; Khorasani, B.; Jeon, M.; Yoon, C.W.; McEwen, J.S.; Scudiero, L.; Ha, S. Improving the electrochemical oxidation of formic acid by tuning the electronic properties of Pd-based bimetallic nanoparticles. Appl. Catal. B 2019, 254, 685–692. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes part III. Intermediates and mechanism on platinum electrodes. J. Electroanal. Chem. 1973, 45, 205–231. [Google Scholar] [CrossRef]
- Rice, C.; Ha, S.; Masel, R.I.; Waszczuk, P.; Wieckowski, A.; Barnard, T. Direct formic acid fuel cells. J. Power Sources 2002, 111, 83–89. [Google Scholar] [CrossRef]
- Lović, J.D.; Tripković, A.V.; Gojković, S.L.; Popović, K.D.; Tripković, D.V.; Olszewski, P.; Kowal, A. Kinetic study of formic acid oxidation on carbon-supported platinum electrocatalyst. J. Electroanal. Chem. 2005, 581, 294–302. [Google Scholar] [CrossRef]
- Gao, W.; Keith, J.A.; Anton, J.; Jacob, T. Theoretical elucidation of the competitive electro-oxidation mechanisms of formic acid on Pt(111). J. Am. Chem. Soc. 2010, 132, 18377–18385. [Google Scholar] [CrossRef]
- Boronat-González, A.; Herrero, E.; Feliu, J.M. Heterogeneous electrocatalysis of formic acid oxidation on platinum single crystal electrodes. Curr. Opin. Electrochem. 2017, 4, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Pickup, P.G. Screening of PdM and PtM catalysts in a multi-anode direct formic acid fuel cell. J. Appl. Electrochem. 2011, 41, 589–597. [Google Scholar] [CrossRef]
- Zhang, B.W.; He, C.L.; Jiang, Y.X.; Chen, M.H.; Li, Y.Y.; Rao, L.; Sun, S.G. High activity of PtBi intermetallics supported on mesoporous carbon towards HCOOH electro-oxidation. Electrochem. Commun. 2012, 25, 105–108. [Google Scholar] [CrossRef]
- Shen, L.; Li, H.; Lu, L.; Luo, Y.; Tang, Y.; Chen, Y.; Lu, T. Improvement and mechanism of electrocatalytic performance of Pd–Ni/C anodic catalyst in direct formic acid fuel cell. Electrochim. Acta 2013, 89, 497–502. [Google Scholar] [CrossRef]
- Jiang, K.; Cai, W.B. Carbon supported Pd-Pt-Cu nanocatalysts for formic acid electrooxidation: Synthetic screening and componential functions. Appl. Catal. B 2014, 147, 185–192. [Google Scholar] [CrossRef]
- Wu, D.; Cao, M.; Shen, M.; Cao, R. Sub-5 nm Pd–Ru nanoparticle alloys as efficient catalysts for formic acid electrooxidation. ChemCatChem. 2014, 6, 1731–1736. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Wang, X.; Manthiram, A. Highly active Pd and Pd–Au nanoparticles supported on functionalized graphene nanoplatelets for enhanced formic acid oxidation. RSC Adv. 2014, 4, 4028–4033. [Google Scholar] [CrossRef]
- Zhang, B.W.; Jiang, Y.X.; Ren, J.; Qu, X.M.; Xu, G.L.; Sun, S.G. PtBi intermetallic and PtBi intermetallic with the Bi-rich surface supported on porous graphitic carbon towards HCOOH electro-oxidation. Electrochim. Acta 2015, 162, 254–262. [Google Scholar] [CrossRef]
- Szumełda, T.; Drelinkiewicz, A.; Lalik, E.; Kosydar, R.; Duraczyńska, D.; Gurgul, J. Carbon-supported Pd100-xAux alloy nanoparticles for the electrocatalytic oxidation of formic acid: Influence of metal particles composition on activity enhancement. Appl. Catal. B 2018, 221, 393–405. [Google Scholar] [CrossRef]
- Yan, X.; Hu, X.; Fu, G.; Xu, L.; Lee, J.M.; Tang, Y. Facile Synthesis of Porous Pd3Pt Half-Shells with Rich “Active Sites” as Efficient Catalysts for Formic Acid Oxidation. Small 2018, 14, 1703940. [Google Scholar] [CrossRef]
- Song, J.; Xiao, Z.; Jiang, Y.; Abdelhafiz, A.A.; Chang, I.; Zeng, J. Surfactant-free room temperature synthesis of PdxPty/C assisted by ultra-sonication as highly active and stable catalysts for formic acid oxidation. Int. J. Hydrog. Energy 2019, 44, 11655–11663. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yu, Z.Y.; Li, G.; Song, Q.T.; Li, G.; Luo, C.X.; Yin, S.H.; Lu, B.A.; Xiao, C.; Xu, B.B.; et al. Intermetallic PtBi Nanoplates with High Catalytic Activity towards Electro-oxidation of Formic Acid and Glycerol. ChemElectroChem 2020, 7, 239–245. [Google Scholar] [CrossRef]
- Watanabe, M.; Horiuchi, M.; Motoo, S. Electrocatalysis by ad-atoms: Part XXIII. Design of platinum ad-electrodes for formic acid fuel cells with ad-atoms of the IVth and the Vth groups. J. Electroanal. Chem. Interfacial Electrochem. 1988, 250, 117–125. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Behm, R.J.; Grgur, B.N.; Markovic, N.M.; Ross, P.N. Formic acid oxidation on pure and Bi-modified Pt(111): Temperature effects. Langmuir 2000, 16, 8159–8166. [Google Scholar] [CrossRef]
- Peng, B.; Wang, J.Y.; Zhang, H.X.; Lin, Y.H.; Cai, W.B. A versatile electroless approach to controlled modification of Sb on Pt surfaces towards efficient electrocatalysis of formic acid. Electrochem. Commun. 2009, 11, 831–833. [Google Scholar] [CrossRef]
- Wang, J.Y.; Kang, Y.Y.; Yang, H.; Cai, W.B. Boron-doped palladium nanoparticles on carbon black as a superior catalyst for formic acid electro-oxidation. J. Phys. Chem. C 2009, 113, 8366–8372. [Google Scholar] [CrossRef]
- Haan, J.L.; Stafford, K.M.; Masel, R.I. Effects of the addition of Antimony, tin, and lead to palladium catalyst formulations for the direct formic acid fuel cell. J. Phys. Chem. C 2010, 114, 11665–11672. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Pb and Sb modified Pt/C catalysts for direct formic acid fuel cells. Electrochim. Acta 2010, 55, 7354–7361. [Google Scholar] [CrossRef]
- Peng, B.; Wang, H.F.; Liu, Z.P.; Cai, W.B. Combined surface-enhanced infrared spectroscopy and first-principles study on electro-oxidation of formic acid at Sb-modified Pt electrodes. J. Phys. Chem. C 2011, 114, 3102–3107. [Google Scholar] [CrossRef]
- Dandan, T.; Wu, B.; Wang, B.; Deng, C.; Gao, Y. A highly active carbon-supported PdSn catalyst for formic acid electrooxidation. Appl. Catal. B 2011, 103, 163–168. [Google Scholar] [CrossRef]
- Si, F.; Ge, J.; Li, C.; Liang, L.; Liu, C.; Xing, W. Investigations of Pt modified Pd/C catalyst synthesized by one-pot galvanic replacement for formic acid electrooxidation. Hydrogen Energy 2014, 39, 2489–2496. [Google Scholar] [CrossRef]
- Beltowska-Brzezinska, M.; Łuczak, T.; Stelmach, J.; Holze, R. The electrooxidation mechanism of formic acid on platinum and on lead ad-atoms modified platinum studied with the kinetic isotope effect. J. Power Sources 2014, 251, 30–37. [Google Scholar] [CrossRef]
- Ferre-Vilaplana, A.; Perales-Rondón, J.V.; Feliu, J.M.; Herrero, E. Understanding the effect of the adatoms in the formic acid oxidation mechanism on Pt(111) electrodes. ACS Catal. 2015, 5, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, A.M.; El-Nagar, G.A.; Al-Akraa, I.M.; El-Deab, M.S.; El-Anadouli, B.E. Towards improving the catalytic activity and stability of platinum-based anodes in direct formic acid fuel cells. Int. J. Hydrog. Energy 2015, 40, 7808–7816. [Google Scholar] [CrossRef]
- Kim, J.; Jung, C.; Rhee, C.K.; Lim, T.H. Electrocatalytic Oxidation of Formic Acid and Methanol on Pt Deposits on Au(111). Langmuir 2007, 23, 10831–10836. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, Y.H.; Xu, B.Q. Pt Flecks on Colloidal Au (Pt∧Au) as Nanostructured Anode Catalysts for Electrooxidation of Formic Acid. J. Phys. Chem. C 2009, 113, 20903–20911. [Google Scholar] [CrossRef]
- Kim, S.; Jung, C.; Kim, J.; Rhee, C.K.; Choi, S.M.; Lim, T.H. Modification of Au Nanoparticles Dispersed on Carbon Support Using Spontaneous Deposition of Pt toward Formic Acid Oxidation. Langmuir 2010, 26, 4497–4505. [Google Scholar] [CrossRef]
- Yin, M.; Huang, Y.; Lv, Q.; Liang, L.; Liao, J.; Liu, C.; Xing, W. Improved direct electrooxidation of formic acid by increasing Au fraction on the surface of PtAu alloy catalyst with heat treatment. Electrochim. Acta 2011, 58, 6–11. [Google Scholar] [CrossRef]
- Chen, G.; Li, Y.; Wang, D.; Zheng, L.; You, G.; Zhong, C.J.; Yang, L.; Cai, F.; Cai, J.; Chen, B.H. Carbon-supported PtAu alloy nanoparticle catalysts for enhanced electrocatalytic oxidation of formic acid. J. Power Sources 2011, 196, 8323–8330. [Google Scholar] [CrossRef]
- Zhang, G.R.; Zhao, D.; Feng, Y.Y.; Zhang, B.; Su, D.S.; Liu, G.; Xu, B.Q. Catalytic Pt-on-Au nanostructures: Why Pt Becomes more active on smaller Au particles. ACS Nano 2012, 6, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Qi, Y.; Deng, M. The ensemble effect of formic acid oxidation on platinum-gold electrode studied by first-principles calculations. J. Power Sources 2015, 278, 203–212. [Google Scholar] [CrossRef]
- Ahn, S.H.; Liu, Y.; Moffat, T.P. Ultrathin Platinum Films for Methanol and Formic Acid Oxidation: Activity as a Function of Film Thickness and Coverage. ACS Catal. 2015, 5, 2124–2136. [Google Scholar] [CrossRef]
- Li, D.; Meng, F.; Wang, H.; Jiang, X.; Zhu, Y. Nanoporous AuPt alloy with low Pt content: A remarkable electrocatalyst with enhanced activity towards formic acid electro-oxidation. Electrochim. Acta 2016, 190, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Duan, T.; Zhang, R.; Ling, L.; Wang, B. Insights into the effect of Pt atomic ensemble on HCOOH oxidation over Pt-decorated Au bimetallic catalyst to maximize Pt utilization. J. Phys. Chem. C 2016, 120, 2234–2246. [Google Scholar] [CrossRef]
- Fedorczyk, A.; Pomorskia, R.; Chmielewski, M.; Ratajczak, J.; Kaszkur, Z.; Skompska, M. Bimetallic Au@Pt nanoparticles dispersed in conducting polymer—A catalyst of enhanced activity towards formic acid electrooxidation. Electrochim. Acta 2017, 246, 1029–1041. [Google Scholar] [CrossRef]
- Cabello, G.; Davoglio, R.A.; Hartl, F.W.; Marco, J.F.; Pereira, E.C.; Biaggio, S.R.; Varela, H.; Cuesta, A. Microwave-assisted synthesis of Pt-Au nanoparticles with enhanced electrocatalytic activity for the oxidation of formic acid. Electrochim. Acta 2017, 224, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Du, C.; Ye, J.; Chen, G.; Du, L.; Yin, G. Selective surface engineering of heterogeneous nanostructures: In situ unraveling of the catalytic mechanism on Pt−Au catalyst. ACS Catal. 2017, 7, 7923–7929. [Google Scholar] [CrossRef]
- Duchesne, P.N.; Li, Z.Y.; Deming, C.P.; Fung, V.; Zhao, X.; Yuan, J.; Regier, T.; Aldalbahi, A.; Almarhoon, Z.; Chen, S.; et al. Golden single-atomic-site platinum electrocatalysts. Nat. Mater. 2018, 17, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.K.; Choi, M.; Yang, S.; Shong, B.; Chung, H.S.; Sohn, Y.; Rhee, C.K. Formic acid electrooxidation activity of Pt and Pt/Au catalysts: Effects of surface physical properties and irreversible adsorption of Bi. Electrochim. Acta 2018, 273, 307–317. [Google Scholar] [CrossRef]
- Pajić, M.N.K.; Stevanović, S.I.; Radmilović, V.V.; Gavrilović-Wohlmuther, A.; Zabinski, P.; Elezović, N.R.; Radmilović, V.R.; Gojković, S.L.; Jovanović, V.M. Dispersion effect in formic acid oxidation on PtAu/C nanocatalyst prepared by water-in-oil microemulsion method. Appl. Catal. B 2019, 243, 585–593. [Google Scholar] [CrossRef]
- Xie, Y.; Dimitrov, N. Ultralow Pt loading nanoporous Au-Cu-Pt thin film as highly active and durable catalyst for formic acid oxidation. Appl. Catal. B 2020, 263, 118366. [Google Scholar] [CrossRef]
- Liang, Z.; Song, L.; Elnabawy, A.O.; Marinkovic, N.; Mavrikakis, M.; Adzic, R.R. Platinum and Palladium monolayer electrocatalysts for formic acid oxidation. Top. Catal. 2020, 63, 742–749. [Google Scholar] [CrossRef]
- Minudri, D.; Tesio, A.Y.; Fungo, F.; Palacios, R.E.; Cappellari, P.S.; Pastor, E.; Planes, G.A. Surface diffusion of poisoning species during CO and formic acid oxidation on PtAu surface. The key role of the active site. J. Power Sources 2021, 483, 229189. [Google Scholar] [CrossRef]
- Lee, J.; Yoo, J.K.; Lee, H.; Kim, S.H.; Sohn, Y.; Rhee, C.K. Formic acid oxidation on Pt deposit model catalysts on Au: Single-layered Pt deposits, plateau-type Pt deposits, and conical Pt deposits. Electrochim. Acta 2019, 310, 38–44. [Google Scholar] [CrossRef]
- Lee, H.; Sohn, Y.; Rhee, C.K. Pt deposits on BiPt NP catalyst for formic acid oxidation: Catalytic enhancement and longer lifetime. Langmuir 2020, 36, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Ahn, C.Y.; Lee, H.; Kim, J.K.; Oh, S.H.; Hwang, W.; Yang, S.; Kim, J.; Kim, O.H.; Choi, I. Bi-modified Pt supported on carbon black as electro-oxidation catalyst for 300 W formic acid fuel cell stack. Appl. Catal. B 2019, 253, 187–195. [Google Scholar] [CrossRef]
- Lee, H.; Kim, Y.J.; Sohn, Y.; Rhee, C.K. Co-deposits of Pt and Bi on Au disk toward formic acid oxidation. J. Solid State Electrochem. 2020, 24, 2535–2542. [Google Scholar] [CrossRef]
- ICP-OES and ICP-MS Detection Limit Guidance. Available online: http://www.nanoscience.co.jp/surface_analysis/pdf/icp-oes-ms-detection-limit-guidance-BR023.pdf (accessed on 21 January 2021).
- Koh, S.; Strasser, P. Electrocatalysis on bimetallic surfaces: Modifying catalytic reactivity for oxygen reduction by voltammetric surface dealloying. J. Am. Chem. Soc. 2007, 129, 12624. [Google Scholar] [CrossRef]
- Peng, Z.; You, H.; Yang, H. An electrochemical approach to PtAg alloy nanostructures rich in Pt at the surface. Adv. Funct. Mater. 2010, 20, 3734. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H. Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat. Chem. 2010, 2, 454. [Google Scholar] [CrossRef]
- Shard, A.G. Detection limits in XPS for more than 6000 binary systems using Al and Mg Kα X-rays. Surf. Interface. Anal. 2014, 46, 175–185. [Google Scholar] [CrossRef]
- Yoo, J.K.; Rhee, C.K. Formic acid oxidation on Bi-modified Pt surfaces: Pt deposits on Au versus bulk Pt. Electrochim. Acta 2016, 216, 16–23. [Google Scholar] [CrossRef]
- Lee, J.; Yoo, J.K.; Kim, J.; Sohn, Y.; Rhee, C.K. Conical multiple-layered Pt deposits on Au and its adsorption stoichiometries of CO and hydrogen. Electrochim. Acta 2018, 290, 244–254. [Google Scholar] [CrossRef]
- Tripkovic’, A.V.; Popovic’, K.D.; Stevanovic´, R.M.; Socha, R.; Kowal, A. Activity of a PtBi alloy in the electrochemical oxidation of formic acid. Electrochem. Commun. 2006, 8, 1492–1498. [Google Scholar] [CrossRef]
- Lović, J.D.; Obradović, M.D.; Tripković, D.V.; Popović, K.D.; Jovanović, V.M.; Gojković, S.L.; Tripković, A.V. High Activity and stability of Pt2Bi Catalyst in Formic Acid Oxidation. Electrocatalysis 2012, 3, 346–352. [Google Scholar] [CrossRef]
- Lović, J.D.; Stevanović, S.I.; Tripković, D.V.; Tripković, V.V.; Stevanović, R.M.; Popović, K.D.; Jovanović, V.M. Formic Acid Oxidation at Platinum-Bismuth Clusters. J. Electrochem. Soc. 2014, 161, H547–H554. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.D.; Riggs, W.M.; Davis, L.E.; Davis, J.F.; Moulder, J.F.; Muilenberg, G.E. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Data for Use in X-Ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Waltham, MA, USA, 1979. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.J.; Lee, H.; Chung, H.-S.; Sohn, Y.; Rhee, C.K. PT-BI Co-Deposit Shell on AU Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation. Catalysts 2021, 11, 1049. https://doi.org/10.3390/catal11091049
Kim YJ, Lee H, Chung H-S, Sohn Y, Rhee CK. PT-BI Co-Deposit Shell on AU Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation. Catalysts. 2021; 11(9):1049. https://doi.org/10.3390/catal11091049
Chicago/Turabian StyleKim, Young Jun, Hyein Lee, Hee-Suk Chung, Youngku Sohn, and Choong Kyun Rhee. 2021. "PT-BI Co-Deposit Shell on AU Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation" Catalysts 11, no. 9: 1049. https://doi.org/10.3390/catal11091049
APA StyleKim, Y. J., Lee, H., Chung, H.-S., Sohn, Y., & Rhee, C. K. (2021). PT-BI Co-Deposit Shell on AU Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation. Catalysts, 11(9), 1049. https://doi.org/10.3390/catal11091049