
In Situ Conditioning of CO2-Rich Syngas during the Synthesis of Methanol

 , and
, and
Abstract
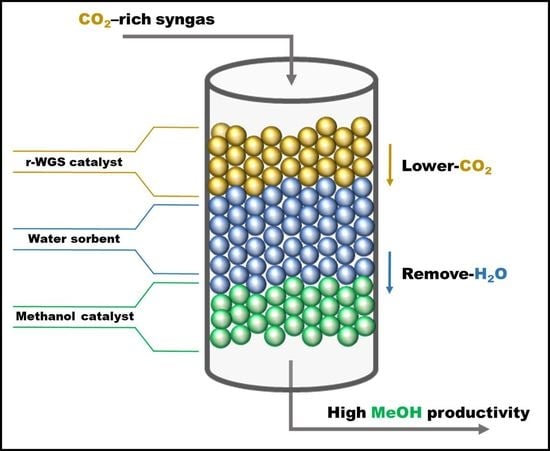
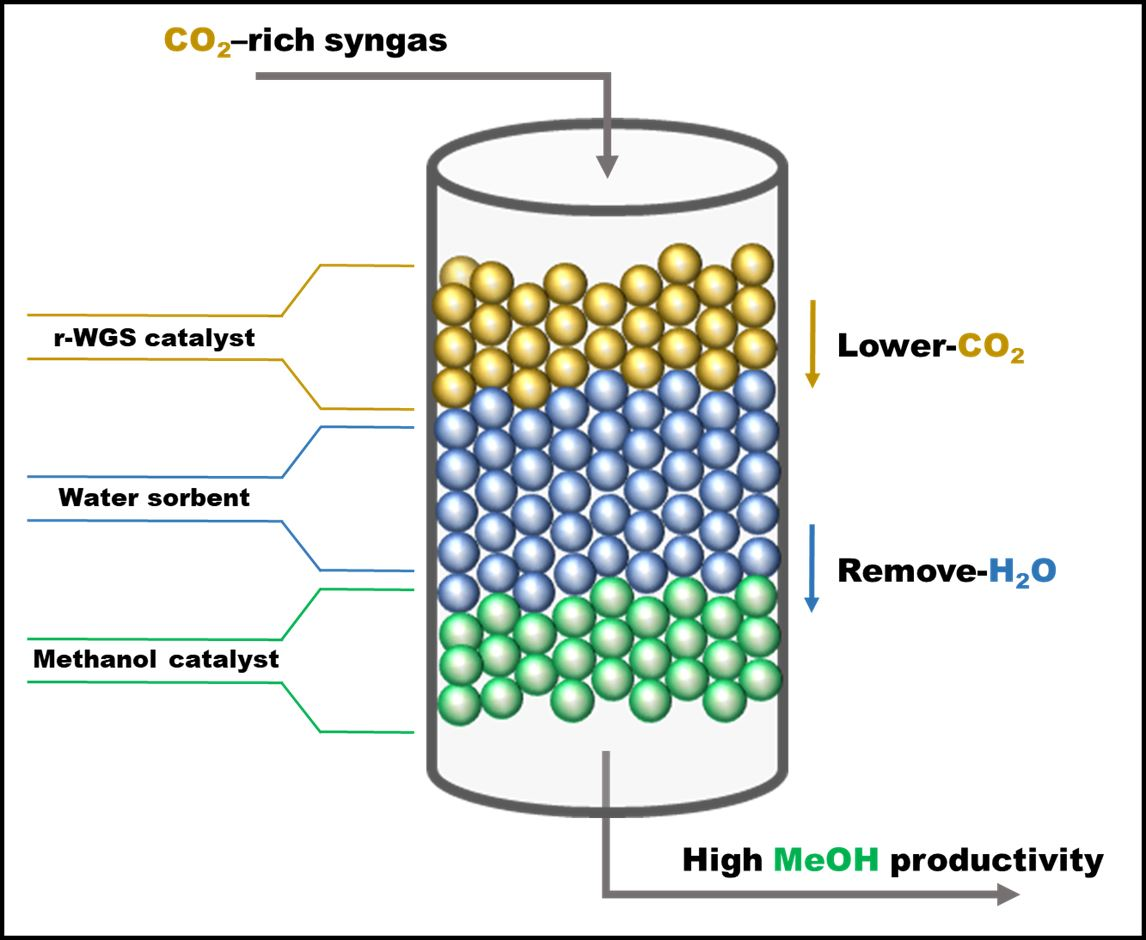
1. Introduction
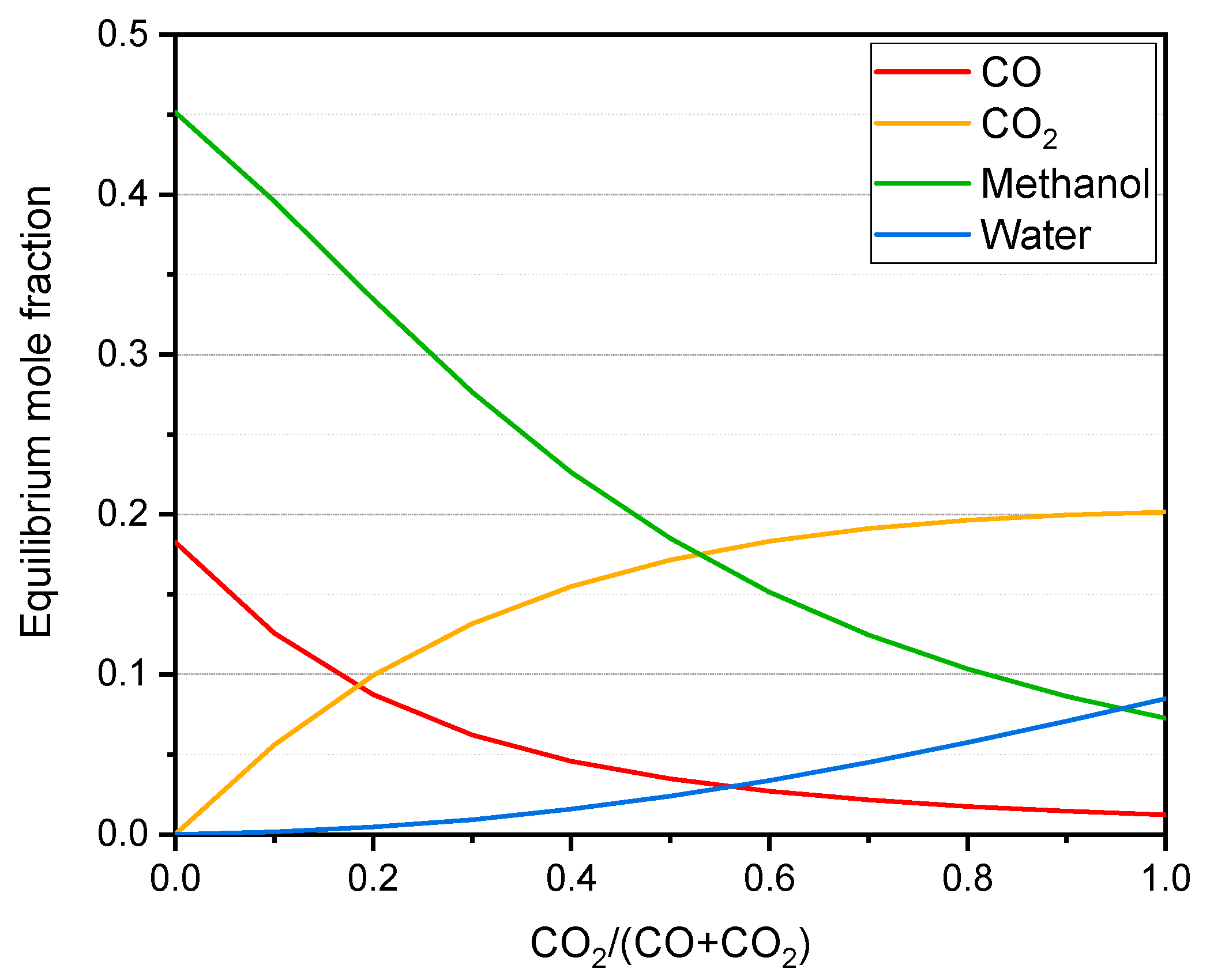
 CH3OH ∆H0 = −90.6 kJ/mol
CH3OH + H2O ∆H0 = −49.5 kJ/mol
CO2 + H2 ∆H0 = −41.1 kJ/mol
CH3OH ∆H0 = −90.6 kJ/mol
CH3OH + H2O ∆H0 = −49.5 kJ/mol
CO2 + H2 ∆H0 = −41.1 kJ/mol
2. Results and Discussion
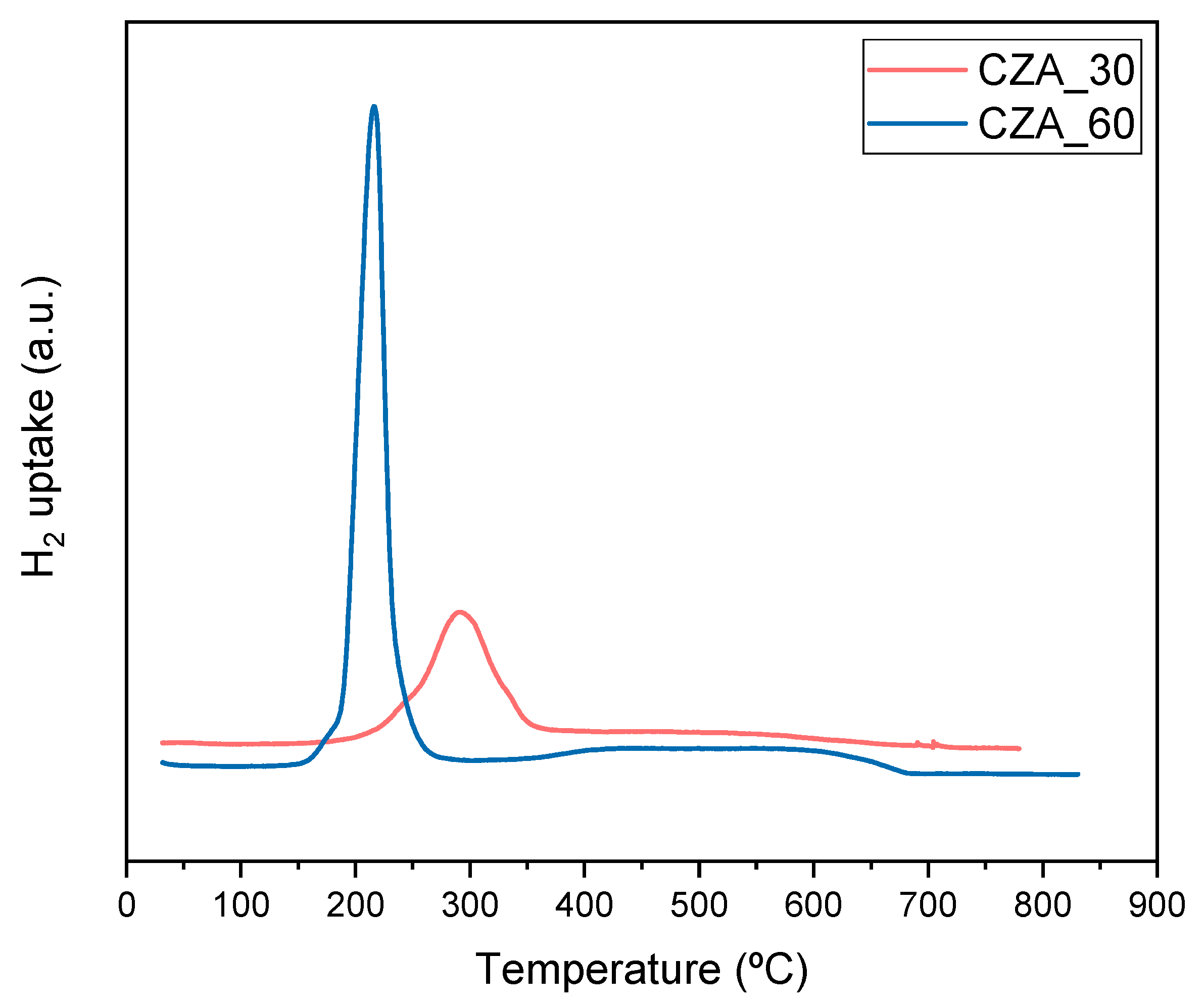
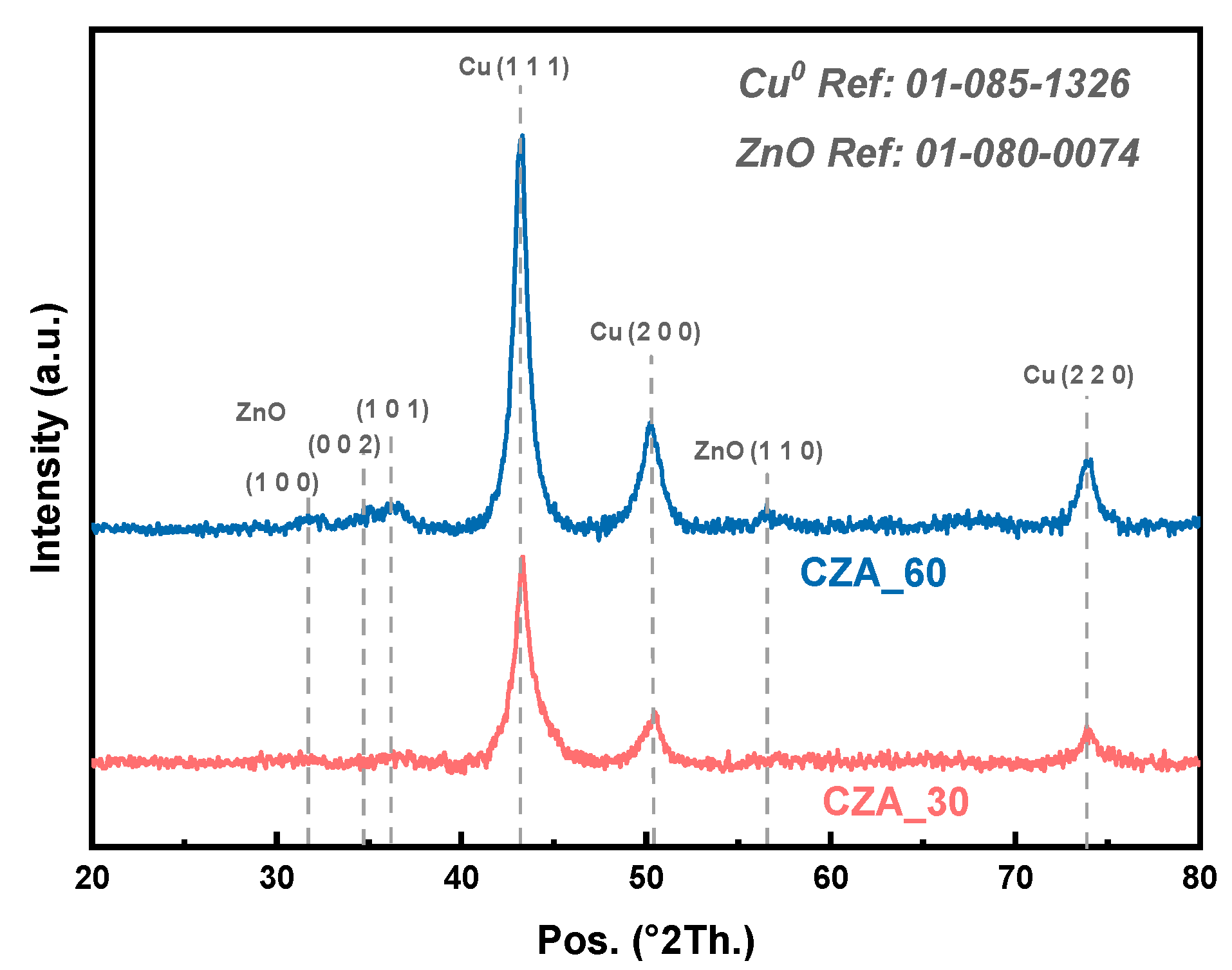
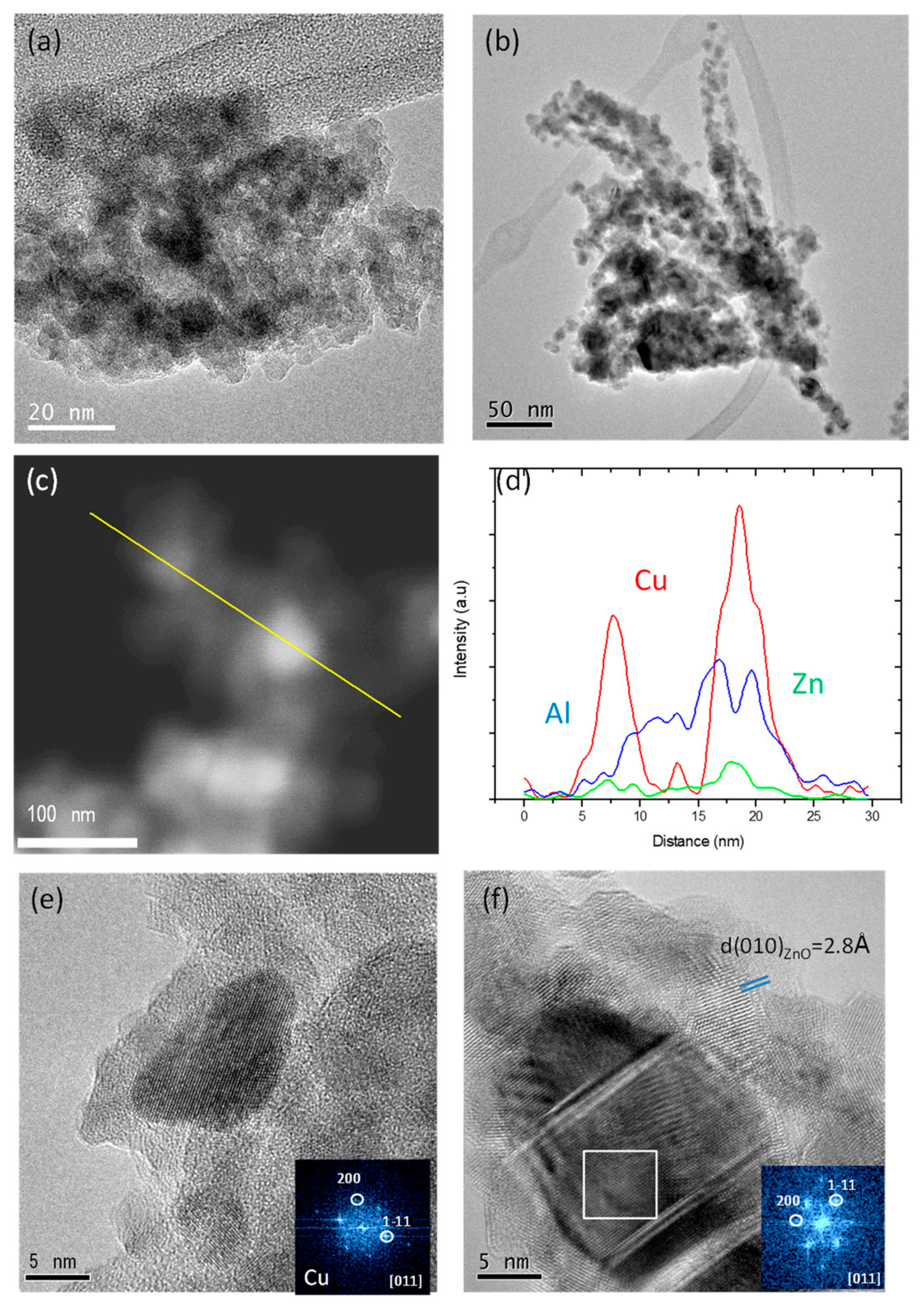
2.1. Characterization Results
2.2. Performance of CZA_30 for the WGS
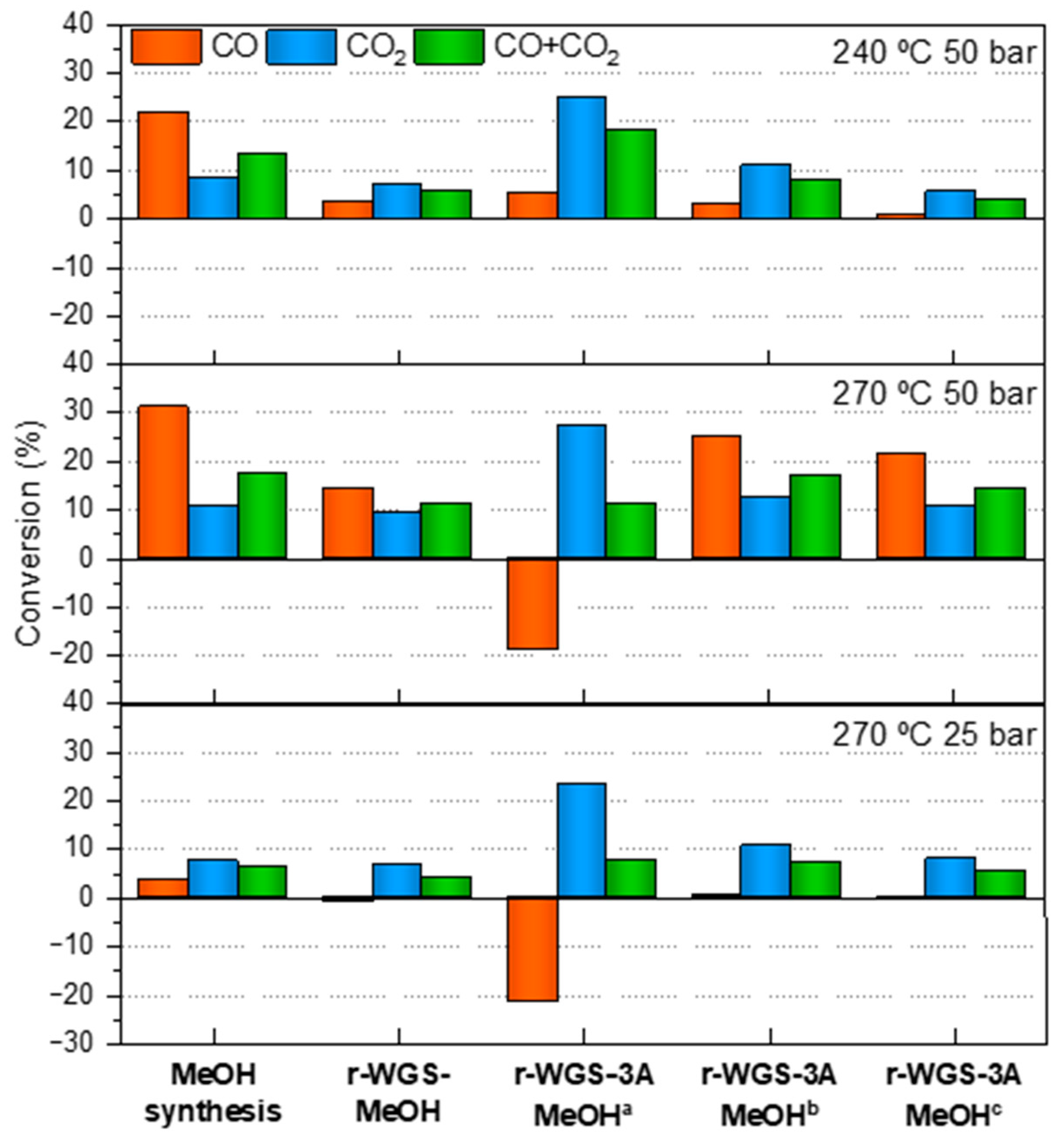
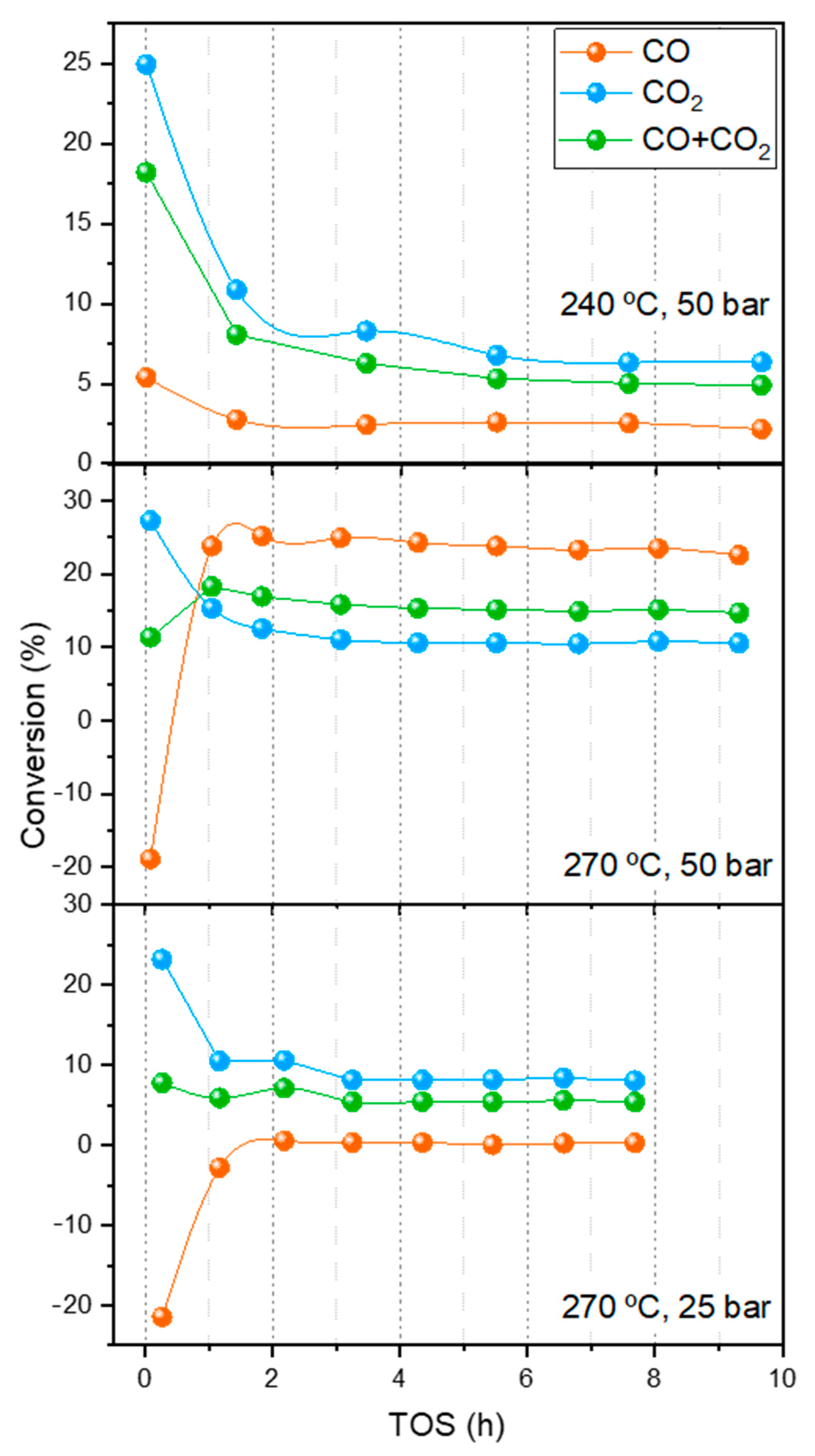
2.3. Synthesis of Methanol in a Single Catalytic Bed
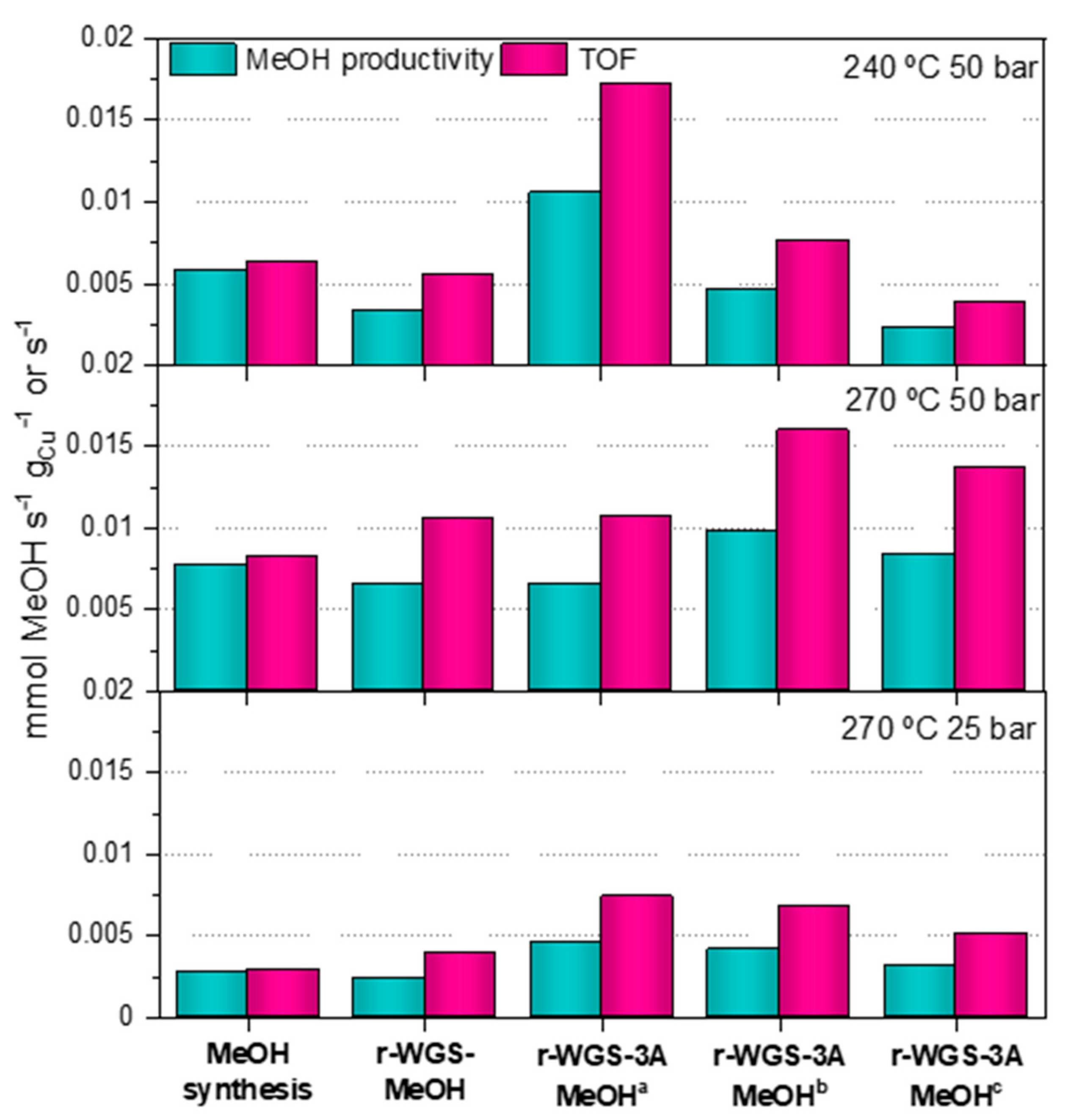
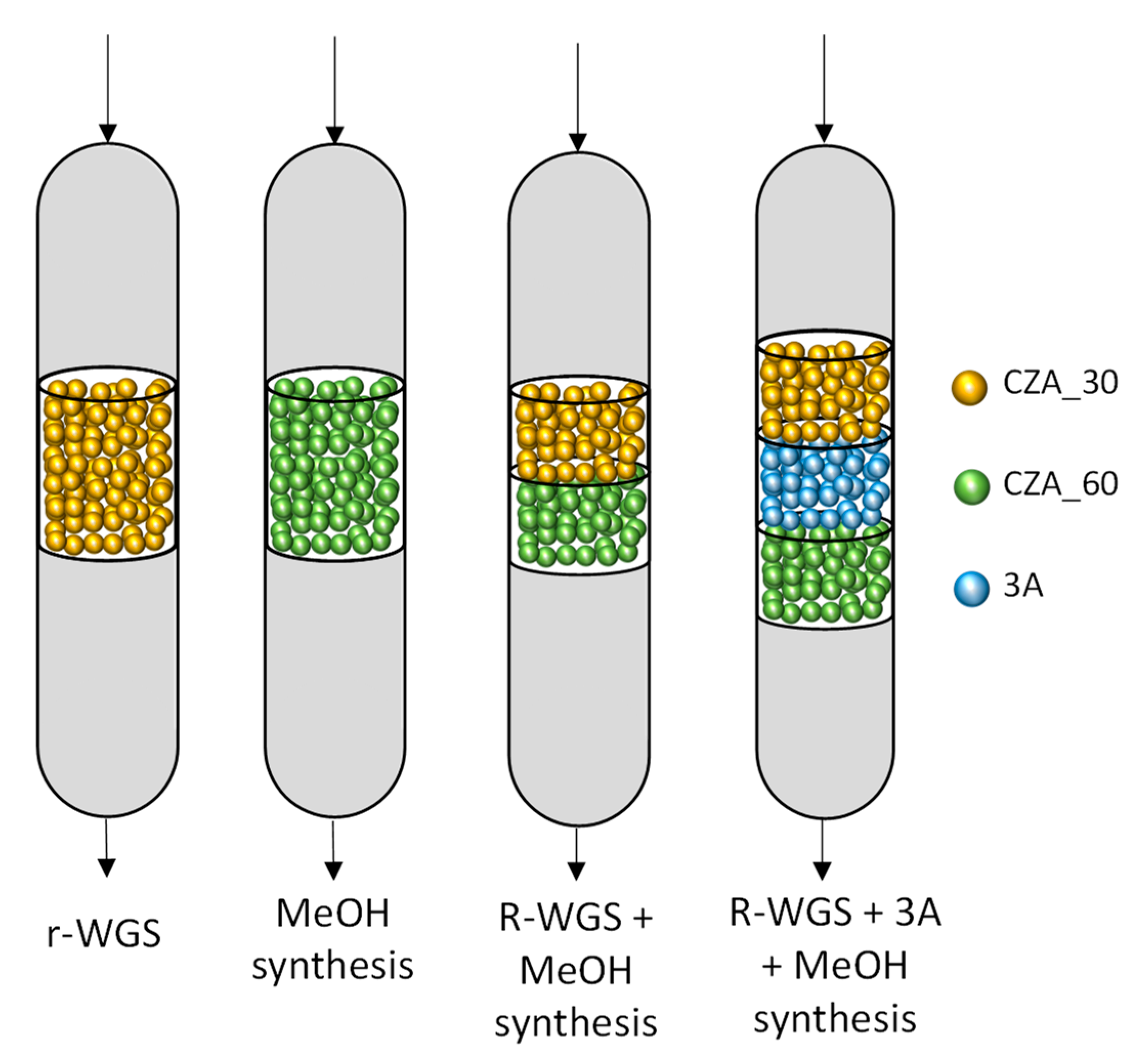
2.4. Synthesis of Methanol in the Double Catalytic Beds
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Characterization
3.3. Catalytic Activity Tests
4. Conclusions
- In general terms, the methanol productivity and TOF can be effectively enhanced when applying the combination of the r-WGS pre-treatment and water sorption strategies in the methanol synthesis from CO2-rich syngas in a single reactor.
- If the CO2/CO ratio after the r-GWS process is of ca. 1.6 or lower, the increase in the TOF is also visible even without the water removal.
- The duration of the period until the zeolite saturates, i.e., when the stationary state is reached, increases with the extent of the r-WGS.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinchen, G.C.; Denny, P.J.; Jennings, J.R.; Spencer, M.S.; Waugh, K.C. Review—Synthesis of Methanol. Appl. Catal. 1988, 36, 1–65. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol synthesis. Catal. Lett. 2012, 142, 1153–1166. [Google Scholar] [CrossRef]
- Ott, J.; Gronemann, V.; Pontzen, F.; Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. Methanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Nestler, F.; Krüger, M.; Full, J.; Hadrich, M.J.; White, R.J.; Schaadt, A. Methanol Synthesis—Industrial Challenges within a Changing Raw Material Landscape. Chemie-Ingenieur-Technik 2018, 90, 1409–1418. [Google Scholar] [CrossRef]
- Klier, K.; Chatikavanij, V.; Herman, R.G.; Simmons, G.W. Catalytic synthesis of methanol from COH2: IV. The effects of carbon dioxide. J. Catal. 1982, 74, 343–360. [Google Scholar] [CrossRef]
- Herman, R.G.; Klier, K.; Simmons, G.W.; Finn, B.P.; Bulko, J.B.; Kobylinski, T.P. Catalytic synthesis of methanol from COH2: I. Phase composition, electronic properties, and activities of the Cu/ZnO/M2O3 catalysts. J. Catal. 1979, 56, 407–429. [Google Scholar] [CrossRef]
- Martin, O.; Pérez-Ramírez, J. New and revisited insights into the promotion of methanol synthesis catalysts by CO2. Catal. Sci. Technol. 2013, 3, 3343–3352. [Google Scholar] [CrossRef]
- Martin, O.; Mondelli, C.; Cervellino, A.; Ferri, D.; Curulla-Ferré, D.; Pérez-Ramírez, J. Operando Synchrotron X-ray Powder Diffraction and Modulated-Excitation Infrared Spectroscopy Elucidate the CO2Promotion on a Commercial Methanol Synthesis Catalyst. Angew. Chemie Int. Ed. 2016, 55, 11031–11036. [Google Scholar] [CrossRef] [PubMed]
- Göransson, K.; Söderlind, U.; He, J.; Zhang, W. Review of syngas production via biomass DFBGs. Renew. Sustain. Energy Rev. 2011, 15, 482–492. [Google Scholar] [CrossRef]
- Swain, P.K.; Das, L.M.; Naik, S.N. Biomass to liquid: A prospective challenge to research and development in 21st century. Renew. Sustain. Energy Rev. 2011, 15, 4917–4933. [Google Scholar] [CrossRef]
- Mondal, P.; Dang, G.S.; Garg, M.O. Syngas production through gasification and cleanup for downstream applications—Recent developments. Fuel Process. Technol. 2011, 92, 1395–1410. [Google Scholar] [CrossRef]
- Molino, A.; Larocca, V.; Chianese, S.; Musmarra, D. Biofuels production by biomass gasification: A review. Energies 2018, 11, 811. [Google Scholar] [CrossRef]
- Martínez, I.; Kulakova, V.; Grasa, G.; Murillo, R. Experimental investigation on sorption enhanced gasification (SEG) of biomass in a fluidized bed reactor for producing a tailored syngas. Fuel 2020, 259, 116252. [Google Scholar] [CrossRef]
- Martínez, I.; Romano, M.C. Flexible sorption enhanced gasification (SEG) of biomass for the production of synthetic natural gas (SNG) and liquid biofuels: Process assessment of stand-alone and power-to-gas plant schemes for SNG production. Energy 2016, 113, 615–630. [Google Scholar] [CrossRef]
- Bae, J.W.; Potdar, H.S.; Kang, S.H.; Jun, K.W. Coproduction of methanol and dimethyl ether from biomass-derived syngas on a Cu-ZnO-Al2O3/γ-A2O3 hybrid catalyst. Energy Fuels 2008, 22, 223–230. [Google Scholar] [CrossRef]
- Jung, J.W.; Lee, Y.J.; Um, S.H.; Yoo, P.J.; Lee, D.H.; Jun, K.W.; Bae, J.W. Effect of copper surface area and acidic sites to intrinsic catalytic activity for dimethyl ether synthesis from biomass-derived syngas. Appl. Catal. B Environ. 2012, 126, 1–8. [Google Scholar] [CrossRef]
- Martínez, I.; Grasa, G.; Callén, M.S.; López, J.M.; Murillo, R. Optimised production of tailored syngas from municipal solid waste (MSW) by sorption-enhanced gasification. Chem. Eng. J. 2020, 401, 126067. [Google Scholar] [CrossRef]
- Clausen, B.S.; Schiøtz, J.; Gråbæk, L.; Ovesen, C.V.; Jacobsen, K.W.; Nørskov, J.K.; Topsøe, H. Wetting/ non-wetting phenomena during catalysis: Evidence from in situ on-line EXAFS studies of Cu-based catalysts. Top. Catal. 1994, 1, 367–376. [Google Scholar] [CrossRef]
- Khassin, A.A.; Minyukova, T.P.; Yurieva, T.M. Genesis of catalysts for methanol synthesis. Mendeleev Commun. 2014, 24, 67–74. [Google Scholar] [CrossRef]
- Gaikwad, R.; Reymond, H.; Phongprueksathat, N.; Rudolf Von Rohr, P.; Urakawa, A. From CO or CO2?: Space-resolved insights into high-pressure CO2 hydrogenation to methanol over Cu/ZnO/Al2O3. Catal. Sci. Technol. 2020, 10, 2763–2768. [Google Scholar] [CrossRef]
- Prašnikar, A.; Pavlišič, A.; Ruiz-Zepeda, F.; Kovač, J.; Likozar, B. Mechanisms of Copper-Based Catalyst Deactivation during CO2 Reduction to Methanol. Ind. Eng. Chem. Res. 2019. [Google Scholar] [CrossRef]
- Wu, J.; Saito, M.; Takeuchi, M.; Watanabe, T. The stability of Cu/ZnO-based catalysts in methanol synthesis from a CO2-rich feed and from a CO-rich feed. Appl. Catal. A Gen. 2001, 218, 235–240. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schlereth, D.; Jacobsen, N.; Kasatkin, I.; Schumann, J.; Behrens, M.; Schlögl, R.; Hinrichsen, O. Kinetics of deactivation on Cu/ZnO/Al2O3 methanol synthesis catalysts. Appl. Catal. A Gen. 2015, 502, 262–270. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Spencer, M.S. Sensitive and insensitive reactions on copper catalysts: The water-gas shift reaction and methanol synthesis from carbon dioxide. Catal. Today 1991, 10, 293–301. [Google Scholar] [CrossRef]
- Hadden, R.A.; Lambert, P.J.; Ranson, C. Relationship between the copper surface area and the activity of CuO/ZnO/Al2O3 water-gas shift catalysts. Appl. Catal. A Gen. 1995, 122, L1. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J. Water gas shift catalysis. Catal. Rev. Sci. Eng. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Kowalik, P.; Próchniak, W.; Konkol, M.; Borowiecki, T. The quantitative description of the effects of cesium doping on the activity and properties of Cu/ZnO/Al2O3 catalyst in low-temperature water-gas shift. Appl. Catal. A Gen. 2012, 423–424, 15–20. [Google Scholar] [CrossRef]
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon Dioxide Hydrogenation To Form Methanol via a Reverse-Water-Gas-Shift Reaction (the CAMERE Process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Okazaki, M.; Kapoor, M.P.; Osaki, T.; Ohashi, F. Oxidative steam reforming of methanol over CuZnAl(Zr)-oxide catalysts for the selective production of hydrogen for fuel cells: Catalyst characterization and performance evaluation. J. Catal. 2000, 194, 373–384. [Google Scholar] [CrossRef]
- van Kampen, J.; Boon, J.; van Sint Annaland, M. Steam adsorption on molecular sieve 3A for sorption enhanced reaction processes. Adsorption 2020. [Google Scholar] [CrossRef]
- Wilmer, H.; Hinrichsen, O. Dynamical Changes in Cu/ZnO/Al2O3 Catalysts. Catal. Lett. 2002, 82, 117–122. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Waugh, K.C.; Whan, D.A. The activity and state of the copper surface in methanol synthesis catalysts. Appl. Catal. 1986, 25, 101–107. [Google Scholar] [CrossRef]
- Pan, W.X.; Cao, R.; Roberts, D.L.; Griffin, G.L. Methanol synthesis activity of Cu ZnO catalysts. J. Catal. 1988, 114, 440–446. [Google Scholar] [CrossRef]
- Ertl, G.; Knözinger, H.; Weitkamp, J. Handbook of Heterogeneous Catalysis; Wiley-VCH: Weinheim, Germany, 2008; Volume 1, ISBN 9783527619474. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Composition, Actual (Expected) | Surface Area | Average Pore Size | DCu | Cu Surface Area | ||
|---|---|---|---|---|---|---|---|
| Cu | ZnO | Al2O3 | m2/gcat | nm | % | m2Cu/gcat | |
| wt. % | wt. % | wt. % | |||||
| CZA_60 | 67.8 (62) | 28.7 (33) | 3.5 (5) | 39 | 10 | 5.9 | 26 |
| CZA_30 | 32.9 (33) | 13.1 (17) | 54.0 (50) | 107 | 7 | 8.7 | 17 |
| B.E. (eV) | Surface Composition (wt. %) | |||
|---|---|---|---|---|
| Cu 2p3/2 | Cu | ZnO | Al2O3 | |
| CZA-60 | 932.4 | 20 | 19 | 61 |
| CZA-30 | 932.5 | 9 | 8 | 83 |
| Pressure | Temperature | Final CO2/CO | Eq. CO2/CO |
|---|---|---|---|
| bar | °C | mole | mole |
| 25 | 270 | 1.6 | 1.5 |
| 50 | 270 | 1.6 | 1.5 |
| 50 | 240 | 1.8 | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peinado, C.; Liuzzi, D.; Sanchís, A.; Pascual, L.; Peña, M.A.; Boon, J.; Rojas, S. In Situ Conditioning of CO2-Rich Syngas during the Synthesis of Methanol. Catalysts 2021, 11, 534. https://doi.org/10.3390/catal11050534
Peinado C, Liuzzi D, Sanchís A, Pascual L, Peña MA, Boon J, Rojas S. In Situ Conditioning of CO2-Rich Syngas during the Synthesis of Methanol. Catalysts. 2021; 11(5):534. https://doi.org/10.3390/catal11050534
Chicago/Turabian StylePeinado, Cristina, Dalia Liuzzi, Alberto Sanchís, Laura Pascual, Miguel A. Peña, Jurriaan Boon, and Sergio Rojas. 2021. "In Situ Conditioning of CO2-Rich Syngas during the Synthesis of Methanol" Catalysts 11, no. 5: 534. https://doi.org/10.3390/catal11050534
APA StylePeinado, C., Liuzzi, D., Sanchís, A., Pascual, L., Peña, M. A., Boon, J., & Rojas, S. (2021). In Situ Conditioning of CO2-Rich Syngas during the Synthesis of Methanol. Catalysts, 11(5), 534. https://doi.org/10.3390/catal11050534