Abstract
Bimetallic AuPd nanoparticles supported on TiO2 are known to catalyze the reduction of NO with CO. Here, we investigated the effects of the addition of lanthanum oxide to a AuPd/TiO2 catalyst with a AuPd particle size of 2.1–2.2 nm. The addition of La2O3 enhanced the catalytic activity; for example, at 250 °C, there was 40.9% NO conversion and 49.3% N2-selectivity for AuPd/TiO2, and 100% NO conversion and 100% N2-selectivity for AuPd-La (1:1)/TiO2. The temperature requiring 100% NO conversion dropped from 400 °C to 200 °C by the simple post-impregnation of La2O3 onto AuPd/TiO2. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) combined with modulation excitation spectroscopy (MES) demonstrated that CO adsorption occurs first on Au atoms and then, within 80 s, moves onto Pd atoms. This transformation between two adsorption sites was facilitated by the addition of La2O3.
1. Introduction
Fundamental discoveries in the 1970s and 1980s [1,2] that gold nanoparticles supported on metal oxides act as a catalyst in a low temperature range is one of the most fascinating observations in chemistry. Before that, bulk Au had been considered inert and unreactive because of its electron negativity (2.54), which is close to the value of carbon (2.55) [3]. Au nanoparticles need to be finely dispersed on the metal oxide support [1,2] and their preparation method greatly influences the catalytic performance of the Au catalysts [4]. In addition, the valence electrons, the nature of support materials, interface/perimeter between Au and metal oxide, and their charge transfer strongly affect the catalytic performance of Au catalysis [5]. Au catalysis covers a wide range of chemical reactions such as oxidation [6], hydrogenation [7], cross-coupling reaction [8], fine chemical synthesis [9], and photocatalysis [10]. Its high potential has opened a path for the industrialization of Au catalysis in biorefinery, fine chemical synthesis, and pharmaceutical industries [11].
Nitric oxide and carbon monoxide emitted from internal combustion engines are dangerous to public health and ecological balance [12]. Noble metals such as Rh, Pt, and Pd supported on metal oxides have been extensively explored for eliminating NO and CO simultaneously [13,14,15,16,17,18,19,20]. Catalytic reduction of NO with CO proceeds via the formation of isocyanate (–NCO) species and their reaction with NO to form N2 and CO2 [14]. Pd-based catalysts have been reported to be an alternative to traditional three-way catalysts [19,21]. However, the limitation of the Pd catalysts is its low selectivity to dinitrogen. There are two main factors to control the N2-selectivity regulated by the following reactions:
2 NO + 2 CO → N2 + 2 CO2
2 NO + CO → N2O + CO2
N2O + CO → N2 + CO2
Reaction (1) is the direct to path to form N2. Reactions (2) and (3) indicate the indirect path via N2O formation and its subsequent reaction with CO. Generally, reaction (3) governs the N2-selectivity [22]. To facilitate reaction (3), a number of attempts have been made for Pd-based catalysts such as the modification of support materials [23,24,25], additives [13,26,27], and alloying [28,29,30]. Alloying Au nanoparticles with Rh has also been reported to facilitate reaction (3) [31]. Among them, bimetallic AuPd nanoparticles supported on TiO2 was reported to catalyze the above reactions efficiently [32]. AuPd (1:1)/TiO2 outperforms monometallic Pd and Au catalysts [32]. In particular, monometallic Au catalysts without any additives cannot promote reaction (3) [33,34]. However, even with highly active bimetallic AuPd catalysts, the full NO conversion requires temperatures as high as 350 °C and the N2-selectivity never reaches 100% [33,34]. Therefore, in this study, we focused on achieving 100% NO conversion and 100% N2-selectivity at lower temperature by modification of the AuPd/TiO2 catalyst.
Lanthanide elements (e.g., La, Ce) were widely used as an additive to improve catalytic performance [15,35,36,37]. For example, the addition of lanthana (La2O3) or ceria (CeO2) does not only improve the catalytic activity of Pd, but also stabilizes its metallic-state nanoparticles [15,36]. The model proposed to describe the Pd–lanthana interaction is based on an assumption of the metal decoration with LaOx (x < 1.5) overlayers migrating from the support material at the Pd–LaOx interface in a strong metal–support interaction (SMSI) under reductive conditions [36]. In this study, we combined bimetallic AuPd nanoparticles and La2O3 supported on TiO2 to harmonize promotional effects for catalytic reduction of NO with CO. AuPd-La2O3/TiO2 catalysts showed high NO conversion and N2-selectivity in the low temperature range. The effects of the La2O3 were characterized by means of spectroscopic techniques.
2. Results and Discussion
Figure 1 shows the annular dark-field transmission electron microscope (ADF-TEM) images of each catalyst and size distribution histograms of bimetallic AuPd nanoparticles. The bimetallic nanoparticles with an average particle size of 2.1–2.2 nm were finely dispersed on the TiO2 support for all samples. The addition of La2O3 had no influence on the size distribution, whereas BET surface area dropped from 52.1 m2/g to 37.0 m2/g as in Table 1. Figure 2a,b shows a focused image of a bimetallic AuPd/TiO2 particle and the corresponding energy-dispersive spectroscopy (EDS) line scan of Au and Pd. Au and Pd elements perfectly overlap each other, which led us to conclude that the bimetallic AuPd nanoparticles were in the form of a well-mixed alloy. Because the size of AuPd nanoparticles is not affected by the AuPd–La ratio, effects of La2O3 addition on catalytic performance can be reasonably evaluated.
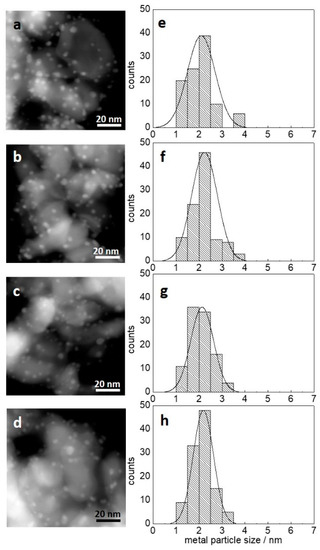
Figure 1.
TEM images and size distribution profiles of (a,e) AuPd/TiO2, (b,f) AuPd-La(2:1)/TiO2, (c,g) AuPd-La(1:1)/TiO2, and (d,h) AuPd-La(1:2)/TiO2.

Table 1.
BET surface area and AuPd particle size of AuPd, AuPd-La, and La supported on TiO2. Catalyst designations are given in the Experimental Section.
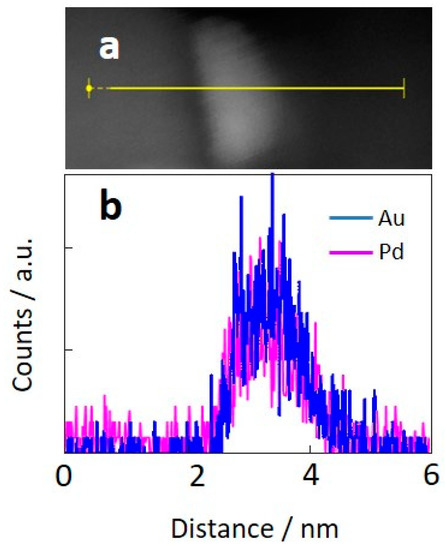
Figure 2.
(a) TEM image of a bimetallic AuPd particle and (b) EDS line scan of AuPd/TiO2.
Figure 3 shows X-ray photoelectron spectroscopy (XPS) spectra of the Au 4f, Pd 3d, and La 3d regions of the catalysts pre-treated with pure hydrogen at 300 °C for 1 h. For the AuPd/TiO2, a peak of Au 4f7/2 centered at 82.8 eV was assigned to metallic Au. Two peaks at 339.9 and 334.6 eV were assigned to metallic Pd [38]. The lower binding energy (BE) for both Pd3/2 and Au 4f7/2 compared to the standard values of corresponding metals arose from the net charge flowing into Au (higher Au s or p electron densities) and Pd (gaining d electrons from Au) [38,39,40], indicating the formation of an alloy through the modification of the electronic structure between Au and Pd. The ratio of Au to Pd (Au/(Au + Pd)) on the surface was 0.43 (Table S1 in the Supplementary Materials), indicating that Pd was slightly more present on the surface of the bimetallic nanoparticles. With the addition of La2O3, the peaks of Au 4f7/2 and Pd 3d5/2 further shifted toward lower BE, implying the formation of electron-enriched Auδ− species. The increased electron density on gold is known to facilitate CO adsorption and thus to enhance catalytic activity [41,42]. Because of a partial coverage of AuPd particles by La2O3, the signals of Au and Pd were attenuated (see Table S1 in the Supplementary Materials). The XPS spectrum of La 3d at 834.6 eV evidenced that La was in the oxidation state of La3+ as La2O3 [43]. Moreover, the La 3d5/2 BE values were close to the value of the finely dispersed La2O3 phase (835.0 eV), but they were substantially higher than the value observed for the pure La2O3 phase (833.5 eV) [44], indicating the well-dispersed La2O3 on the catalyst surface. This accounts for a reason as to why the crystalline phase of La2O3 was not observed by XRD (Figure S1).
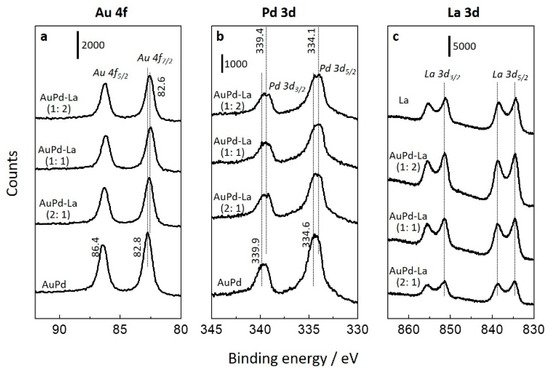
Figure 3.
XPS spectra of (a) Au 4f, (b) Pd 3d, and (c) La 3d regions.
Figure 4 shows the catalytic performance for the reduction of NO with CO (2000 ppm NO, 2000 ppm CO, He balance: 150 mL/min). AuPd/TiO2 only showed low NO conversion and N2-selectivity. In addition, it required 400 °C for 100% NO conversion with 75% N2-selectivity. Hence, the unpromoted catalyst was proven to be inefficient for the reduction of N2O with CO (Reaction (3)). On the other hand, the addition of La2O3 to AuPd/TiO2 drastically changed the catalytic performance. The best performance was achieved with AuPd-La(1:1)/TiO2: 100% NO conversion and 63.7% N2-selectivity at 200 °C; and 100% NO conversion and 100% N2-selectivity at 250 °C. Further addition of La2O3 (i.e., AuPd-La(1:2)/TiO2), deteriorated the catalytic performance probably due to covering the AuPd nanoparticles by the La2O3 layers. For the catalysts other than AuPd-La(1:1)/TiO2, the conversion and selectivity dropped in the temperature range of 250–350 °C (Figure 4). This occasionally happens for the catalytic reduction of NO with CO due to switching between two different mechanisms: the bimolecular mechanism at low temperature and the dissociative mechanism at high temperature [29]. As clearly seen in Figure 4, La2O3/TiO2 (dotted line) showed poor activity, confirming the unique synergistic effect of bimetallic AuPd and La2O3.
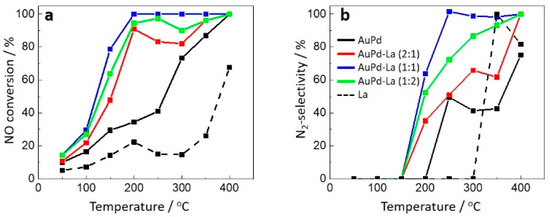
Figure 4.
(a) NO conversion and (b) N2-selectivity during catalytic reduction of NO with CO; Gas mixture: 2000 ppm NO, 2000 ppm CO, He balance; Total flow rate: 150 mL/min; GHSV: 12,000 h−1; Reaction temperature: 50–400 °C.
To further study the surface properties of bimetallic AuPd nanoparticles and their synergy with La2O3, carbon monoxide was used as a probe molecule. Its adsorption-desorption behavior was followed by using in situ diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) in tandem with modulation excitation spectroscopy (MES). Figure 5 displays the phase-domain (a, c, e, g) and time-domain (b, d, f, h) DRIFTS spectra taken during repeated cycles of CO adsorption-desorption at 50 °C. In Figure 5a,b (AuPd/TiO2), a broad IR band at 2150–2220 cm−1 was assigned to CO adsorbed on TiO2 [45,46]. A sharp and intense band at 2108 cm−1 was assignable to CO linearly-adsorbed on metallic Au [38,47]. Both bands were detected at the same in-phase angle of 310° and, hence, these bands appeared at the same time. Interestingly, the band at 2108 cm−1 shifted gradually and reached its maximum at 2080 cm−1 at an in-phase angle of 260°. This unambiguous shift observed in the time-domain spectra is rationalized by CO changing its binding partner from Au to Pd. This transformation completed within 80 s. This new band is characteristic of linearly-adsorbed CO on Pd [38,47]. The addition of La2O3 accelerated this transformation and enhanced CO adsorption property on Pd revealed by the in-phase angle of Au-CO at 2108 cm−1 increasing by 30° (310° → 340°). This indicates that CO adsorption on Au occurred more rapidly, and its change in binding partner from Au-CO to Pd-CO was also accelerated. AuPd-La(1:2)/TiO2 showed intense band of Pd-CO, and its desorption was considerably delayed compared to the catalysts with a lower amount of La2O3 addition. The excess addition of La2O3 appeared to strengthen the Pd–CO bond. As evidenced from the XPS spectra (Figure 3), electron-enrichment of AuPd surfaces were induced by the addition of La2O3. Electron-enriched metal surfaces are known to facilitate electron back-donation to 2 pi (π) anti-bonding molecular orbital of C–O, leading to strong affinity of adsorbed CO to the metal surfaces. This characteristic affinity was confirmed by the time-resolved MES-DRIFTS spectra (Figure 5h). This would account for the deteriorated catalytic performance with AuPd-La(1:2)/TiO2. On the other hand, MES-DRIFTS revealed no influence of the addition of La2O3 on NO adsorption as shown in Figure S2 in the Supplementary Materials. Au nanoparticles were reported to adsorb almost no NO molecules on the surface [45]. Therefore, the role of bimetallic AuPd nanoparticles would be a separation of the adsorption site; NO adsorbs preferentially on Pd sites while CO first adsorbs on Au sites, which migrates to Pd sites to react with adsorbed NO. The synergistic effect by La2O3 seems to enhance this effect.
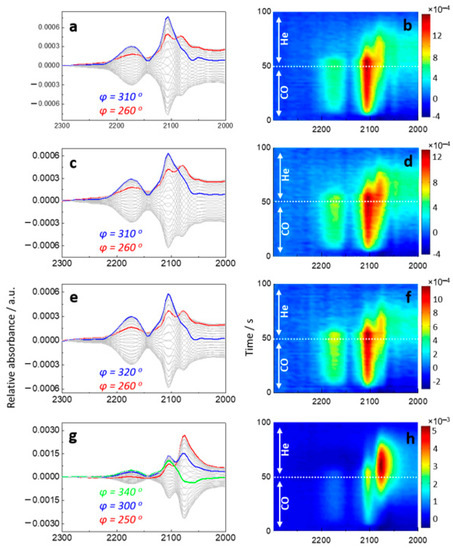
Figure 5.
(a,c,e,g) Phase-domain and (b,d,f,h) time-domain DRIFTS spectra during CO adsorption and desorption on (a,b) AuPd/TiO2, (c,d) AuPd-La(2:1)/TiO2, (e,f) AuPd-La(1:1)/TiO2, and (g,h) AuPd-La(1:2)/TiO2 at 50 °C.
Figure 6 shows in situ time-domain and phase-domain DRIFTS spectra during NO + CO reaction (CO-concentration modulation) at 300 °C. At such high temperature, the band at 2108 cm−1 (Au-CO) was not observed while the band at 2065 cm−1 (Pd-CO), together with a broad band of bridged CO at 1850–1980 cm−1, directly appeared. This indicates that at high temperature, CO-transfer from Au to Pd sites occurs rapidly. Compared to the CO adsorption spectra (Figure 5), an obvious redshift (2081 cm−1 → 2065 cm−1) of CO adsorption on Pd was observed in the presence of NO at 300 °C. The band at around 2065 cm−1 was reported to be relevant to small Pd clusters covered with O atoms [45]. NO molecules adsorbed on Pd dissociated to N and O atoms, resulting in the Pd clusters covered with O atoms. This is consistent with our assumption that CO and NO share the same active sites. After saturation of the surface with adsorbed CO, a new band at 2204 cm−1 gradually emerged with a phase-delay of ca. 30° compared to adsorbed CO (in-phase angle of 352°). This IR band is characteristic of the stretching vibration of isocyanate species (−NCO) on the TiO2 surface [34,46]. The formation of isocyanate and its reaction with NO is an important intermediate step to produce N2 and CO2 [18]. The addition of La2O3 shifted the band position of isocyanate to lower wavenumbers of 2183–2175 cm−1, which was assigned to the isocyanate adsorbed on metal surfaces [46,48]. This might have promoted further reaction of –NCO with NO to form N2 and CO2 on the bimetallic particles. The fact that the addition of La2O3 doubled the amount of isocyanate detected on the surface supports this assumption. Nonetheless, its reaction with NO occurred in the almost same in-phase angle of 318–324°, indicating that the reaction rate of isocyanate with NO also doubled. Interestingly, by adding La2O3, the band of isocyanate species on TiO2 (2202 cm−1) completely disappeared. The presence of La2O3 might have suppressed the formation of isocyanate on TiO2 and facilitated its formation only on bimetallic nanoparticles.
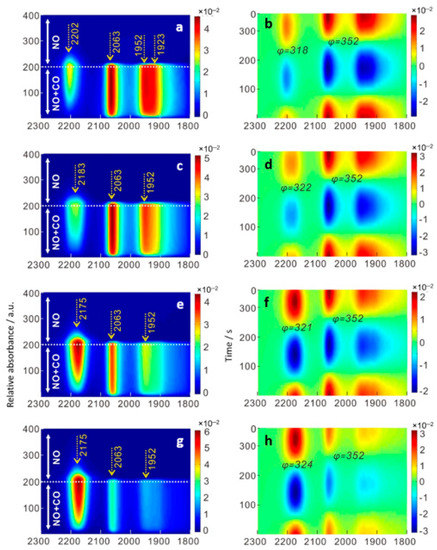
Figure 6.
(a,c,e,g) Time-domain and (b,d,f,h) phase-domain DRIFTS spectra during catalytic reduction of NO with CO on (a,b) AuPd/TiO2, (c,d) AuPd-La(2:1)/TiO2, (e,f) AuPd-La(1:1)/TiO2, and (g,h) AuPd-La(1:2)/TiO2 at 300 °C.
3. Materials and Methods
3.1. Catalyst Preparation
Titanium(IV) oxide (Sigma-Aldrich, Burlington, USA, a mixture of anatase and rutile, 99.5%) was used as a catalytic material support. Chloroauric acid (HAuCl4·H2O, Xi’an Wonder Energy Chemical Co., Ltd., Xi’an, China, 98%), palladium(II) sodium chloride (Na2PdCl4, Xi’an Wonder Energy Chemical Co., Ltd., Xi’an, China, Pd ≥ 35.5%), sodium hydroxide (J&K Scientific Ltd., Beijing, China, 1 M), and tetra (hydroxymethyl) phosphonium chloride (Sigma-Aldrich, Burlington, USA) were used for the synthesis of bimetallic AuPd nanoparticles. Lanthanum(III) nitrate hexahydrate (LaN3O9·6H2O, Sigma-Aldrich, Burlington, USA, ≥99%) was used for post-impregnation of La2O3 as a promoter.
Bimetallic AuPd nanoparticles supported by TiO2 were prepared according to a report by Duff et al. as follows [47,49]: HAuCl4 and Na2PdCl4 as Au and Pd precursors and THPC as a reducing agent were dissolved in deionized water. The molar ratios of Au to Pd were adjusted to 1:1. The ratio of NaOH, THPC, and the metals (Au + Pd) was adjusted to 6:1.4:1. The resulting colloidal solution was stirred vigorously at room temperature for 1 h, and then added into a suspension of TiO2 in 100 mL of deionized water. The total metal loading amounts to 2 wt%. After the pH value of the suspension was adjusted at 2.0 with nitric acid, the suspension was filtered and washed with 3 L of deionized water. The obtained material was dried overnight at 80 °C in air.
AuPd-La/TiO2 with different AuPd:La molar ratios (2:1, 1:1, 1:2) were prepared by impregnating the lanthanum precursor onto the above prepared AuPd(1:1)/TiO2 catalyst. The lanthanum(III) nitrate hexahydrate was dissolved in deionized water and stirred for 30 min. Then, AuPd(1:1)/TiO2 powders were added and stirred for another 1 h. The suspension was aged for 1 h at 60 °C in a rotary evaporator and dried at 35 °C under vacuum. The obtained powders were further dried at 60 °C in an oven overnight. A reference catalyst, La/TiO2, whose La molar amount corresponded to the AuPd-La(1:1)/ TiO2 catalyst, was synthesized in the same impregnation manner. Even though La was present as La2O3, the promoted catalysts are denoted as AuPd-La/ TiO2 for simplicity.
3.2. Catalytic Reaction
The catalytic activity test was carried out in a fixed bed micro-flow reactor system. A total of 400 mg of catalyst powder was loaded into a stainless-steel tube reactor with an inner diameter of 8.2 mm. Prior to catalytic reaction, the catalysts were pre-treated at 300 °C for 1 h under pure H2 (flow rate of 100 mL/min) with a ramping rate of 5 °C/min. To remove gaseous H2 in the gas lines and adsorbed hydrogen atoms on the catalyst surface, the catalyst was then kept in a pure He stream at 300 °C for 10 min. Subsequently, the reactor was cooled down to 50 °C and the reaction mixture (2000 ppm of CO, 2000 ppm of NO, balanced with He) was introduced at the total flow rate of 150 mL/min, corresponding to a gas-hourly space velocity (GHSV) of 12,000 h−1. Catalytic performance in the temperature range of 50–400 °C was evaluated with a gas chromatograph (NINGBO HaiShu Sklon Electronic Instrument Co.,Ltd., Ningbo, China, 9750 GC) equipped with a thermal conductivity detector (TCD) and a capillary column (Molecular Sieve 5A) and a NO-NO2-NOx analyzer (Thermo Fischer Scientific, Waltham, USA, 42i-HL). All data were recorded after waiting for 30–70 min until the gaseous compositions reached a steady state at each measurement temperature. The same experiments were repeated at least twice to ensure the reproducibility of the data. A blank test without any catalyst showed no catalytic activity by the inner wall of the stainless-steel tube reactor. The mass balance of carbon and nitrogen atoms were almost 100% for all the catalysts tested in the entire temperature range.
3.3. Catalyst Characterization
BET surface area was measured by the adsorption of nitrogen molecules at liquid nitrogen temperature by employing a Quadrasorb SI instrument (Quantachrome, Boynton, FL, USA). Prior to each of the measurements, the sample powders were vacuumed at 150 °C for 1 h to remove the adsorbed molecules (H2O and CO2) on the catalyst surface and in the pores of the TiO2 support. X-ray diffraction (XRD) patterns were measured with a PANalytical X’Pert diffractometer (Malvern Panalytical, Malvern, UK) equipped with Pd-filtered Cu Kα radiation at 60 kV and 55 mA. X-ray photoelectron spectroscopy (XPS) was used to record the surface compositions and oxidation state of each element using a Thermo ESCALAB 250Xi (Thermo Fischer Scientific, Waltham, USA). Al Kα radiation (1486.6 eV, 15 kV, 10.8 mA) was selected to excite photoelectrons at the 50 eV pass energy. Transmission electron microscopy (TEM, JEOL, Tokyo, Japan, JEM-2100, 200 kv) was used to analyze the mean size of bimetallic AuPd nanoparticles. The EDS line scan was applied to gain information about the elemental distribution of Au and Pd in the nanoparticles. In situ infrared studies were performed with an infrared spectroscopy (Bruker Corporation, Billerica, MA, USA, Vertex 70) equipped with an MCT detector at the spectral resolution of 4 cm−1. A total of 20 mg of the sample was loaded into a porous ceramic cup and placed in an in situ diffuse reflectance FTIR spectroscopic (DRIFTS) cell (ST Japan, Tokyo, Japan). Prior to each measurement, the samples were pre-treated with pure H2 at the flow rate of 100 mL/min for 1 h at 300 °C (ramping rate: 5 °C/min). Modulation-excitation spectroscopy (MES) was coupled with in situ DRIFTS by periodically changing between two different gas mixtures: (i) CO (2000 ppm) in He and pure He (CO adsorption-desorption modulation); and (ii) CO (2000 ppm) and NO (2000 ppm) in He and NO (2000 ppm) in He (CO-modulation during NO-CO reaction). The spectra of the initial three cycles were disposed to obtain stable responses. Afterward, the five cycles were averaged as one cycle to increase the signal-to noise (S/N) ratio. Phase-domain spectra were obtained by a mathematical treatment of the time-domain spectra according to the following equation:
where T represents the length of a cycle; ω is the frequency of modulation; is the phase angle used for demodulation; k is the demodulation index (k = 1 in this study); and A(t) and Ak are the responses of the active species in the time- and phase-domains, respectively.
4. Conclusions
This work showed a promotional effect of bimetallic AuPd nanoparticles supported on TiO2 by the addition of La2O3 in terms of both NO conversion and N2-selectivity for the catalytic reduction of NO with CO. XPS study revealed electron-enrichment of bimetallic AuPd nanoparticles in the presence of La2O3, leading to a beneficial CO adsorption property. In situ MES-DRIFTS demonstrated that CO first adsorbs on Au sites and then gradually move to Pd sites. This site transformation was considerably amplified by the addition of La2O3. Another critical role of La2O3 is to promote the formation of the isocyanate intermediate. This effect seems critical to facilitate the sequential reaction of isocyanate with NO to produce N2 and CO2 efficiently. In this study, we focused on the effect of La2O3 addition to the AuPd/TiO2 catalyst under model gas conditions (only NO and CO in the feed gas). There were some reports that under industry-relevant conditions, Pd can be easily oxidized [50,51]. In light of this, the effect of La2O3 addition onto the behavior of AuPd alloys should be the next scope of this research direction.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11080916/s1, Table S1: XPS results of AuPd, AuPd-La, La supported on TiO2, Figure S1: XRD patterns, Figure S2: Phase-domain IR spectra during NO adsorption-desorption cycles at 50 °C.
Author Contributions
Conceptualization, N.M.; Investigation, X.W.; Writing of original draft preparation, X.W.; Writing of review and editing, X.W., N.M., and D.M.M.; Supervision, N.M.; Project administration, N.M.; Funding acquisition, D.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded internally by ZHAW.
Acknowledgments
Yan Xie from the Gold Catalysis Research Center (GCRC) of the Dalian Institute of Chemical Physics (DICP) is acknowledged for the XRD, XPS and TEM measurements. Jinsuo Gao from the Dalian University of Technology (DUT) is acknowledged for the BET measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bond, G.C.; Thompson, D.T. Catalysis by Gold. Catal. Rev. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, F.; Song, C.; Jia, J.; Xu, Z. Recent Advances of the Combination of Au/Acid Catalysis. Chin. J. Chem. 2014, 32, 937–956. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. The influence of the preparation methods on the catalytic activity of platinum and gold supported on TiO2 for CO oxidation. Catal. Lett. 1997, 44, 83–87. [Google Scholar] [CrossRef]
- Ishida, T.; Murayama, T.; Taketoshi, A.; Haruta, M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464–525. [Google Scholar] [CrossRef]
- Yamazoe, S.; Koyasu, K.; Tsukuda, T. Nonscalable Oxidation Catalysis of Gold Clusters. Acc. Chem. Res. 2014, 47, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.B.; Jin, R.C. Heterogeneous catalysis by gold and gold-based bimetal nanoclusters. Nano Today 2018, 18, 86–102. [Google Scholar] [CrossRef]
- Akram, M.O.; Banerjee, S.; Saswade, S.S.; Bedi, V.; Patil, N.T. Oxidant-free oxidative gold catalysis: The new paradigm in cross-coupling reactions. Chem. Commun. 2018, 54, 11069–11083. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, L.; Liu, Y.M.; Cao, Y. Supported Gold Catalysis: From Small Molecule Activation to Green Chemical Synthesis. Acc. Chem. Res. 2014, 47, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Zidan, M.; Rohe, S.; McCallum, T.; Barriault, L. Recent advances in mono and binuclear gold photoredox catalysis. Catal. Sci. Technol. 2018, 8, 6019–6028. [Google Scholar] [CrossRef]
- Ciriminna, R.; Falletta, E.; Della Pina, C.; Teles, J.H.; Pagliaro, M. Industrial Applications of Gold Catalysis. Angew. Chem. Int. Ed. 2016, 55, 14209–14216. [Google Scholar] [CrossRef]
- Kim, K.H.; Jahan, S.A.; Kabir, E. A review of breath analysis for diagnosis of human health. Trends Anal. Chem. 2012, 33, 1–8. [Google Scholar] [CrossRef]
- Dallago, R.M.; Baibich, I.M. Pd-W and Pd-Mo catalysts for NO decomposition and NO/CO reduction reactions. J. Braz. Chem. Soc. 2009, 20, 873–879. [Google Scholar] [CrossRef][Green Version]
- Rasko, J.; Szabo, Z.; Bansagi, T.; Solymosi, F. FTIR study of the photo-induced reaction of NO + CO on Rh/TiO2. PCCP 2001, 3, 4437–4443. [Google Scholar] [CrossRef]
- Hirano, T.; Ozawa, Y.; Sekido, T.; Ogino, T.; Miyao, T.; Naito, S. Marked effect of In, Pb and Ce addition upon the reduction of NO by CO over SiO2 supported Pd catalysts. Catal. Commun. 2007, 8, 1249–1254. [Google Scholar] [CrossRef]
- Nováková, J.; Kubelková, L. Contribution to the mechanism of NO reduction by CO over Pt/NaX zeolite. Appl. Catal. B Environ. 1997, 14, 273–286. [Google Scholar] [CrossRef]
- Almusaiteer, K.; Chuang, S.S.C. Dynamic Behavior of Adsorbed NO and CO under Transient Conditions on Pd/Al2O3. J. Catal. 1999, 184, 189–201. [Google Scholar] [CrossRef]
- Xiao, P.; Davis, R.C.; Ouyang, X.; Li, J.; Thomas, A.; Scott, S.L.; Zhu, J. Mechanism of NO reduction by CO over Pt/SBA-15. Catal. Commun. 2014, 50, 69–72. [Google Scholar] [CrossRef]
- Beck, D.D.; Sommers, J.W.; DiMaggio, C.L. Impact of sulfur on model palladium-only catalysts under simulated three-way operation. Appl. Catal. B Environ. 1994, 3, 205–227. [Google Scholar] [CrossRef]
- Hoost, T.E.; Otto, K.; Laframboise, K.A. FTIR Spectroscopy of Nitric Oxide Adsorption on Pd/Al2O3: Evidence of Metal-Support Interaction. J. Catal. 1995, 155, 303–311. [Google Scholar] [CrossRef]
- Debeila, M.A.; Coville, N.J.; Scurrell, M.S.; Hearne, G.R. Direct observation of thermally activated NO adsorbate species on Au/TiO2: DRIFTS studies. J. Mol. Catal. A Chem. 2004, 219, 131–141. [Google Scholar] [CrossRef]
- Xu, Z.C.; Li, Y.R.; Lin, Y.T.; Zhu, T.Y. A review of the catalysts used in the reduction of NO by CO for gas purification. Environ. Sci. Pollut. Res. 2020, 27, 6723–6748. [Google Scholar] [CrossRef]
- Ferrer, V.; Finol, D.; Solano, R.; Moronta, A.; Ramos, M. Reduction of NO by CO using Pd-CeTb and Pd-CeZr catalysts supported on SiO2 and La2O3-Al2O3. J. Environ. Sci. 2015, 27, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Higo, T.; Omori, Y.; Shigemoto, A.; Ueno, K.; Ogo, S.; Sekine, Y. Promotive effect of H2O on low-temperature NO reduction by CO over Pd/La0.9Ba0.1AlO3-delta. Catal. Today 2020, 352, 192–197. [Google Scholar] [CrossRef]
- Salker, A.V.; Desai, M.S.F. Catalytic activity and mechanistic approach of NO reduction by CO over M0.05Co2.95O4 (M = Rh, Pd & Ru) spinel system. Appl. Surf. Sci. 2016, 389, 344–353. [Google Scholar] [CrossRef]
- Schmal, M.; Baldanza, M.A.S.; Vannice, M.A. Pd-xMo/Al2O3 catalysts for NO reduction by CO. J. Catal. 1999, 185, 138–151. [Google Scholar] [CrossRef]
- Stoyanova, D.; Georgieva, P.; Kasabova, N. Pd-containing catalysts promoted by NiO designed for the reduction of NO with CO at stoichiometric NO/CO ratio. React. Kinet. Mech. Catal. 2013, 108, 391–402. [Google Scholar] [CrossRef]
- De Sarkar, A.; Khanra, B. CO oxidation and NO reduction over supported Pt-Rh and Pd-Rh nanocatalysts: A comparative study. J. Mol. Catal. A Chem. 2005, 229, 25–29. [Google Scholar] [CrossRef]
- Hirano, T.; Ozawa, Y.; Sekido, T.; Ogino, T.; Miyao, T.; Naito, S. The role of additives in the catalytic reduction of NO by CO over Pd-In/SiO2 and Pd-Pb/SiO2 catalysts. Appl. Catal. A Gen. 2007, 320, 91–97. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, H.J.; Maeda, N.; Baiker, A. Structure and Catalytic Behavior of Alumina Supported Bimetallic Au-Rh Nanoparticles in the Reduction of NO by CO. Catalysts 2019, 9, 937. [Google Scholar] [CrossRef]
- Wang, X.W.; Maeda, N.; Meier, D.M.; Baiker, A. Potassium Titanate Nanobelts: A Unique Support for Au and AuRh Nanoparticles in the Catalytic Reduction of NO with CO. Chemcatchem 2021, 13, 438–444. [Google Scholar] [CrossRef]
- Shin, H.U.; Lolla, D.; Nikolov, Z.; Chase, G.G. Pd-Au nanoparticles supported by TiO2 fibers for catalytic NO decomposition by CO. J. Ind. Eng. Chem. 2016, 33, 91–98. [Google Scholar] [CrossRef]
- Wang, X.W.; Maeda, N.; Baiker, A. Synergistic Effects of Au and FeOx Nanocomposites in Catalytic NO Reduction with CO. ACS Catal. 2016, 6, 7898–7906. [Google Scholar] [CrossRef]
- Wang, X.W.; Wu, X.L.; Maeda, N.; Baiker, A. Striking activity enhancement of gold supported on Al-Ti mixed oxide by promotion with ceria in the reduction of NO with CO. Appl. Catal. B Environ. 2017, 209, 62–68. [Google Scholar] [CrossRef]
- Samed, A.J.; Tanaka, T.; Ikeue, K.; Machida, M. NO–H2–CO–O2 Reactions Over Pt Catalysts Supported on Ln-incorporated FSM-16 (Ln = La, Ce and Pr). Top. Catal. 2010, 53, 591–596. [Google Scholar] [CrossRef]
- Barrera, A.; Viniegra, M.; Bosch, P.; Lara, V.H.; Fuentes, S. Pd/Al2O3-La2O3 catalysts prepared by sol–gel: Characterization and catalytic activity in the NO reduction by H2. Appl. Catal. B Environ. 2001, 34, 97–111. [Google Scholar] [CrossRef]
- Vidal, H.; Bernal, S.; Baker, R.T.; Finol, D.; Pérez Omil, J.A.; Pintado, J.M.; Rodríguez-Izquierdo, J.M. Characterization of La2O3/SiO2 mixed oxide catalyst supports. J. Catal. 1999, 183, 53–62. [Google Scholar] [CrossRef]
- Xu, J.; White, T.; Li, P.; He, C.; Yu, J.; Yuan, W.; Han, Y.-F. Biphasic Pd−Au Alloy Catalyst for Low-Temperature CO Oxidation. J. Am. Chem. Soc. 2010, 132, 10398–10406. [Google Scholar] [CrossRef]
- Deki, S.; Akamatsu, K.; Hatakenaka, Y.; Mizuhata, M.; Kajinami, A. Synthesis and characterization of nano-sized gold-palladium bimetallic particles dispersed in polymer thin film matrix. Nanostruct. Mater. 1999, 11, 59–65. [Google Scholar] [CrossRef]
- José-Yacamán, M.; Mejía-Rosales, S.; Pérez-Tijerina, E.; Blom, D.A.; Allard, L.F. Imaging Au-Pd Nanoparticles with the Aberration-Corrected STEM/TEM. Microsc. Microanal. 2006, 12, 772–773. [Google Scholar] [CrossRef][Green Version]
- Horváth, D.; Toth, L.; Guczi, L. Gold nanoparticles: Effect of treatment on structure and catalytic activity of Au/Fe2O3 catalyst prepared by co-precipitation. Catal. Lett. 2000, 67, 117–128. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Andreeva, D.; Tabakova, T. FTIR Study of the Low-Temperature Water–Gas Shift Reaction on Au/Fe2O3 and Au/TiO2 Catalysts. J. Catal. 1999, 188, 176–185. [Google Scholar] [CrossRef]
- Fu, C.; Wang, J.; Yang, M.; Su, X.; Xu, J.; Jiang, B. Effect of La doping on microstructure of SnO2 nanopowders prepared by co-precipitation method. J. Non-Cryst. Solids 2011, 357, 1172–1176. [Google Scholar] [CrossRef]
- Kim, D.H.; Woo, S.I.; Lee, J.M.; Yang, O.B. The role of lanthanum oxide on Pd-only three-way catalysts prepared by co-impregnation and sequential impregnation methods. Catal. Lett. 2000, 70, 35–41. [Google Scholar] [CrossRef]
- Spezzati, G.; Su, Y.; Hofmann, J.P.; Benavidez, A.D.; DeLaRiva, A.T.; McCabe, J.; Datye, A.K.; Hensen, E.J.M. Atomically Dispersed Pd–O Species on CeO2(111) as Highly Active Sites for Low-Temperature CO Oxidation. ACS Catal. 2017, 7, 6887–6891. [Google Scholar] [CrossRef]
- Solymosi, F.; Bánsági, T.; Zakar, T.S. Infrared Study of the NO + CO Interaction over Au/TiO2 Catalyst. Catal. Lett. 2003, 87, 7–10. [Google Scholar] [CrossRef]
- Marx, S.; Krumeich, F.; Baiker, A. Surface Properties of Supported, Colloid-Derived Gold/Palladium Mono- and Bimetallic Nanoparticles. J. Phys. Chem. C 2011, 115, 8195–8205. [Google Scholar] [CrossRef]
- Grunwaldt, J.-D.; Kiener, C.; Wögerbauer, C.; Baiker, A. Preparation of Supported Gold Catalysts for Low-Temperature CO Oxidation via “Size-Controlled” Gold Colloids. J. Catal. 1999, 181, 223–232. [Google Scholar] [CrossRef]
- Duff, D.G.; Baiker, A.; Edwards, P.P. A new hydrosol of gold clusters. 1. Formation and particle size variation. Langmuir 1993, 9, 2301–2309. [Google Scholar] [CrossRef]
- Morlang, A.; Neuhausen, U.; Klementiev, K.V.; Schutze, F.W.; Miehe, G.; Fuess, H.; Lox, E.S. Bimetallic Pt/Pd diesel oxidation catalysts—Structural characterisation and catalytic behaviour. Appl. Catal. B Environ. 2005, 60, 191–199. [Google Scholar] [CrossRef]
- Schutz, J.; Stormer, H.; Lott, P.; Deutschmann, O. Effects of Hydrothermal Aging on CO and NO Oxidation Activity over Monometallic and Bimetallic Pt-Pd Catalysts. Catalysts 2021, 11, 300. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).