
Critical Role of Al Pair Sites in Methane Oxidation to Methanol on Cu-Exchanged Mordenite Zeolites

 , and
, and
Abstract
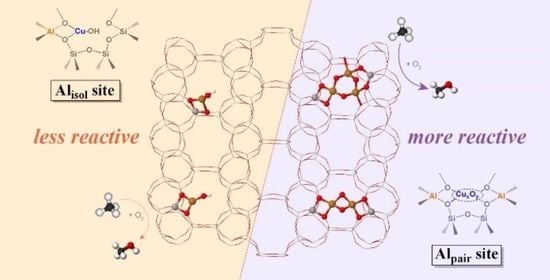
1. Introduction
2. Results and Discussion
2.1. Preparation and Characterization of Cu-MOR Catalysts
2.2. Reactivity of Cu-MOR Catalysts for Methane Oxidation
2.3. In Situ UV-Vis Characterization of Methane Oxidation on Cu-MOR
2.4. Theoretical Assessment of Methane Oxidation on Cu-MOR
3. Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Testing of Activity for Methane Oxidation to Methanol
3.4. Ultraviolet-Visible Spectroscopy Measurements
3.5. Periodic Density Functional Theory Treatments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Saha, D.; Grappe, H.A.; Chakraborty, A.; Orkoulas, G. Postextraction Separation, On-Board Storage, and Catalytic Conversion of Methane in Natural Gas: A Review. Chem. Rev. 2016, 116, 11436–11499. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Guan, N.; Li, L. Methane Activation and Utilization: Current Status and Future Challenges. Energy Technol. 2019, 8, 1900826. [Google Scholar] [CrossRef]
- Bao, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Significant Advances in C1 Catalysis: Highly Efficient Catalysts and Catalytic Reactions. ACS Catal. 2019, 9, 3026–3053. [Google Scholar] [CrossRef]
- Lian, M.; Wang, Y.; Zhao, M.; Li, Q.; Weng, W.; Xia, W.; Wan, H. Stability of Ni/SiO2 in Partial Oxidation of Methane: Effects of W Modification. Acta Phys. Chim. Sin. 2019, 35, 607–615. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic Zeolite Modification for in situ Peroxide Formation in Methane Oxidation to Methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Wang, C.; Xie, Z.; Guan, N.; Li, L. Water-Involved Methane-Selective Catalytic Oxidation by Dioxygen over Copper Zeolites. Chem 2021, 7, 1557–1568. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.E.R.; Bols, M.L.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev. 2018, 118, 2718–2768. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Yoshizawa, K. Methane selective oxidation to methanol by metal-exchanged zeolites: A review of active sites and their reactivity. Catal. Sci. Technol. 2019, 9, 1744–1768. [Google Scholar] [CrossRef]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: A critical examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef] [PubMed]
- Que, L., Jr.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Bozbag, S.E.; Alayon, E.M.C.; Pechacek, J.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Methane to methanol over copper mordenite: Yield improvement through multiple cycles and different synthesis techniques. Catal. Sci. Technol. 2016, 6, 5011–5022. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective oxidation of methane by the bis(mu-oxo)dicopper core stabilized on ZSM-5 and mordenite zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Grundner, S.; Luo, W.; Sanchez-Sanchez, M.; Lercher, J.A. Synthesis of single-site copper catalysts for methane partial oxidation. Chem. Commun. 2016, 52, 2553–2556. [Google Scholar] [CrossRef] [PubMed]
- Grundner, S.; Markovits, M.A.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Gobel, C.; Schlesiger, C.; Malzer, W.; Trunschke, A.; Schomacker, R.; Thomas, A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Roman-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2018, 57, 8906–8910. [Google Scholar] [CrossRef]
- Wang, G.R.; Chen, W.; Huang, L.; Liu, Z.Q.; Sun, X.Y.; Zheng, A.M. Reactivity descriptors of diverse copper-oxo species on ZSM-5 zeolite towards methane activation. Catal. Today 2019, 338, 108–116. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci.USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.-J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Knorpp, A.; Pinar, A.B.; Baerlocher, C.; McCusker, L.B.; Casati, N.; Newton, M.A.; Checchia, S.; Meyet, J.; Palagin, D.; van Bokhoven, J.A. Paired copper monomers in zeolite omega: The active site for methane-to-methanol conversion. Angew. Chem. Int. Ed. 2021, 60, 5854–5858. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Palagin, D.; Knorpp, A.J.; Pinar, A.B.; Ranocchiari, M.; van Bokhoven, J.A. Assessing the relative stability of copper oxide clusters as active sites of a CuMOR zeolite for methane to methanol conversion: Size matters? Nanoscale 2017, 9, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Dědeček, J.; Kaucký, D.; Wichterlová, B. Co2+ ion siting in pentasil-containing zeolites, part 3. Microporous Mesoporous Mater. 2000, 35–36, 483–494. [Google Scholar] [CrossRef]
- Dědeček, J.; Kaucký, D.; Wichterlová, B. Al distribution in ZSM-5 zeolites: An experimental study. Chem. Commun. 2001, 970–971. [Google Scholar] [CrossRef]
- Dědeček, J.; Kaucký, D.; Wichterlová, B.; Gonsiorová, O. Co2+ions as probes of Al distribution in the framework of zeolites. ZSM-5 study. Phys. Chem. Chem. Phys. 2002, 4, 5406–5413. [Google Scholar] [CrossRef]
- Karcz, R.; Dedecek, J.; Supronowicz, B.; Thomas, H.M.; Klein, P.; Tabor, E.; Sazama, P.; Pashkova, V.; Sklenak, S. TNU-9 Zeolite: Aluminum Distribution and Extra-Framework Sites of Divalent Cations. Chem. Eur. J. 2017, 23, 8857–8870. [Google Scholar] [CrossRef] [PubMed]
- Mlekodaj, K.; Dedecek, J.; Pashkova, V.; Tabor, E.; Klein, P.; Urbanova, M.; Karcz, R.; Sazama, P.; Whittleton, S.R.; Thomas, H.M.; et al. Al Organization in the SSZ-13 Zeolite. Al Distribution and Extraframework Sites of Divalent Cations. J. Phys. Chem. C 2018, 123, 7968–7987. [Google Scholar] [CrossRef]
- Indovina, V.; Campa, M.C.; Pietrogiacomi, D. Isolated Co2+ and [Co−O−Co]2+ Species in Na-MOR Exchanged with Cobalt to Various Extents: An FTIR Characterization by CO Adsorption of Oxidized and Prereduced Samples. J. Phys. Chem. C 2008, 112, 5093–5101. [Google Scholar] [CrossRef]
- Gounder, R.; Iglesia, E. The catalytic diversity of zeolites: Confinement and solvation effects within voids of molecular dimensions. Chem. Commun. 2013, 49, 3491–3509. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal cyclic conversion of methane into methanol over copper-exchanged zeolite at low temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef]
- Brezicki, G.; Kammert, J.D.; Gunnoe, T.B.; Paolucci, C.; Davis, R.J. Insights into the speciation of Cu in the Cu-H-mordenite catalyst for the oxidation of methane to methanol. ACS Catal. 2019, 9, 5308–5319. [Google Scholar] [CrossRef]
- Ikuno, T.; Grundner, S.; Jentys, A.; Li, G.; Pidko, E.; Fulton, J.; Sanchez-Sanchez, M.; Lercher, J.A. Formation of Active Cu-oxo Clusters for Methane Oxidation in Cu-Exchanged Mordenite. J. Phys. Chem. C 2019, 123, 8759–8769. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic definition of the copper active sites in mordenite: Selective methane oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef]
- Dědeček, J.; Sklenak, S.; Li, C.; Wichterlová, B.; Gábová, V.; Brus, J.; Sierka, M.; Sauer, J. Effect of Al−Si−Al and Al−Si−Si−Al Pairs in the ZSM-5 Zeolite Framework on the 27Al NMR Spectra. A Combined High-Resolution 27Al NMR and DFT/MM Study. J. Phys. Chem. C 2009, 113, 1447–1458. [Google Scholar] [CrossRef]
- Takaishi, T.; Kato, M.; Itabashi, K. Determination of the ordered distribution of aluminum atoms in a zeolitic framework. Part II. Zeolites 1995, 15, 21–32. [Google Scholar] [CrossRef]
- Takaishi, T.; Kato, M.; Itabashi, K. Stability of the Al-O-Si-O-Al Linkage in a Zeolitic Framework. J. Phys. Chem. 1994, 98, 5742–5743. [Google Scholar] [CrossRef]
- Arvidsson, A.A.; Zhdanov, V.P.; Carlsson, P.-A.; Grönbeck, H.; Hellman, A. Metal dimer sites in ZSM-5 zeolite for methane-to-methanol conversion from first-principles kinetic modelling: Is the [Cu–O–Cu]2+ motif relevant for Ni, Co, Fe, Ag, and Au? Catal. Sci. Technol. 2017, 7, 1470–1477. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B Condens. Matter 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Si/Al | Alacce (%) b | Alpair (%) b | Alisol (%) b | Alpair/Alisol |
|---|---|---|---|---|---|
| MOR-A | 14 | 57 | 50 | 7 | 7.1 |
| MOR-B | 20 | 31 | 14 | 17 | 0.82 |
| MOR-C | 40 | 65 | 46 | 19 | 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, P.; Zhang, Z.; Chen, Z.; Lin, J.; Wan, S.; Wang, Y.; Wang, S. Critical Role of Al Pair Sites in Methane Oxidation to Methanol on Cu-Exchanged Mordenite Zeolites. Catalysts 2021, 11, 751. https://doi.org/10.3390/catal11060751
Han P, Zhang Z, Chen Z, Lin J, Wan S, Wang Y, Wang S. Critical Role of Al Pair Sites in Methane Oxidation to Methanol on Cu-Exchanged Mordenite Zeolites. Catalysts. 2021; 11(6):751. https://doi.org/10.3390/catal11060751
Chicago/Turabian StyleHan, Peijie, Zhaoxia Zhang, Zheng Chen, Jingdong Lin, Shaolong Wan, Yong Wang, and Shuai Wang. 2021. "Critical Role of Al Pair Sites in Methane Oxidation to Methanol on Cu-Exchanged Mordenite Zeolites" Catalysts 11, no. 6: 751. https://doi.org/10.3390/catal11060751
APA StyleHan, P., Zhang, Z., Chen, Z., Lin, J., Wan, S., Wang, Y., & Wang, S. (2021). Critical Role of Al Pair Sites in Methane Oxidation to Methanol on Cu-Exchanged Mordenite Zeolites. Catalysts, 11(6), 751. https://doi.org/10.3390/catal11060751