
Bio-DEE Synthesis and Dehydrogenation Coupling of Bio-Ethanol to Bio-Butanol over Multicomponent Mixed Metal Oxide Catalysts

 , , , , ,
, , , , ,
Abstract
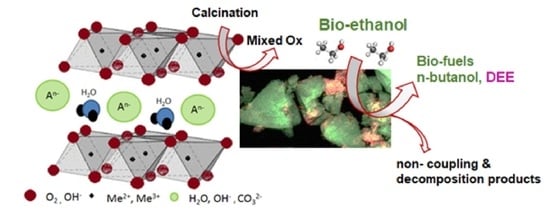
1. Introduction
2. Results
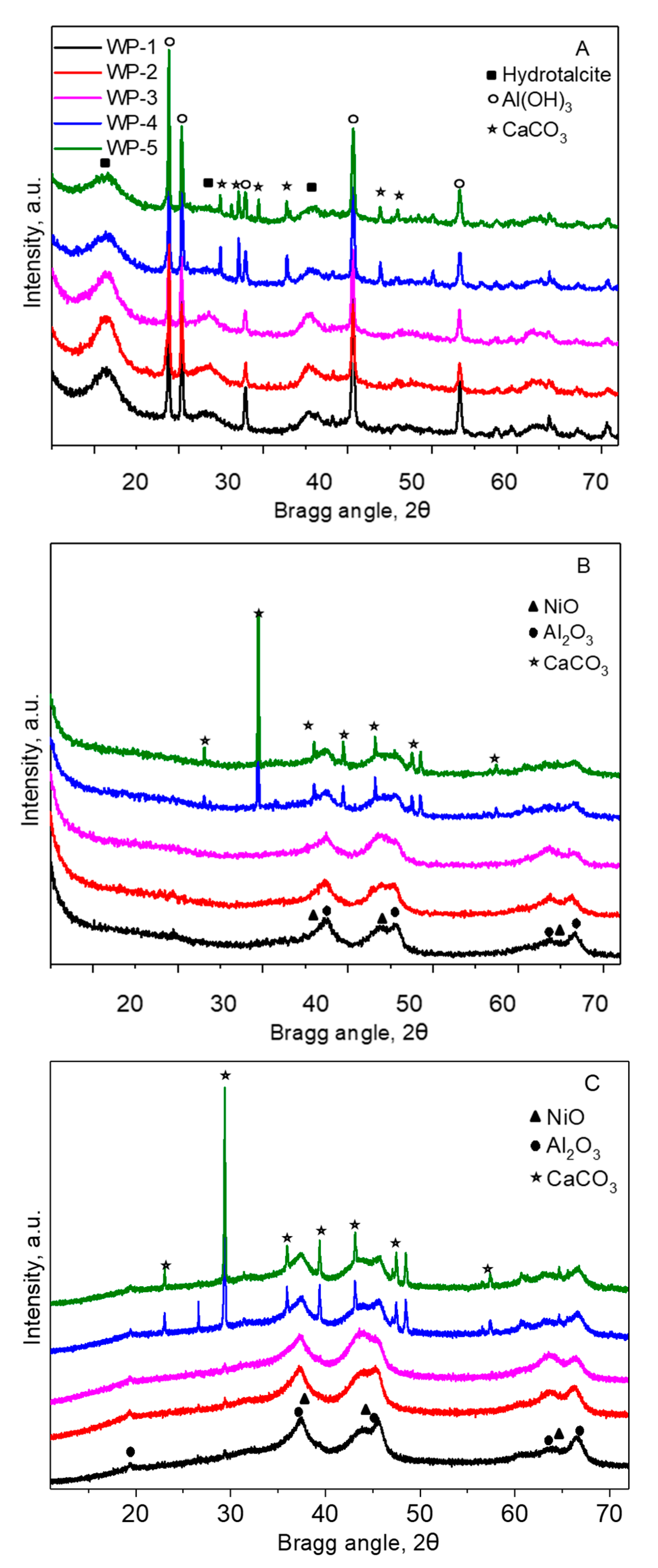
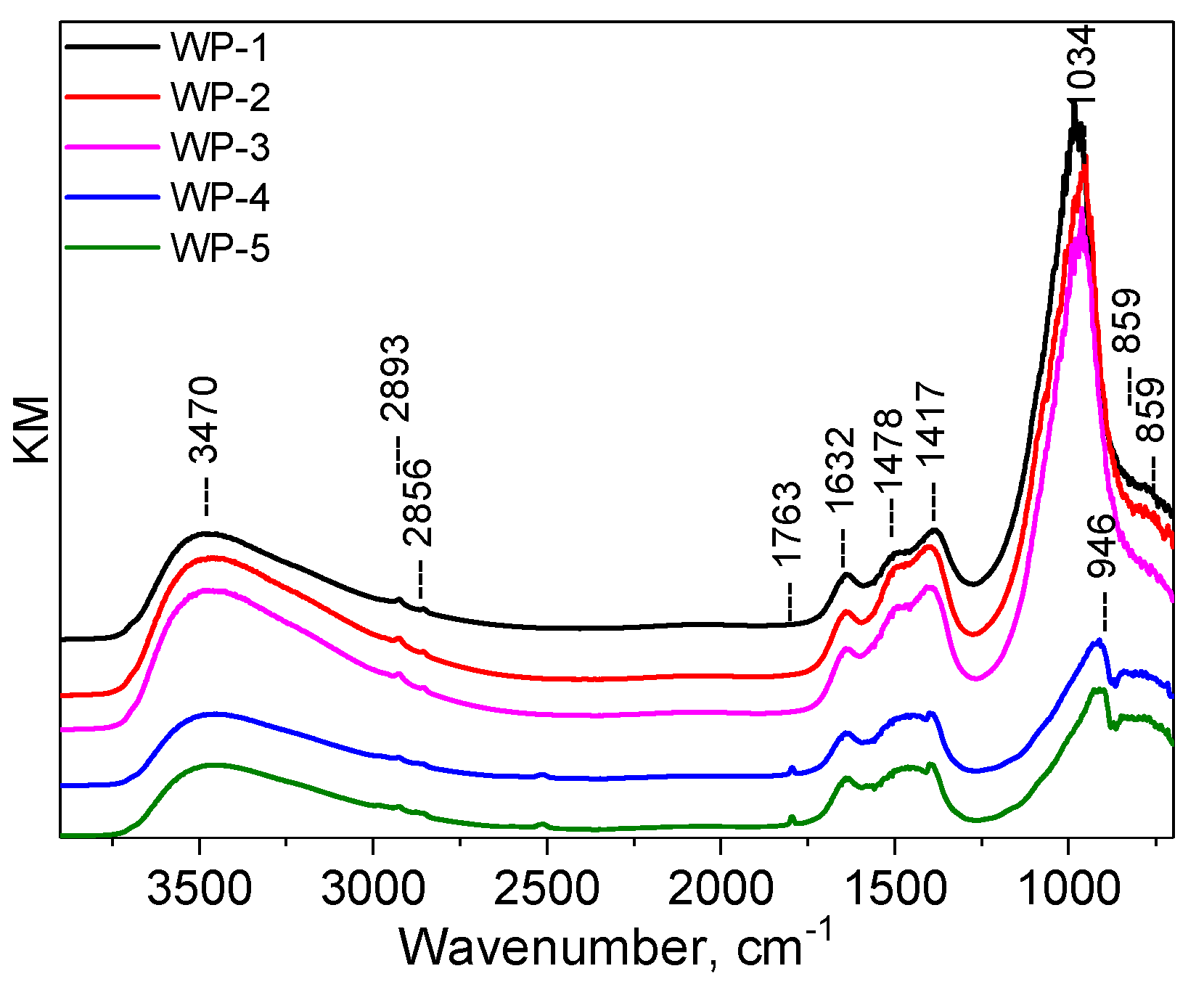
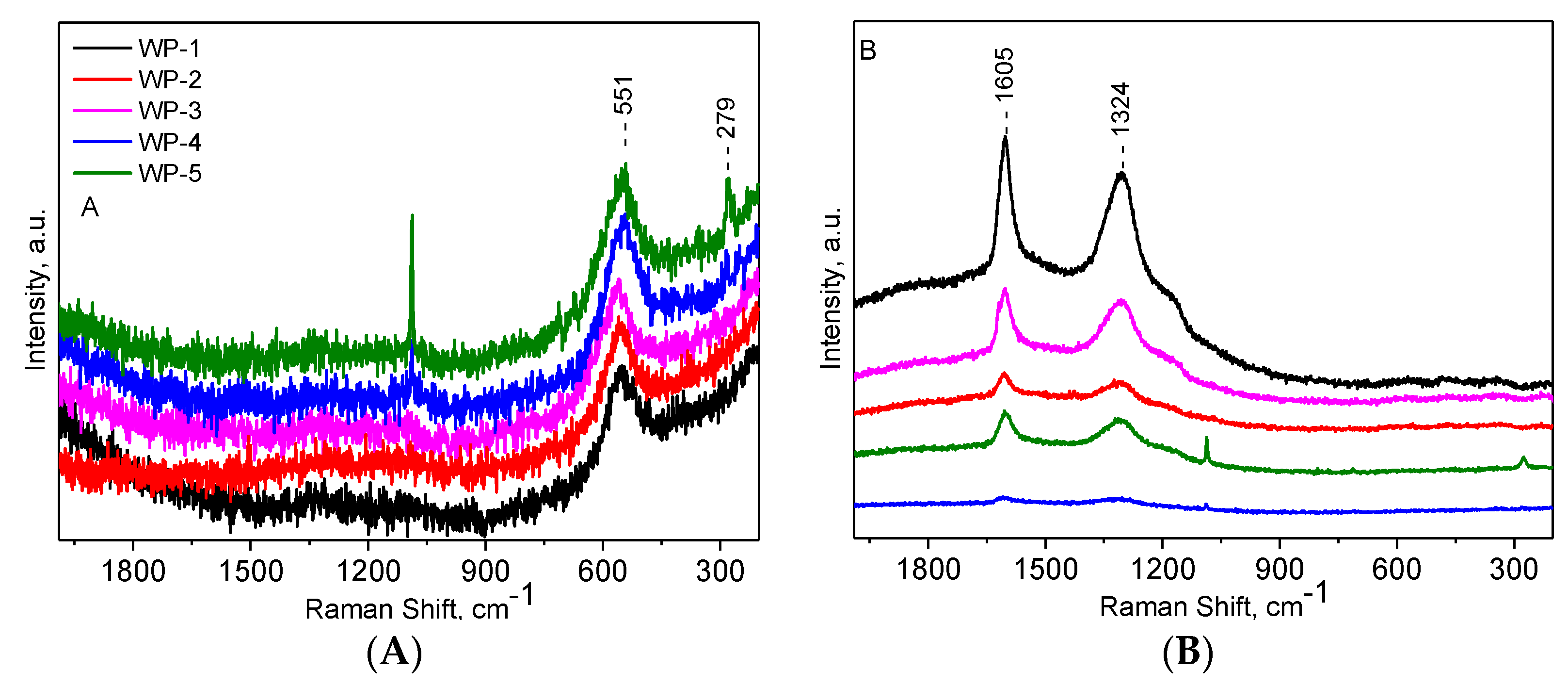
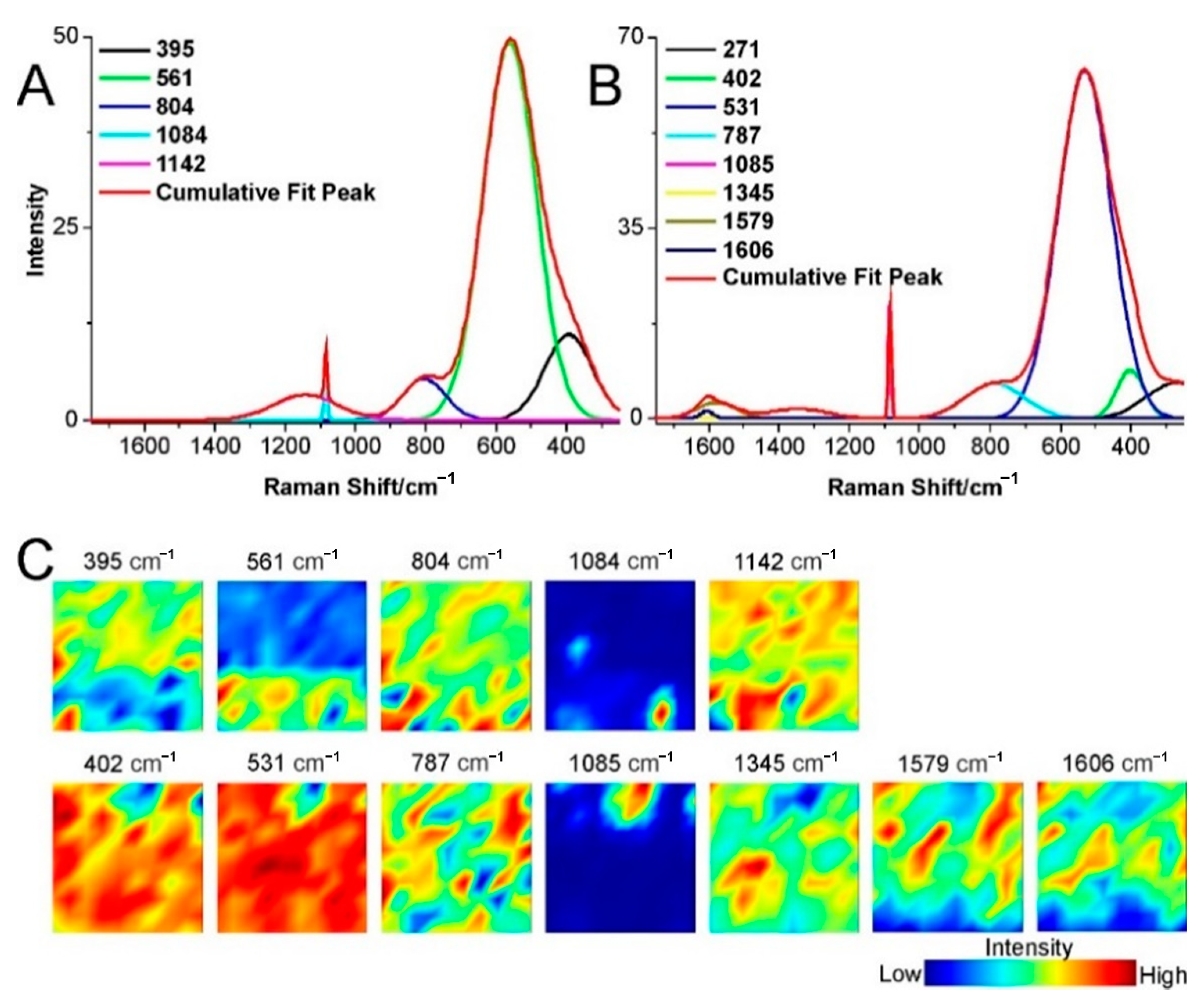
2.1. Catalyst Characterization
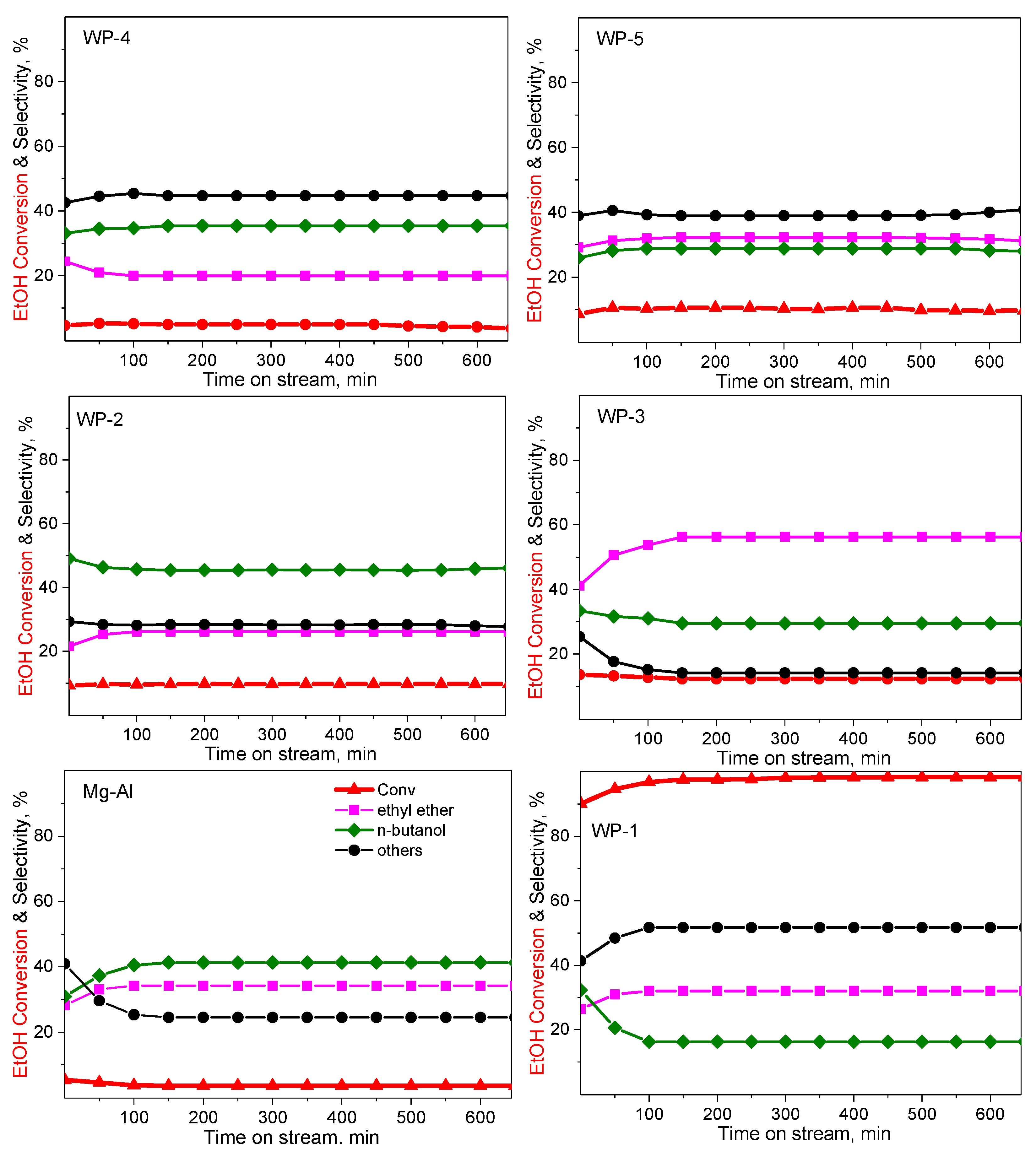
2.2. Catalytic Performance
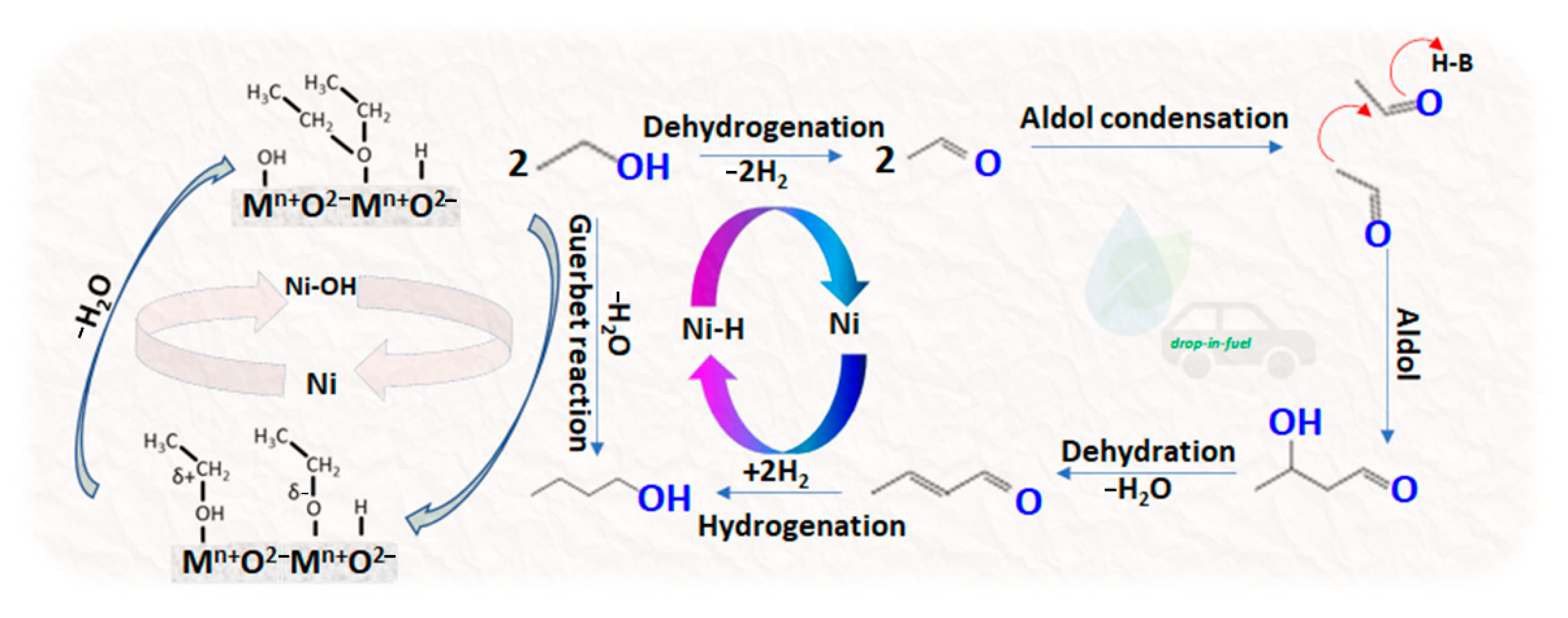
- Alcohol oxidation (dehydrogenation) to aldehyde:2RCH2CH2OH ↔ 2RCH2CHO + 2H2
- Aldol condensation of aldehydes with the elimination of water (C-C bond-forming) to an aldehyde:2RCH2CHO ↔ RCH2CH = CRCHO + H2O
- Additionally, allylic aldehyde reduction (hydrogenation) to alcohol:RCH2CH = CRCHO + 2H2 ↔ RCH2CH2CHRCH2OH
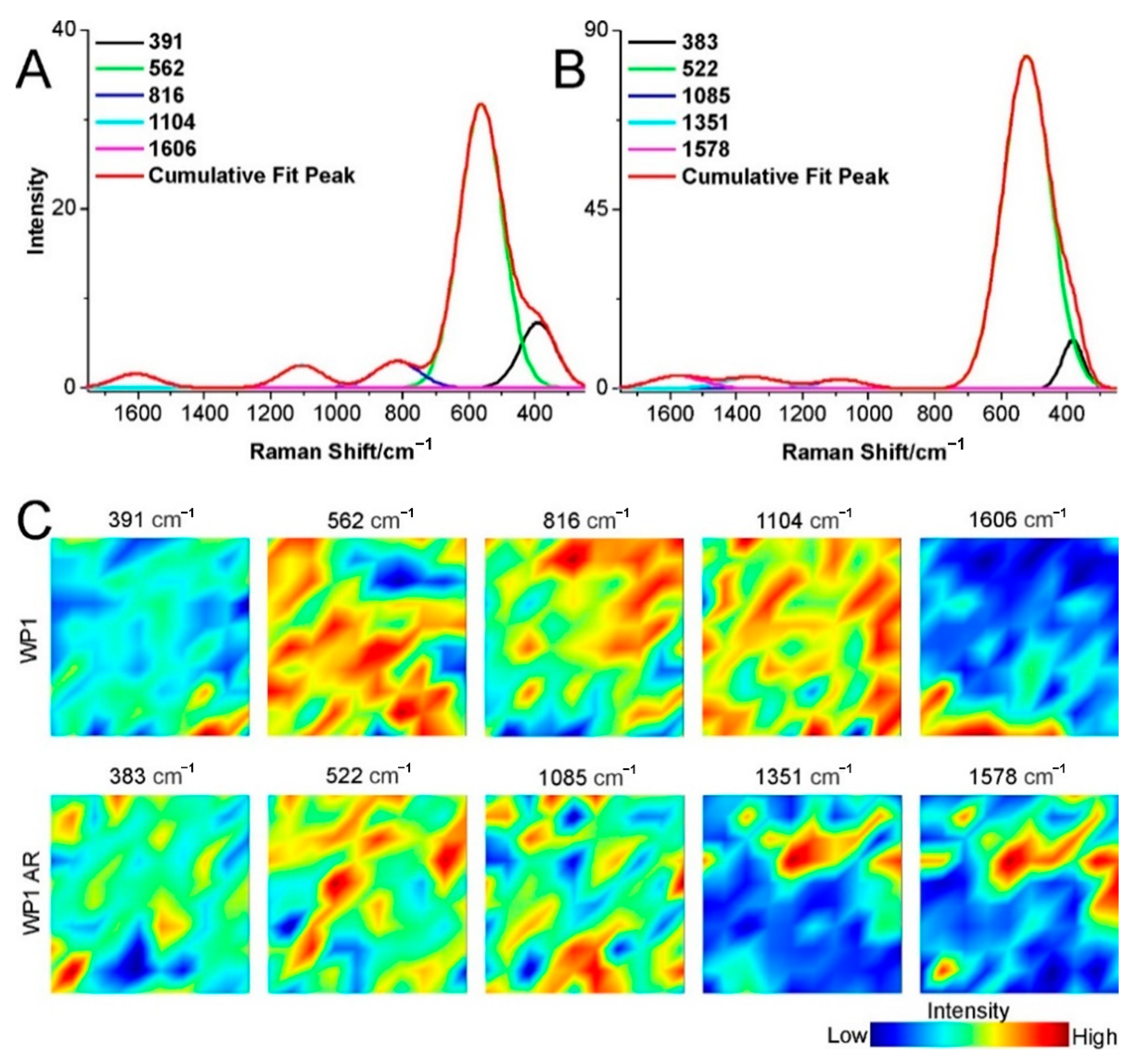
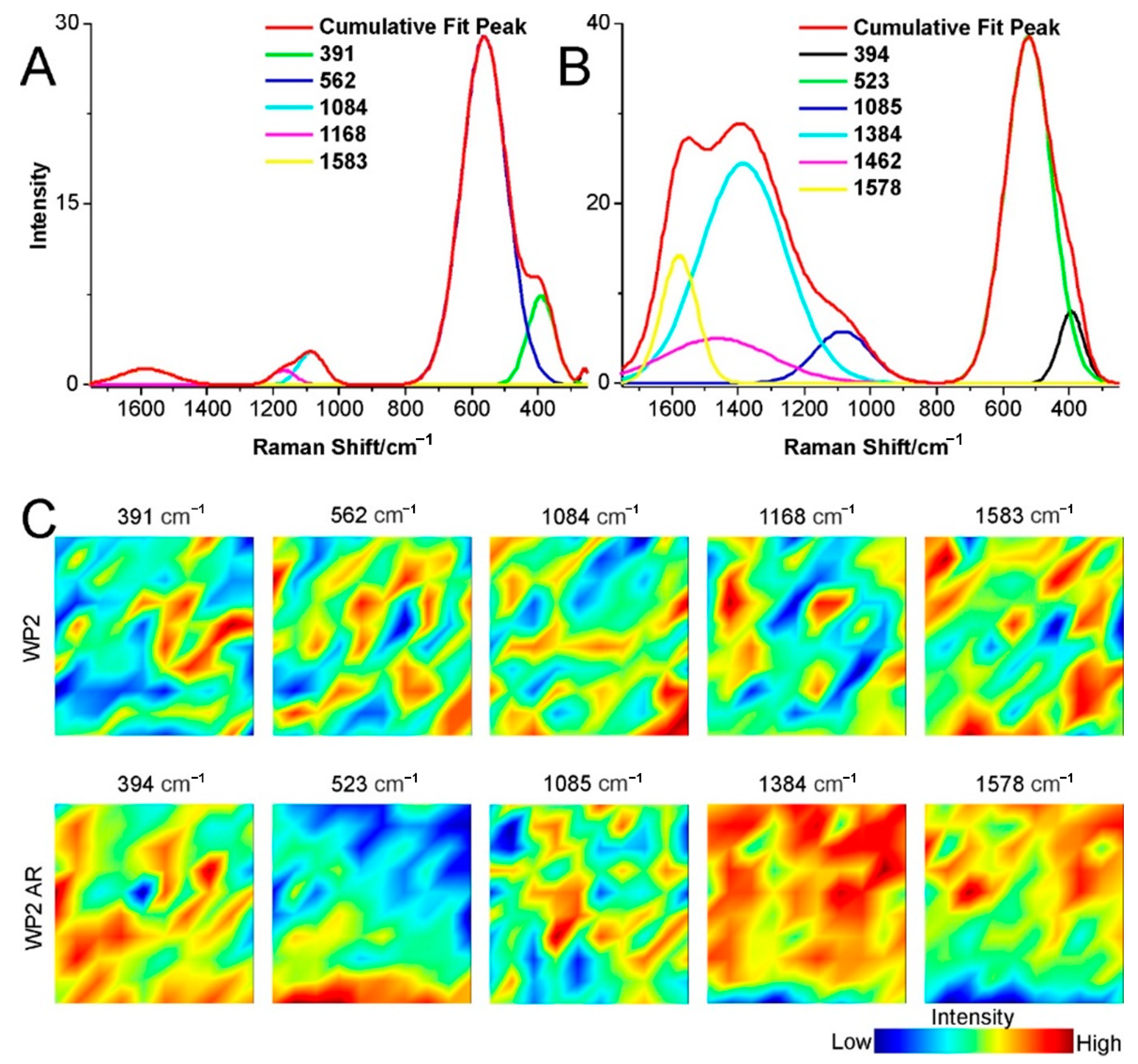
2.3. Structural Changes of Catalyst
3. Materials and Methods
3.1. Materials and Reagents
3.2. Synthesis and Characterization
3.3. Catalytic Tests
3.4. Raman Spectroscopy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Houghton, J.T.; Jenkins, G.J.; Ephraums, J.J. Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 1990; ISBN 978-0-521-40360-3. [Google Scholar]
- Colmenares, J.C.; Colmenares Quintero, R.F.; Pieta, I.S. Catalytic Dry Reforming for Biomass-Based Fuels Processing: Progress and Future Perspectives. Energy Technol. 2016, 4, 881–890. [Google Scholar] [CrossRef]
- Gawande, M.B.; Goswami, A.; Felpin, F.X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysi. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Rathi, A.K.; Aparicio, C.; Tomanec, O.; Petr, M.; Pocklanova, R.; Gawande, M.B.; Varma, R.S.; Zboril, R. In situ generation of Pd–Pt core–shell nanoparticles on reduced graphene oxide (Pd@ Pt/rGO) using microwaves: Applications in dehalogenation reactions and reduction of olefins. ACS Appl. Mater. Interfaces 2017, 9, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-X.; Wang, A.-J.; Fang, K.-M.; Yuan, J.; Feng, J.-J. One-Pot Seedless Aqueous Synthesis of Reduced Graphene Oxide (rGO)-Supported Core–Shell Pt@Pd Nanoflowers as Advanced Catalysts for Oxygen Reduction and Hydrogen Evolution. ACS Sustain. Chem. Eng. 2017, 5, 8675–8683. [Google Scholar] [CrossRef]
- Gibson, G.; Winne, S.; Levin, S.; Skinner, I.; Tsamis, A.; Twisse, F.; Biedka, M.; Brannigan, C. Transport. State of the Art onAlternative FuelsTransport Systemsin the European Union; Update 2020; European Commission: Brussels, Belgium, 2020. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- European Environment Agency. “Energy”. Available online: https://www.eea.europa.eu/themes/energy/intro (accessed on 15 November 2020).
- EU Agency, Transport Environment. Roadmap to De-Carbonising European Shipping. Available online: https://www.transportenvironment.org/sites/te/files/publications/2018_11_Roadmap_decarbonising_European_shipping.pdf (accessed on 15 November 2020).
- Kowalik, P.; Antoniak-Jurak, K.; Błesznowski, M.; Herrera, M.C.; Larrubia, M.A.; Alemany, L.J.; Pieta, I.S. Biofuel steam reforming catalyst for fuel cell application. Catal. Today 2015, 254, 129. [Google Scholar] [CrossRef]
- Zinoviev, S.; Müller-Langer, F.; Das, P.; Bertero, N.; Fornasiero, P.; Kaltschmitt, M.; Centi, G.; Miertus, S. Next-generation biofuels: Survey of emerging technologies and sustainability issues. ChemSusChem 2010, 3, 1106–1133. [Google Scholar] [CrossRef]
- Pieta, I.S.; Epling, W.S.; Kazmierczuk, A.; Lisowski, P.; Nowakowski, R.; Serwicka, E. Waste into Fuel-Catalyst and Process Development for MSW Valorisation. Catalysts 2018, 8, 113–128. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Sattler, C. A review on solar thermal syngas production via redox pair-based water/carbon dioxide splitting thermochemical cycles. Renew. Sustain. Energy Rev. 2015, 42, 254–285. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J. Syngas in Perspective. Catal. Today 2002, 71, 243–247. [Google Scholar] [CrossRef]
- EU. Status Review of Renewable Support. Schemes in Europe for 2016 and 2017; Ref: C18-SD-63-0314; Council of European Energy Regulators: Brussels, Belgium, 2018. [Google Scholar]
- Matsakas, L.; Gao, Q.; Jansson, S.; Rova, U.; Christakopoulos, P. Green conversion of municipal solid wastes into fuels and chemicals. Electron. J. Biotechnol. 2017, 26, 69–83. [Google Scholar] [CrossRef]
- Chum, H.L.; Warner, E.; Seabra, J.E.A.; Macedo, I.C. A comparison of commercial ethanol production systems from Brazilian sugarcane and US corn. Biofuels Bioprod. Biorefining 2014, 8, 205–223. [Google Scholar] [CrossRef]
- Susmozas, A.; Martin-Sampedro, R.; Ibarra, D.; Eugenio, M.E.; Iglesias, R.; Manzanares, P.; Moreno, A.D. Process Strategies for the Transition of 1G to Advanced Bioethanol Production. Processes 2020, 8, 1310. [Google Scholar] [CrossRef]
- Kraleva, E.; Rodrigues, C.P.; Pohl, M.M.; Ehrich, H.; Noronh, F.B. Syngas production by partial oxidation of ethanol on PtNi/SiO2–CeO2 catalysts. Catal. Sci. Technol. 2019, 9, 634. [Google Scholar] [CrossRef]
- Kraleva, E.; Sokolov, S.; Schneider, M.; Ehrich, H. Support effects on the properties of Co and Ni catalysts for the hydrogen production from bio-ethanol partial oxidation. Int. J. Hydr. Energy 2013, 38, 4380–4388. [Google Scholar] [CrossRef]
- Busic, A.; Mardetko, N.; Kundas, S.; Morzak, G.; Belskaya, H.; Ivancic Santek, M.; Komes, D.; Novak, S.; Santek, B. Bioethanol Production from Renewable Raw Materials and Its Separation and Purification: A Review. Food Technol Biotechnol. 2018, 56, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Karatzos, S.; McMillan, J.; Saddler, J. The Potential and Challenges of “Drop in” Biofuels International Energy Agency Bioenergy. Available online: http://www.ieabioenergy.com/wp-content/uploads/2014/05/P02-The-potential-and-challenges-of-drop-in-biofuels-Karatzos.pdf (accessed on 15 November 2020).
- León, M.; Díaz, E.; Ordónez, S. Ethanol catalytic condensation over Mg-Al mixed oxides derived from hydrotalcites. Catal. Today 2011, 164, 436–442. [Google Scholar] [CrossRef]
- Bravo-Suárez, J.J.; Subramaniam, B.; Chaudhari, V.R. Vapor-phase methanol and ethanol coupling reactions on CuMgAl mixed metal oxides. Appl. Catal. A 2013, 455, 234–246. [Google Scholar] [CrossRef]
- Yang, C.; Meng, Z.Y. Bimolecular condensation of ethanol to 1-butanol catalyzed by Alkali Cation Zeolites. J. Catal. 1993, 142, 37–44. [Google Scholar] [CrossRef]
- Ndou, A.S.; Plint, N.; Coville, N.J. Dimerisation of ethanol to butanol over solid-base catalysts. Appl. Catal. A 2003, 251, 337–345. [Google Scholar] [CrossRef]
- Gautam, M.; Martin, D.W. Combustion characteristics of higher-alcohol/gasoline blends. Proc. Inst. Mech. Eng. 2000, 214, 497–511. [Google Scholar] [CrossRef]
- Herman, R.G. Advances in catalytic synthesis and utilization of higher alcohols. Catal. Today 2000, 55, 233–245. [Google Scholar] [CrossRef]
- Ndaba, B.; Chiyanzu, I.; Marx, S. n-Butanol derived from biochemical and chemical routes: A review. Biotechnol. Rep. 2015, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Colley, S.W.; Fawcett, C.R.; Rathmell, C.; Tuck, M.W. Process for the Preparation of Ethylacetate. Patent USOO68092.17B1, 26 October 2004. [Google Scholar]
- Matthey, J. Davy Technologies. Available online: http://www.davyprotech.com/ (accessed on 15 November 2020).
- Duan, X.; Evans, D.G. Layered Double Hydroxides; Springer: Berlin, Germany; New York, NY, USA, 2005. [Google Scholar]
- Debek, R.; Zubek, K.; Motak, M.; Costa, P.D.; Grzybek, T. Effect of nickel incorporation into hydrotalcite-based catalyst systems for dry reforming of methane. Res. Chem. Intermed. 2015, 41, 9485–9495. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chang, V.W.; Schumacher, D.J. CO2 reforming of methane to syngas I: Evaluation of hydrotalcite clay-derived catalysts. Appl. Clay Sci. 1998, 13, 317–328. [Google Scholar] [CrossRef]
- Koo, K.Y.; Lee, J.H.; Jung, U.H.; Kim, S.H.; Yoon, W.L. Combined H2O and CO2 reforming of coke oven gas over Ca-promoted Ni/MgAl2O4 catalyst for direct reduced iron production. Fuel 2015, 153, 303–309. [Google Scholar] [CrossRef]
- Koo, K.Y.; Lee, S.H.; Jung, U.H.; Roh, H.S.; Yoon, W.L. Syngas production via combined steam and carbon dioxide reforming of methane over Ni-Ce/MgAl2O4 catalysts with enhanced coke resistance. Fuel Process. Technol. 2014, 119, 151–157. [Google Scholar] [CrossRef]
- Jang, W.-J.; Jeong, D.-W.; Shim, J.-O.; Kim, H.-M.; Roh, H.-S.; Son, I.H.; Lee, S.J. Combined steam and carbon dioxide reforming of methane and side reactions: Thermodynamic equilibrium analysis and experimental application. Appl. Energy 2016, 173, 80–91. [Google Scholar] [CrossRef]
- Frost, R.L.; Palmer, S.J.; Theiss, F. Synthesis and Raman spectroscopic characterisation of hydrotalcites based on the formula Ca6Al2(CO3)(OH)16 4H2O. J. Raman Spectrosc. 2011, 42, 1163–1167. [Google Scholar] [CrossRef]
- Jabbour, K.; Massiani, P.; Davidson, A.; Casale, S.; Hassan, N.E. Ordered mesoporous “one-pot” synthesized Ni-Mg(Ca)-Al2O3 as effective and remarkably stable catalysts for combined steam and dry reforming of methane (CSDRM). Appl. Catal. B Environ. 2017, 201, 527–542. [Google Scholar] [CrossRef]
- Kong, X.; Zheng, R.; Zhu, Y.; Ding, G.; Zhu, Y.; Li, Y.W. Rational design of Ni-based catalysts derived from hydrotalcite for selective hydrogenation of 5-hydroxymethylfurfural. Green Chem. 2015, 17, 2504–2514. [Google Scholar] [CrossRef]
- Lin, X.; Li, R.; Lu, M.; Chen, C.; Li, D.; Zhan, Y.; Jiang, L. Carbon dioxide reforming of methane over Ni catalysts prepared from Ni-Mg-Al layered double hydroxides: Influence of Ni loadings. Fuel 2015, 162, 271–280. [Google Scholar] [CrossRef]
- Villegas, J.C.; Giraldo, O.H.; Laubernds, K.; Suib, S.L. New layered double hydroxides containing intercalated manganese oxide species: Synthesis and characterization. Inorg. Chem. 2003, 42, 5621–5631. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, N.; Chu, W.; Liu, M. Carbon dioxide reforming of methane for syngas production over La-promoted NiMgAl catalysts derived from hydrotalcites. Chem. Eng. J. 2012, 209, 623–632. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, N.; Wei, W.; Sun, Y. Partial oxidation of methane over Ni/Mg/Al/La mixed oxides prepared from layered double hydrotalcites. Int. J. Hydrog. Energy 2010, 35, 11776–11786. [Google Scholar] [CrossRef]
- GSTC. Technology: Downstream Conversion Processes; Gasification & Syngas Technologies Council: Arlington, VA, USA, 2016; Available online: http://www.gasification-syngas.org/technology/downstream-conversion-processes/ (accessed on 15 November 2020).
- Hernández, W.Y.; Lauwaert, J.; van der Voort, P.; Verberckmoes, A. Recent advances on the utilization of layered double hydroxides (LDHs) and related heterogeneous catalysts in a lignocellulosic-feedstock biorefinery scheme. Green Chem. 2017, 19, 5269–5302. [Google Scholar] [CrossRef]
- Serrano-Lotina, A.; Rodríguez, L.; Muñoz, G.; Martin, A.J.; Folgado, M.A.; Daza, L. Biogas reforming over La-NiMgAl catalysts derived from hydrotalcite-like structure: Influence of calcination temperature. Catal. Commun. 2011, 12, 961–967. [Google Scholar] [CrossRef]
- Olszówka, J.E.; Karcz, R.; Michalik-Zym, A.; Napruszewska, B.D.; Bielańska, E.; Kryściak-Czerwenka, J.; Socha, R.P.; Nattich-Rak, M.; Krzan, M.; Klimek, A.; et al. Effect of grinding on the physico-chemical properties of Mg-Al hydrotalcite and its performance as a catalyst for Baeyer-Villiger oxidation of cyclohexanone. Catal. Today 2019, 33, 147–153. [Google Scholar] [CrossRef]
- Michalik, A.; Napruszewska, B.D.; Walczyk, A.; Krysciak-Czerwenka, J.; Duraczynska, D.; Serwicka, E.M. Synthesis of Nanocrystalline Mg-Al Hydrotalcites in the Presence of Starch-the Effect on Structure and Composition. Materials 2020, 13, 602. [Google Scholar] [CrossRef]
- Napruszewska, B.D.; Michalik-Zym, A.; Dula, R.; Bielanska, E.; Rojek, W.; Machej, T.; Socha, R.P.; Litynska-Dobrzynska, L.; Bahranowski, K.; Serwicka, E.M. Composites derived from exfoliated Laponite and Mn-Al hydrotalcite prepared in inverse microemulsion: A new strategy for design of robust VOCs combustion catalysts. Appl. Cat. B 2017, 211, 46–56. [Google Scholar] [CrossRef]
- Napruszewska, B.D.; Michalik-Zym, A.; Dula, R.; Duraczyńska, D.; Rojek, W.; Socha, W.; Lityńska-Dobrzyńska, L.; Bahranowski, K.; Serwicka, E.M. VOCs combustion catalysts based on composites of exfoliated organo-Laponite and multimetallic (Mn, Al, Zr, Ce) hydrotalcites prepared by inverse microemulsion. Catal. Today 2019, 333, 182–189. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Ebitani, K.; Yoshida, T.; Yoshida, H.; Kaneda, K. Mg-Al Mixed Oxides as Highly Active Acid-BaseCatalysts for Cycloaddition of Carbon Dioxide to Epoxides. J. Am. Chem. Soc. 1999, 121, 4526–4527. [Google Scholar] [CrossRef]
- Costantino, U.; Marmottini, F.; Nocchetti, M.; Vivani, R. New Synthetic Routes to Hydrotalcite-Like Compounds− Characterisation and Properties of the Obtained Materials. Eur. J. Inorg. Chem. 1998, 10, 1439–1446. [Google Scholar] [CrossRef]
- Galvita, V.; Siddiqi, G.; Sun, P.; Bell, A.T. Ethane dehydrogenation on Pt/Mg(Al)O and PtSn/Mg(Al)O catalysts. J. Catal. 2010, 271, 209–219. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- NIST. Available online: https://www.nist.gov/pml/productsservices/physical-reference-data (accessed on 17 November 2020).
- Konopacka-Łyskawa, D. Synthesis Methods and Favorable Conditions forSpherical Vaterite Precipitation: A Review. Crystals 2019, 9, 223–239. [Google Scholar] [CrossRef]
- Popescu, M.A.; Isopescu, R.; Matei, C.; Fagarasan, G.; Plesu, V. Thermal decomposition of calcium carbonate polymorphs precipitatedin the presence of ammonia and alkylamines. Adv. Powder Technol. 2015, 25, 500–507. [Google Scholar] [CrossRef]
- ’Cabrera, M.M.; Granados, M.L.; Fierro, J.L.G. Thermal decomposition of a hydrotalcite-containing Cu–Zn–Alprecursor: Thermal methods combined with anin situDRIFT study. Phys. Chem. Chem. Phys. 2002, 4, 3122–3127. [Google Scholar] [CrossRef]
- Turco, M.; Bagnasco, G.; Costantino, U.; Marmottini, F.; Montanari, T.; Ramis, G.; Buscac, G. Production of hydrogen from oxidative steam reforming of methanol I. Preparation and characterization of Cu/ZnO/Al2O3 catalysts from a hydrotalcite-like LDH precursor. J. Catal. 2004, 228, 43–55. [Google Scholar] [CrossRef]
- Rives, V.; Kannan, S. Layered double hydroxides with the hydrotalcite-type structurecontaining Cu2z, Ni2z and Al3z J. Mater. Chem. 2000, 10, 489–495. [Google Scholar] [CrossRef]
- Tamri, S.; Sta, I.; Jlassi, M.; Hajji, M.; Ezzaouia, H. Fabrication of ZnO-NiO nanocomposite thin films and experimental study of the effect of the NiO, ZnO concentration on its physical properties. Mat. Sci. Semicond. Process. 2017, 71, 310–320. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Y.; Xua, C.; Zheng, C.; Wang, G. Synthesis of NiOnanorods by a novel simple precursor thermal decomposition approach. Chem. Phys. Lett. 2002, 362, 119–122. [Google Scholar] [CrossRef]
- Jang, M.; Yoon, M.; Lee, S.; Kim, H.; Onodera, A.; Kojima, S. A study on the Raman spectra of Al-doped and Ga-doped ZnO ceramics. Curr. Appl. Phys. 2009, 9, 651–657. [Google Scholar] [CrossRef]
- Pieta, I.S.; Rathi, A.; Pieta, P.; Nowakowski, R.; Hołdynski, M.; Pisarek, M.; Kaminska, A.; Gawande, M.B.; Zboril, R. Electrocatalytic methanol oxidation over Cu, Ni and bimetallic Cu-Ni nanoparticles supported on graphitic carbon nitride. Appl. Catal. B 2019, 244, 272–283. [Google Scholar] [CrossRef]
- Lewalska-Graczyk, A.; Pieta, P.; Garbarino, G.; Busca, G.; Holdynski, M.; Kalisz, G.; Sroka-Bartnicka, A.; Nowakowski, R.; Naushad, M.; Gawande, M.B.; et al. Graphitic Carbon Nitride–Nickel Catalyst: From Material Characterization to Efficient Ethanol Electrooxidation. ACS Sustain. Chem. Eng. 2020, 18, 7244–7255. [Google Scholar] [CrossRef]
- Kouketsu, Y.; Mizukami, T.; Mori, H.; Endo, S.; Aoya, M.; Hara, H.; Nakamura, D.; Wallis, A. A new approach to develop the Raman carbonaceous materialgeothermometer for low-grade metamorphism using peak width. Isl. Arc. 2014, 23, 33–50. [Google Scholar] [CrossRef]
- di Cosimo, J.I.; Díez, V.K.; Xu, M.; Iglesia, E.; Apesteguía, C.R. Structure and surface and catalytic properties of Mg-Al basic oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Apesteguía, C.R.; Ginés, M.J.L.; Iglesia, E. Structural requirements and reaction pathways in condensation reactions of alcohols on MgyAlOx catalysts. J. Catal. 2000, 190, 261–275. [Google Scholar] [CrossRef]
- Gabriels, D.; Hernández, W.Y.; Sels, B.; van der Voort, P.; Verberckmoes, A. Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal. Sci. Technol. 2015, 5, 3876–3902. [Google Scholar] [CrossRef]
- Chistyakov, A.V.; Nikolaev, S.A.; Zharova, P.A.; Tsodikov, M.V.; Manenti, F. Linear α-alcohols production from supercritical ethanol over Cu/Al2O3 catalyst. Energy 2019, 166, 569–576. [Google Scholar] [CrossRef]
- Gallo, J.M.R.; Bueno, J.M.C.; Schuchardt, U. Catalytic Transformations of Ethanol for Biorefineries. J. Braz. Chem. Soc. 2014, 25, 2229–2243. [Google Scholar] [CrossRef]
- Hu, X.X.; Wang, G.; Qin, C.; Xie, X.; Zhang, C.; Xu, W.; Liu, Y. Ligandless nickel-catalyzed transfer hydrogenation of alkenes and alkynes using water as the hydrogen donor. Org. Chem. Front. 2019, 6, 2619–2623. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; An, Z.; Zhu, Y.; Shu, X.; Song, H.; Xiang, X.; He, J. Acid-Base Promoted Dehydrogenation Coupling of Ethanol on Supported Ag Particles. Ind. Eng. Chem. Res. 2020, 59, 3342–3350. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Atomic Ratios from Synthesis (Measured by EDS) | ||||||
|---|---|---|---|---|---|---|---|
| Ni | Mg | Ca | Mn | Cr | Zr | Al | |
| WP-1: Ni0.35Al-CO3 | 0.35 (0.44) | - | - | - | - | - | 1.00 (1.0) |
| WP-2: Ni0.35Mg0.13 Al-CO3 | 0.35 (0.51) | 0.13 (0.15) | - | - | - | - | 1.00 (1.0) |
| WP-3: Ni0.35Mg0.13 Mn0.0018 Cr0.0017 Zr0.0054 Al-CO3 | 0.35 (0.41) | 0.13 (0.22) | - | 0.0018 (0.0098) | 0.0017 (0.0088) | 0.0054 (0.0062) | 1.00 (1.0) |
| WP-4: Ni0.35Ca0.13 Al-CO3 | 0.35 (0.43) | - | 0.13 (0.14) | - | - | - | 1.00 (1.0) |
| WP-5: Ni0.35Ca0.13 Mn0.0018 Cr0.0017 Zr0.0054 Al-CO3 | 0.35 (0.41) | - | 0.13 (0.22) | 0.0018 (0.0098) | 0.0017 (0.0088) | 0.0054 (0.0062) | 1.00 (1.0) |
| Element | Ionic Radii, pm (Ion Coordination) | |
|---|---|---|
| Crystal | Effective | |
| Mg | 86 (+2) | 72 (+2) |
| Ca | 114 (+2) | 100 (+2) |
| Al | 67.5 (+2) | 53.5 (+2) |
| Ni | 83 (+2), 70 (+3) | 69 (+2), 56 (+3) |
| Cr | 87 (+2), 75.5 (+3), 69 (+4) | 73 (+2), 61.5 (+3), 55 (+4) |
| Zr | 86 (+4) | 72 (+4) |
| Mn | 81 (+2), 72 (+3), 67 (+4) | 67 (+2), 58 (+3), 53 (+4) |
| Catalyst | Selectivity, mol% | |||||
|---|---|---|---|---|---|---|
| ABET, m2g−1 | Conv, % | Ethyl Ether | n-BuOH | Others 1 | C Balance, mol% | |
| WP-1: Ni0.35Al-CO3 | 223 | 97.5 | 32.1 | 16.3 | 51.7 | 93.4 |
| WP-2: Ni0.35Mg0.13 Al-CO3 | 216 | 9.5 | 26.1 | 45.7 | 28.2 | 95.1 |
| WP-3: Ni0.35Mg0.13 Mn0.0018 Cr0.0017 Zr0.0054 Al-CO3 | 230 | 12.8 | 53.7 | 31.1 | 15.2 | 94.2 |
| WP-4: Ni0.35Ca0.13 Al-CO3 | 207 | 5.1 | 19.9 | 34.7 | 45.4 | 84.1 |
| WP-5: Ni0.35Ca0.13 Mn0.0018 Cr0.0017 Zr0.0054 Al-CO3 | 212 | 10.3 | 31.9 | 28.8 | 39.2 | 80.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieta, I.S.; Michalik, A.; Kraleva, E.; Mrdenovic, D.; Sek, A.; Wahaczyk, E.; Lewalska-Graczyk, A.; Krysa, M.; Sroka-Bartnicka, A.; Pieta, P.; et al. Bio-DEE Synthesis and Dehydrogenation Coupling of Bio-Ethanol to Bio-Butanol over Multicomponent Mixed Metal Oxide Catalysts. Catalysts 2021, 11, 660. https://doi.org/10.3390/catal11060660
Pieta IS, Michalik A, Kraleva E, Mrdenovic D, Sek A, Wahaczyk E, Lewalska-Graczyk A, Krysa M, Sroka-Bartnicka A, Pieta P, et al. Bio-DEE Synthesis and Dehydrogenation Coupling of Bio-Ethanol to Bio-Butanol over Multicomponent Mixed Metal Oxide Catalysts. Catalysts. 2021; 11(6):660. https://doi.org/10.3390/catal11060660
Chicago/Turabian StylePieta, Izabela S., Alicja Michalik, Elka Kraleva, Dusan Mrdenovic, Alicja Sek, Ewa Wahaczyk, Agnieszka Lewalska-Graczyk, Mikolaj Krysa, Anna Sroka-Bartnicka, Piotr Pieta, and et al. 2021. "Bio-DEE Synthesis and Dehydrogenation Coupling of Bio-Ethanol to Bio-Butanol over Multicomponent Mixed Metal Oxide Catalysts" Catalysts 11, no. 6: 660. https://doi.org/10.3390/catal11060660
APA StylePieta, I. S., Michalik, A., Kraleva, E., Mrdenovic, D., Sek, A., Wahaczyk, E., Lewalska-Graczyk, A., Krysa, M., Sroka-Bartnicka, A., Pieta, P., Nowakowski, R., Lew, A., & Serwicka, E. M. (2021). Bio-DEE Synthesis and Dehydrogenation Coupling of Bio-Ethanol to Bio-Butanol over Multicomponent Mixed Metal Oxide Catalysts. Catalysts, 11(6), 660. https://doi.org/10.3390/catal11060660