
Biocatalytic Silylation: The Condensation of Phenols and Alcohols with Triethylsilanol


 ,
, 
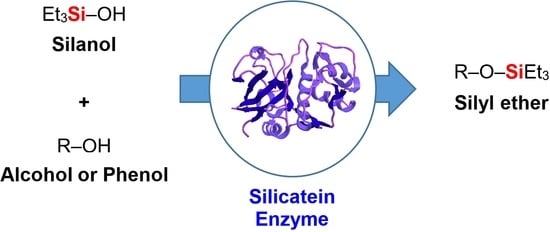
Abstract
:
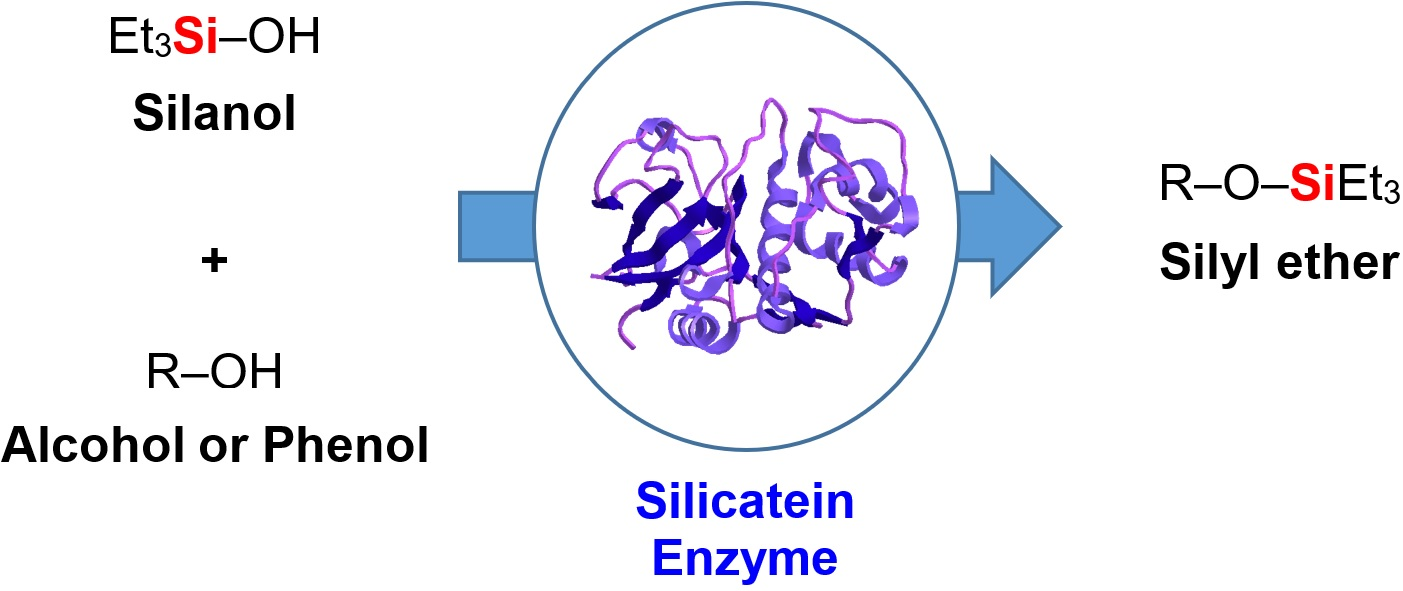
1. Introduction
2. Results and Discussion
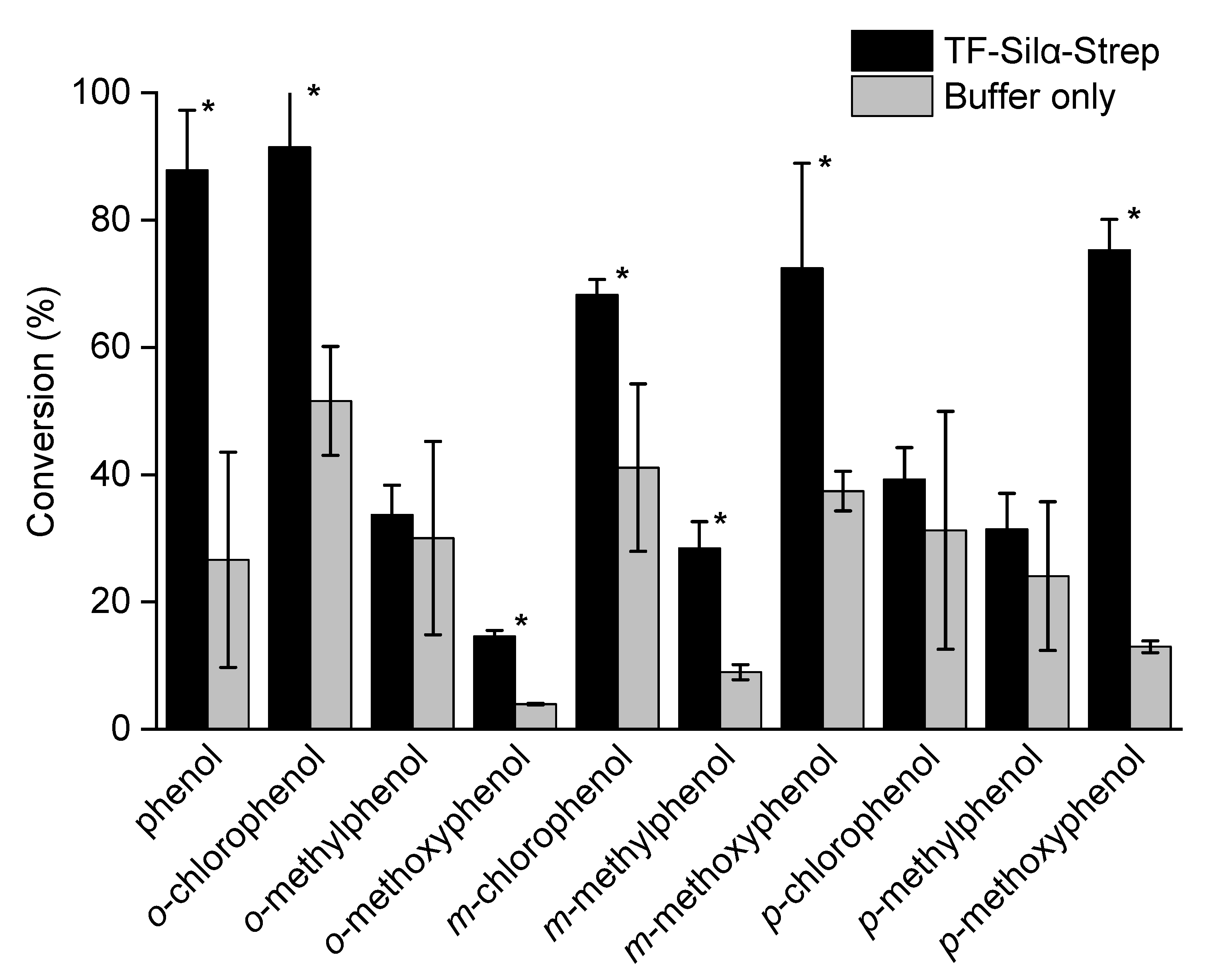
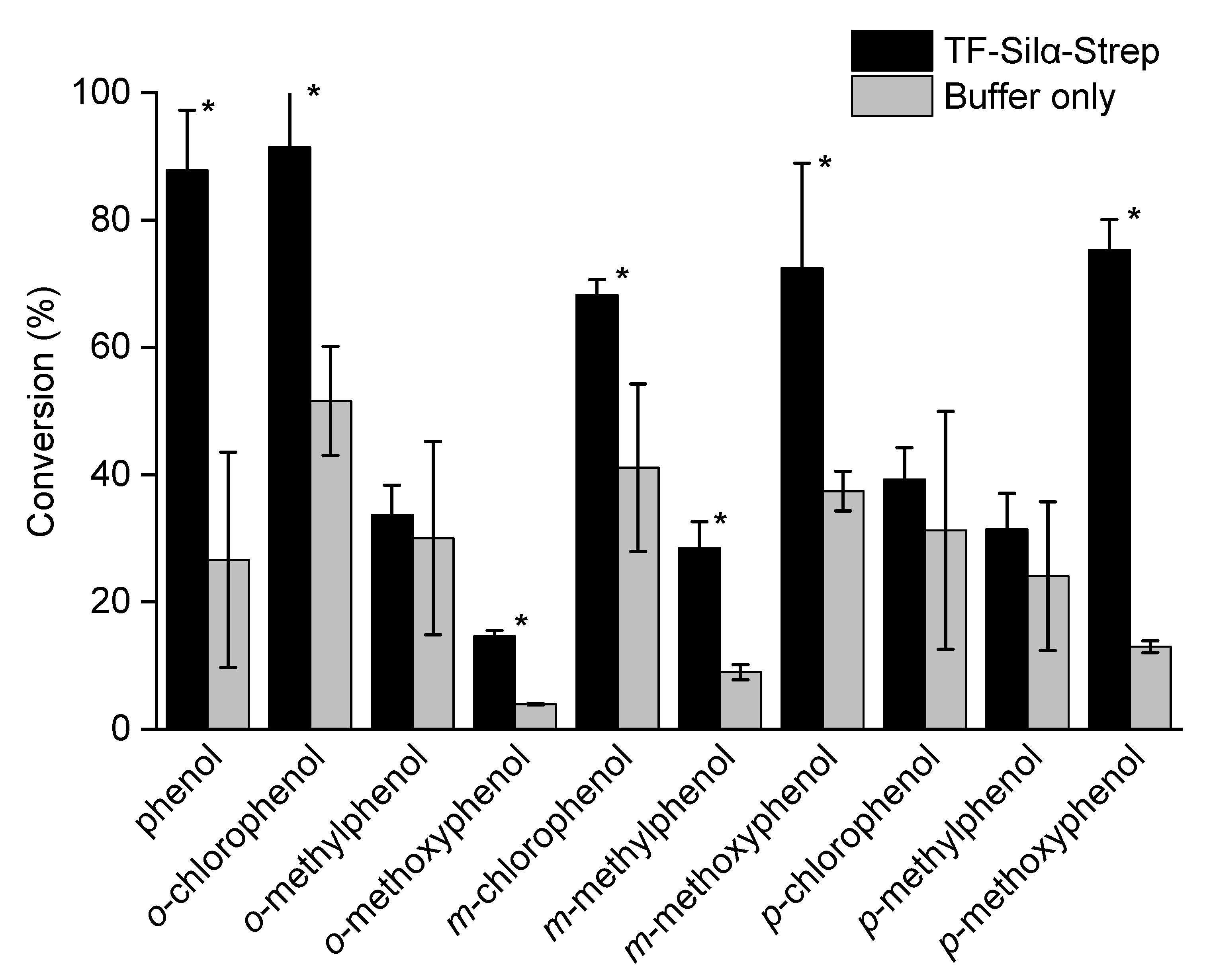
2.1. Triethylsilylation of Phenols
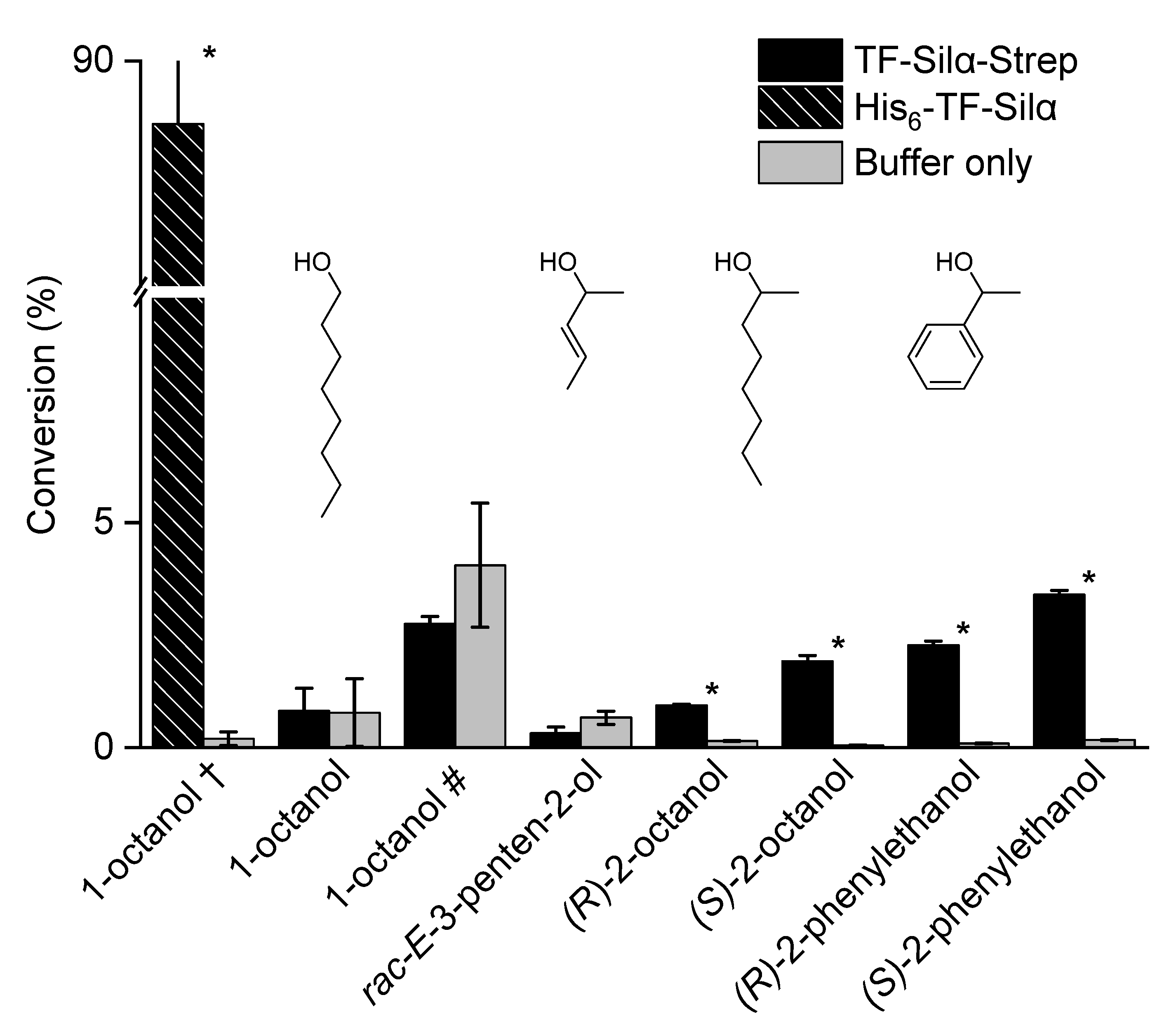
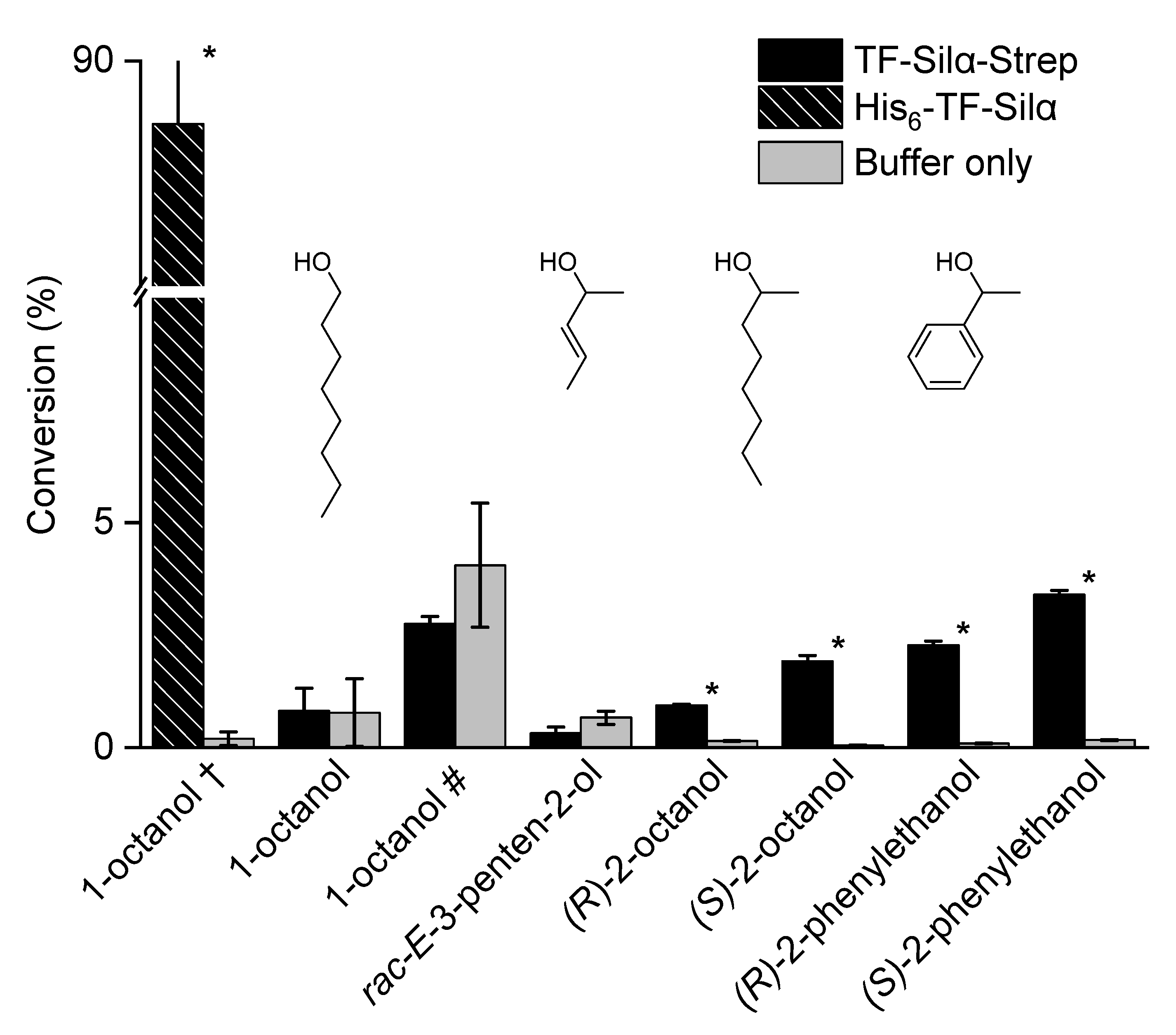
2.2. Triethylsilylation of Aliphatic Alcohols
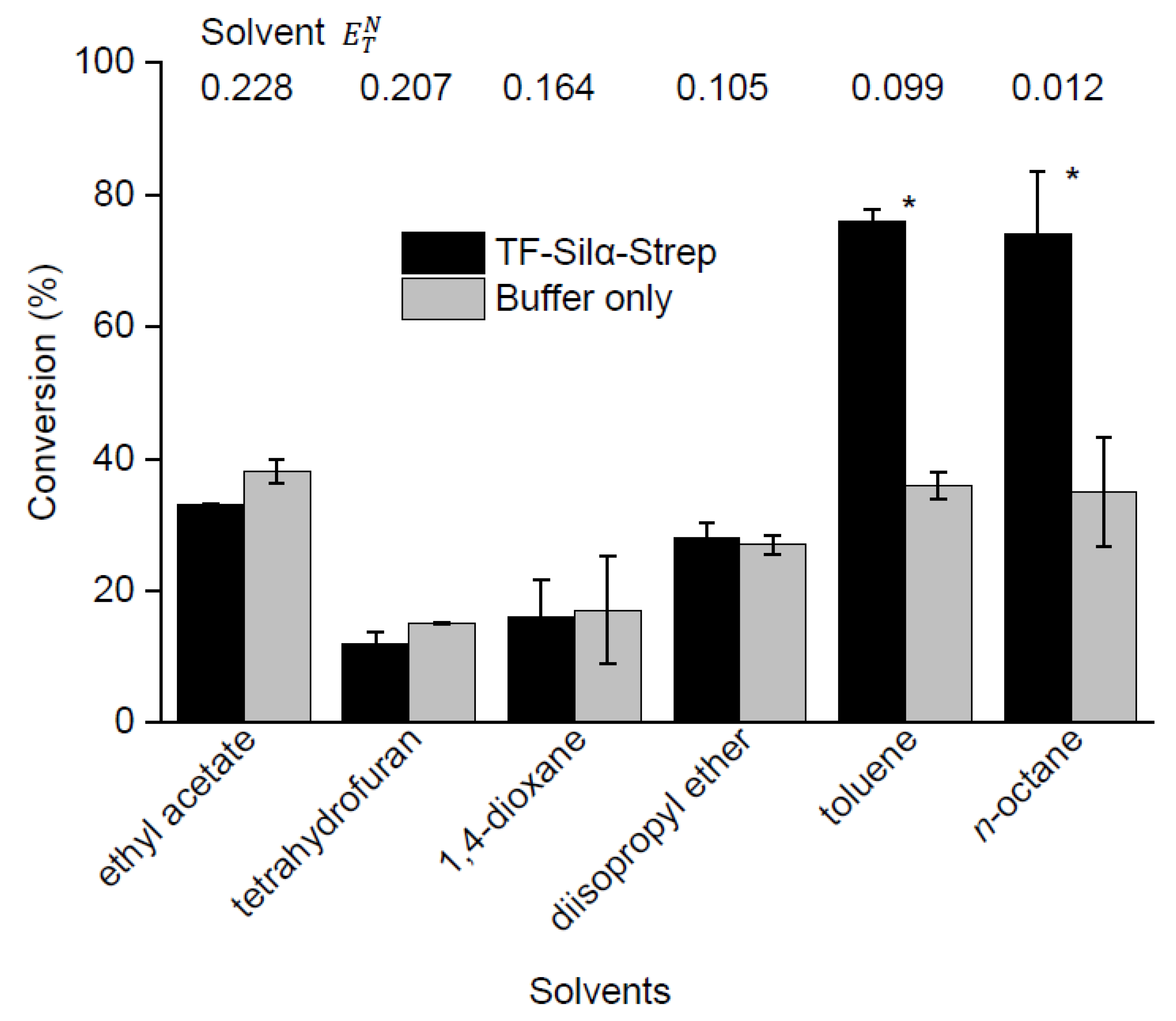
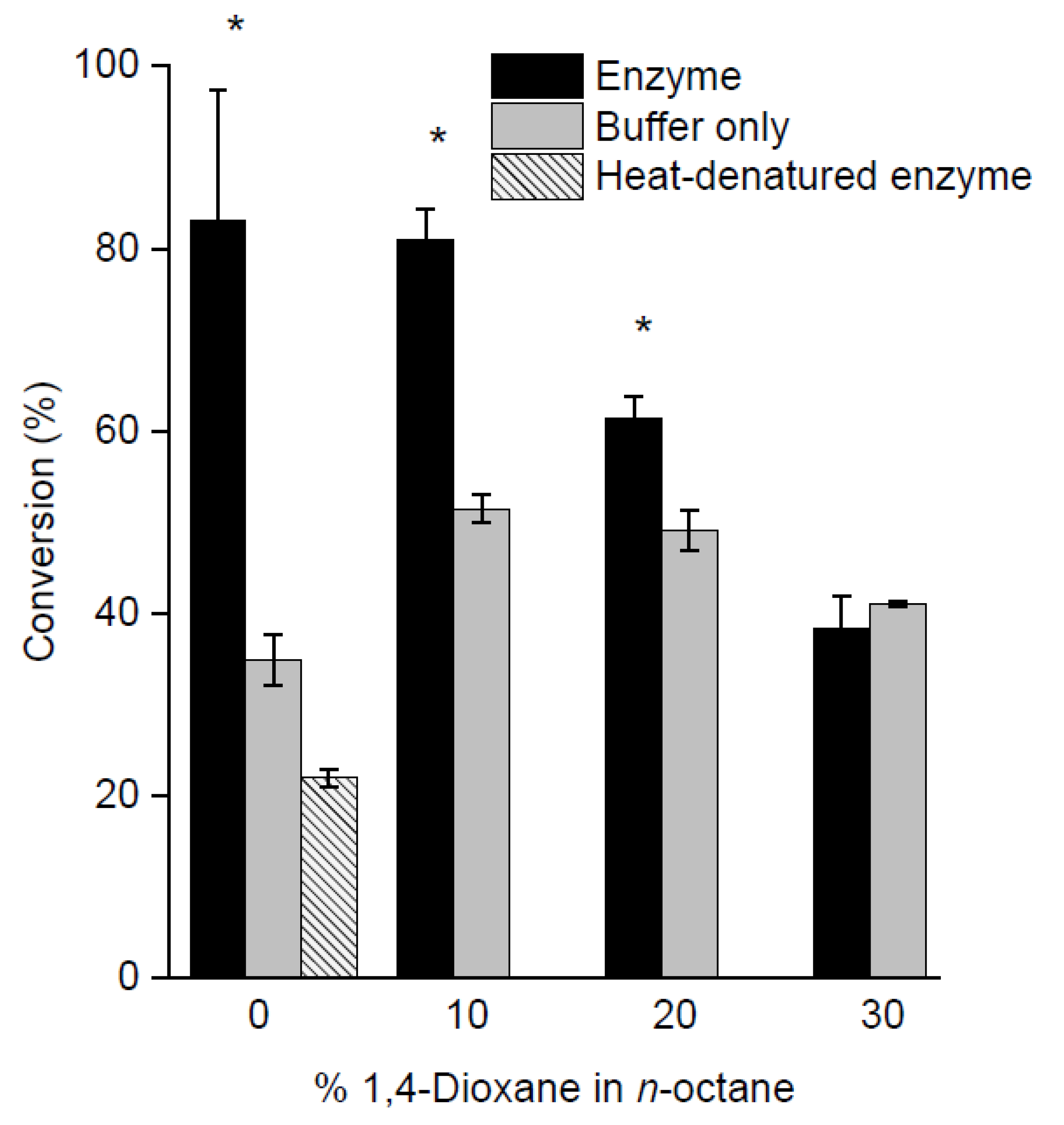
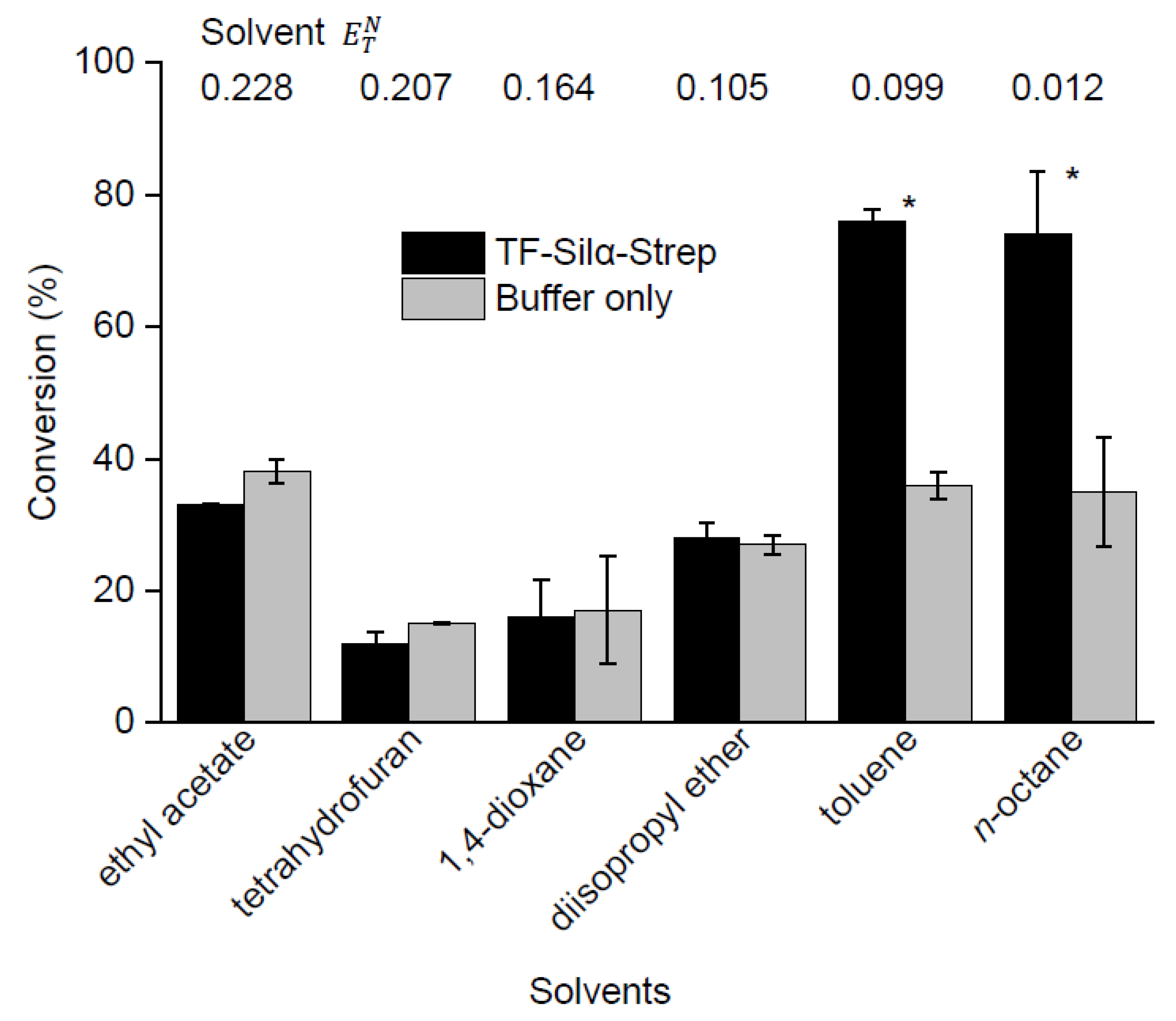
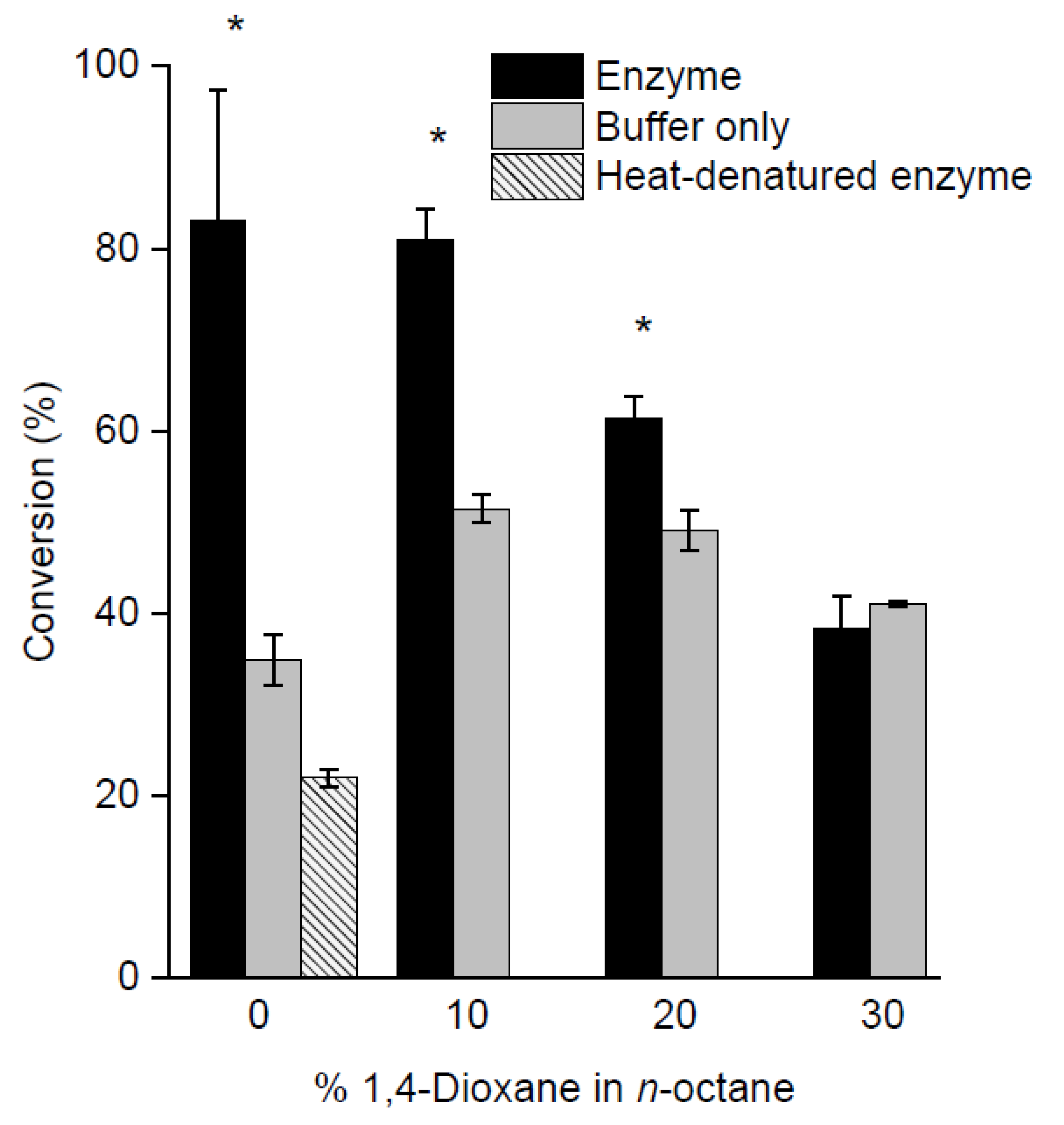
2.3. Screening of Reaction Media
3. Materials and Methods
3.1. Materials and Equipment
3.2. Preparation of Lyophilised Enzyme and Matrix
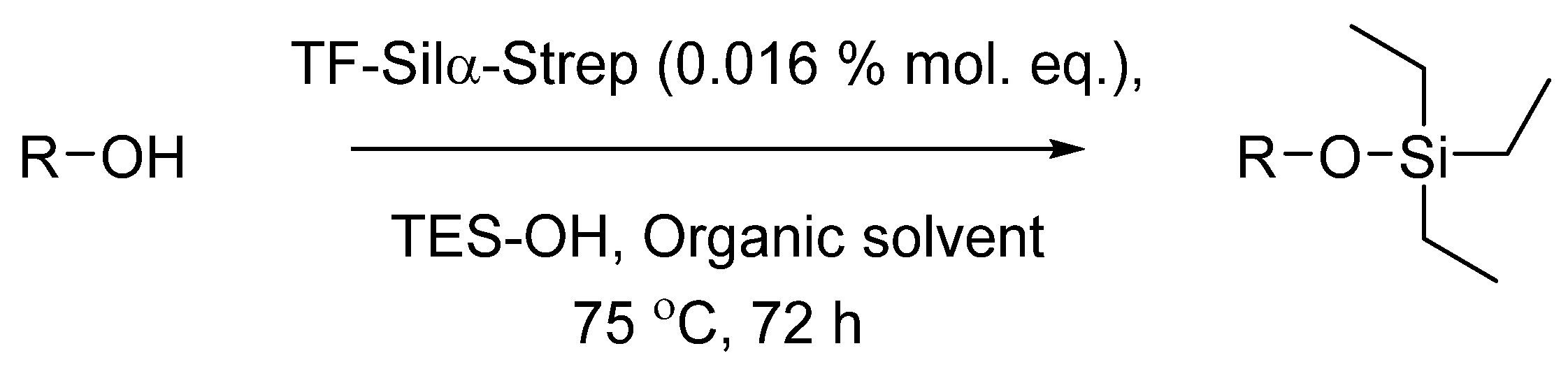
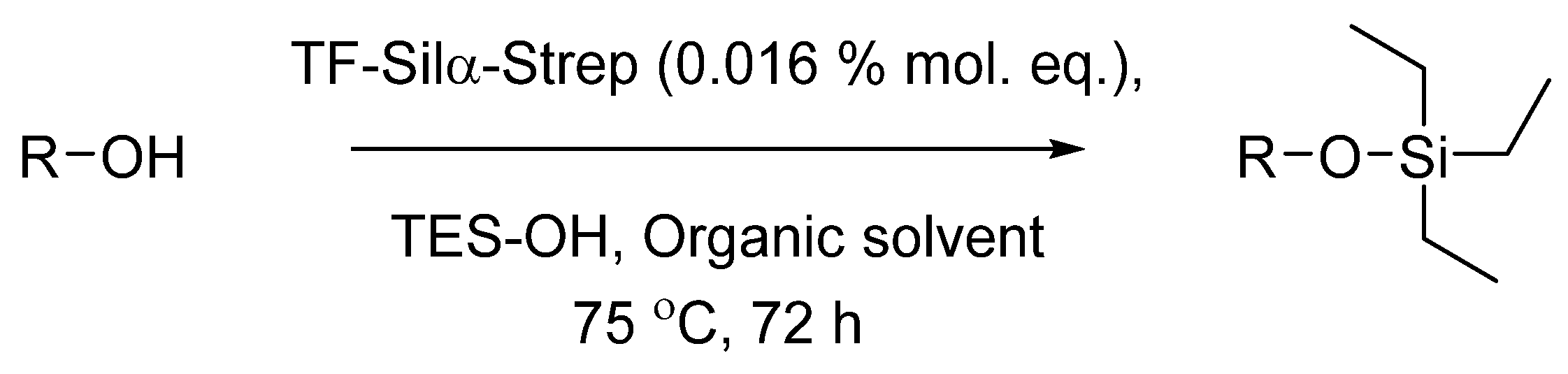
3.3. Enzymatic Condensation Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Smith, A.B.; Tomioka, T.; Risatti, C.A.; Sperry, J.B.; Sfouggatakis, C. Gram-scale synthesis of (+)-spongistatin 1: Development of an improved, scalable synthesis of the f-ring subunit, fragment union, and final elaboration. Org. Lett. 2008, 10, 4359–4362. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hansen, T.M.; Wang, T.; Wu, D.; Weyer, L.; Ying, L.; Engler, M.M.; Sanville, M.; Leitheiser, C.; Christmann, M.; et al. Total synthesis of phorboxazole a via de novo oxazole formation: Strategy and component assembly. J. Am. Chem. Soc. 2011, 133, 1484–1505. [Google Scholar] [CrossRef]
- Fürstner, A.; Bouchez, L.C.; Funel, J.-A.; Liepins, V.; Porée, F.-H.; Gilmour, R.; Beaufils, F.; Laurich, D.; Tamiya, M. Total syntheses of amphidinolide h and g. Angew. Chem. Int. Ed. 2007, 46, 9265–9270. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Jiang, X.; Lindsay-Scott, P.J.; Corbu, A.; Yamashiro, S.; Bacconi, A.; Fowler, V.M. Total synthesis and biological evaluation of monorhizopodin and 16-epi-monorhizopodin. Angew. Chem. Int. Ed. 2011, 50, 1139–1144. [Google Scholar] [CrossRef] [Green Version]
- Fortner, K.C.; Kato, D.; Tanaka, Y.; Shair, M.D. Enantioselective synthesis of (+)-cephalostatin 1. J. Am. Chem. Soc. 2010, 132, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, H.; Kikuchi, S.; Nakamura, Y.; Yamagami, Y.; Kishi, T.; Kadota, I.; Yamamoto, Y. Total synthesis of brevenal. Org. Lett. 2009, 11, 2531–2534. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohanna, J.; Zhou, J.; Iyer, K.; Rainier, J.D. Total synthesis of brevenal. J. Am. Chem. Soc. 2011, 133, 3208–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouch, R.D. Recent advances in silyl protection of alcohols. Synth. Commun. 2013, 43, 2265–2279. [Google Scholar] [CrossRef]
- Stalling, D.L.; Gehrke, C.W.; Zumwalt, R.W. A new silylation reagent for amino acids bis (trimethylsilyl)trifluoroacetamide (bstfa). Biochem. Biophys. Res. Commun. 1968, 31, 616–622. [Google Scholar] [CrossRef]
- Verboom, W.; Visser, G.W.; Reinhoudt, D.N. N,N′-bis[trimethylsilyl]-urea: A useful silylating agent for alcohols and carboxylic acids. Synthesis 1981, 1981, 807–809. [Google Scholar] [CrossRef]
- Smith, E.D.; Sheppard, H. Quantitative gas chromatography of amino-acids as trimethylsilyl derivatives. Nature 1965, 208, 878–880. [Google Scholar] [CrossRef]
- Kim, B.-h.; Woo, H.-G. Dehydrocoupling, redistributive coupling, and addition of main group 4 hydrides. Adv. Organomet. Chem. 2004, 52, 143–174. [Google Scholar] [CrossRef]
- Lukevics, E.; Dzintara, M. The alcoholysis of hydrosilanes. J. Organomet. Chem. 1985, 295, 265–315. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Foster, K.L.; Beck, V.H.; Piers, W.E. B(c6f5)3-catalyzed silation of alcohols: A mild, general method for synthesis of silyl ethers. J. Org. Chem. 1999, 64, 4887–4892. [Google Scholar] [CrossRef]
- Clive, D.L.J.; Kellner, D. A new method for silylation of hydroxylic compounds: Reaction of phenols and alcohols with silanols mediated by diethyl azodicarboxylate and triphenylphosphine. Tetrahedron Lett. 1991, 32, 7159–7160. [Google Scholar] [CrossRef]
- Nishiwaki, H.; Kiyomori, A.; Kubota, T. Silylation of Hydroxyl Groups. U.S. Patent US20030139619A1, 24 July 2003. [Google Scholar]
- Ran, N.; Zhao, L.; Chen, Z.; Tao, J. Recent applications of biocatalysis in developing green chemistry for chemical synthesis at the industrial scale. Green Chem. 2008, 10, 361–372. [Google Scholar] [CrossRef]
- Cha, J.N.; Shimizu, K.; Zhou, Y.; Christiansen, S.C.; Chmelka, B.F.; Stucky, G.D.; Morse, D.E. Silicatein filaments and subunits from a marine sponge direct the polymerization of silica and silicones in vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Cha, J.; Stucky, G.D.; Morse, D.E. Silicatein α: Cathepsin l-like protein in sponge biosilica. Proc. Natl. Acad. Sci. USA 1998, 95, 6234–6238. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei Dakhili, S.Y.; Caslin, S.A.; Faponle, A.S.; Quayle, P.; de Visser, S.P.; Wong, L.S. Recombinant silicateins as model biocatalysts in organosiloxane chemistry. Proc. Natl. Acad. Sci. USA. 2017, 114, E5285–E5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparkes, E.I.; Kettles, R.A.; Egedeuzu, C.S.; Stephenson, N.L.; Caslin, S.A.; Tabatabaei Dakhili, S.Y.; Wong, L.S. Improved production and biophysical analysis of recombinant silicatein-α. Biomolecules 2020, 10, 1209. [Google Scholar] [CrossRef] [PubMed]
- White, D.P.; Anthony, J.C.; Oyefeso, A.O. Computational measurement of steric effects: The size of organic substituents computed by ligand repulsive energies. J. Org. Chem. 1999, 64, 7707–7716. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Reichardt, C.; Harbusch-Görnert, E. Über pyridinium-n-phenolat-betaine und ihre verwendung zur charakterisierung der polarität von lösungsmitteln, x. Erweiterung, korrektur und neudefinition der et-lösungsmittelpolaritätsskala mit hilfe eines lipophilen penta-tert-butyl-substituierten pyridinium-n-phenolat-betainfarbstoffes. Liebigs Ann. Der Chem. 1983, 1983, 721–743. [Google Scholar] [CrossRef]
- Klibanov, A.M. Enzymatic catalysis in anhydrous organic solvents. Trends Biochem. Sci. 1989, 14, 141–144. [Google Scholar] [CrossRef]
- Stepankova, V.; Bidmanova, S.; Koudelakova, T.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. Strategies for stabilization of enzymes in organic solvents. Acs Catal. 2013, 3, 2823–2836. [Google Scholar] [CrossRef]
- Arnold, F.H. Design by directed evolution. Acc. Chem. Res. 1998, 31, 125–131. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Setting |
|---|---|
| Instrument | Agilent 5975 Series MSD |
| Carrier Gas | 9.9995% ultra-high-purity helium |
| GC Inlet, Split | 240 °C, split flow 100 mL min−1, split ratio 50 |
| MS Ionisation | Electron ionisation |
| GC Temperature Program | 50 °C (2 min)→240 °C (8.5 min) at 30 °C min−1, 10.7 min total run time |
| GC Column | VF-5ht |
| Volume Injected | 1 μL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparkes, E.I.; Egedeuzu, C.S.; Lias, B.; Sung, R.; Caslin, S.A.; Tabatabaei Dakhili, S.Y.; Taylor, P.G.; Quayle, P.; Wong, L.S. Biocatalytic Silylation: The Condensation of Phenols and Alcohols with Triethylsilanol. Catalysts 2021, 11, 879. https://doi.org/10.3390/catal11080879
Sparkes EI, Egedeuzu CS, Lias B, Sung R, Caslin SA, Tabatabaei Dakhili SY, Taylor PG, Quayle P, Wong LS. Biocatalytic Silylation: The Condensation of Phenols and Alcohols with Triethylsilanol. Catalysts. 2021; 11(8):879. https://doi.org/10.3390/catal11080879
Chicago/Turabian StyleSparkes, Emily I., Chisom S. Egedeuzu, Billie Lias, Rehana Sung, Stephanie A. Caslin, S. Yasin Tabatabaei Dakhili, Peter G. Taylor, Peter Quayle, and Lu Shin Wong. 2021. "Biocatalytic Silylation: The Condensation of Phenols and Alcohols with Triethylsilanol" Catalysts 11, no. 8: 879. https://doi.org/10.3390/catal11080879
APA StyleSparkes, E. I., Egedeuzu, C. S., Lias, B., Sung, R., Caslin, S. A., Tabatabaei Dakhili, S. Y., Taylor, P. G., Quayle, P., & Wong, L. S. (2021). Biocatalytic Silylation: The Condensation of Phenols and Alcohols with Triethylsilanol. Catalysts, 11(8), 879. https://doi.org/10.3390/catal11080879