
Influence of Cs Loading on Pt/m-ZrO2 Water–Gas Shift Catalysts



 ,
, 
Abstract
1. Introduction
2. Results and Discussion
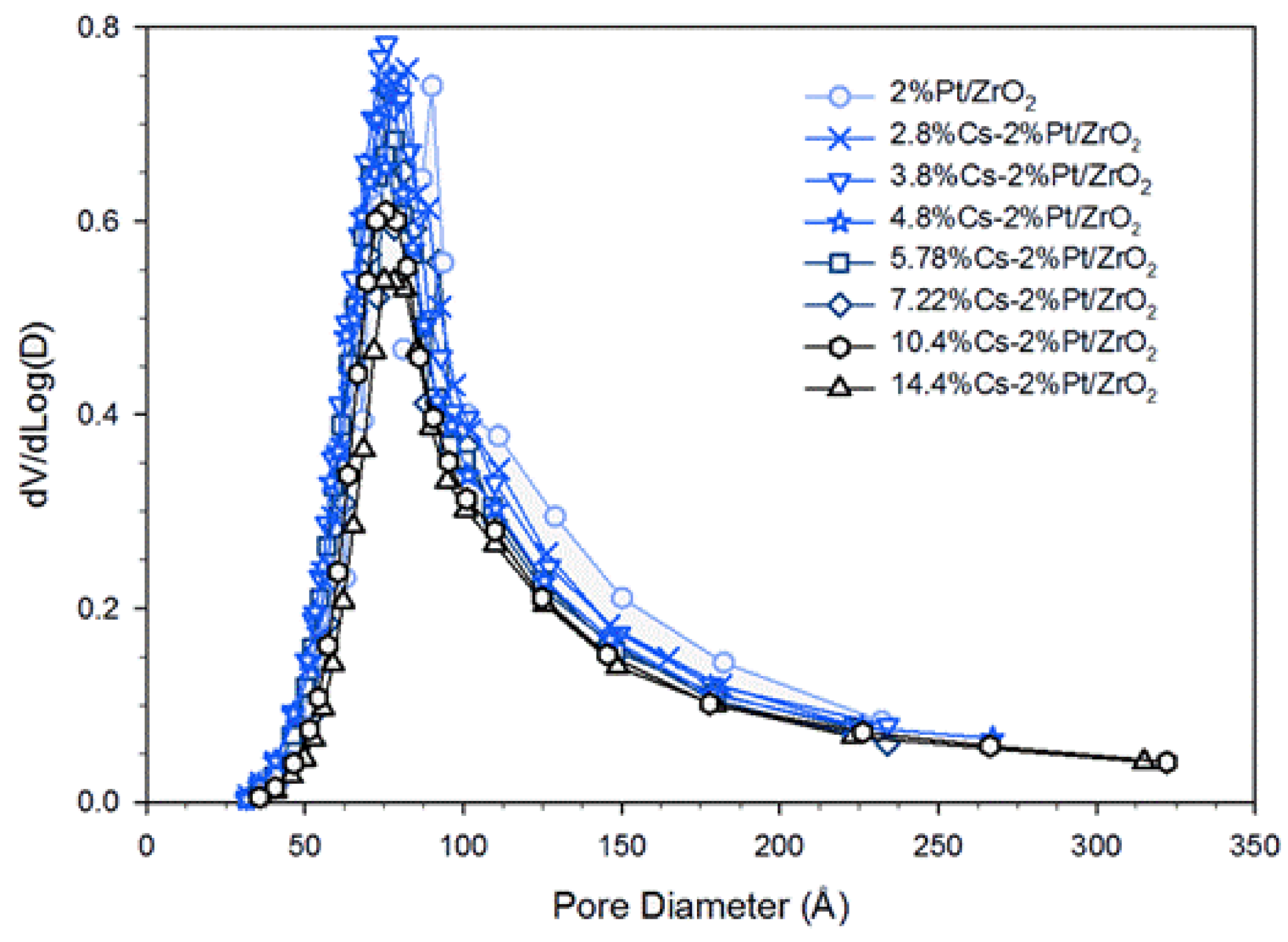
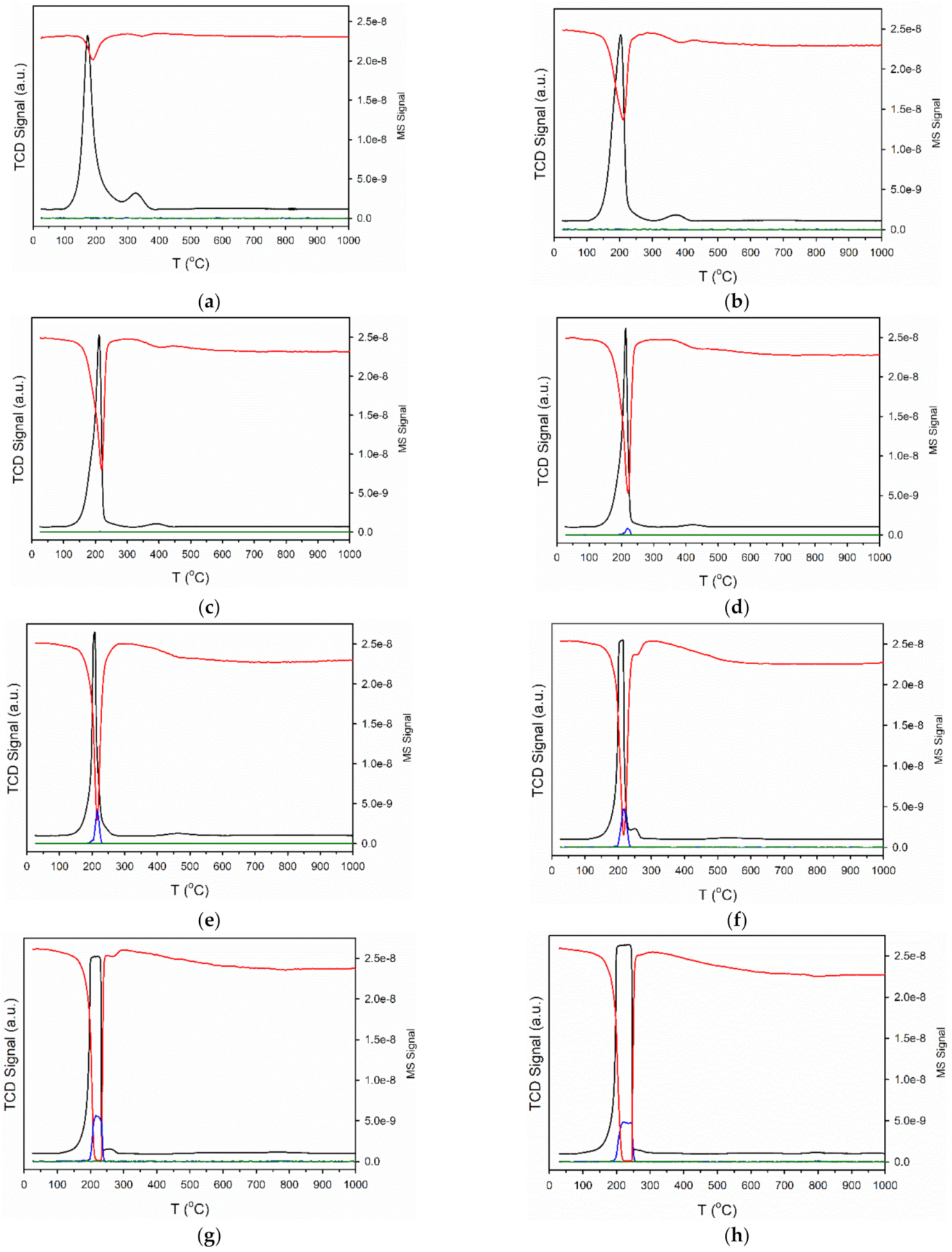
2.1. Catalyst Characterization
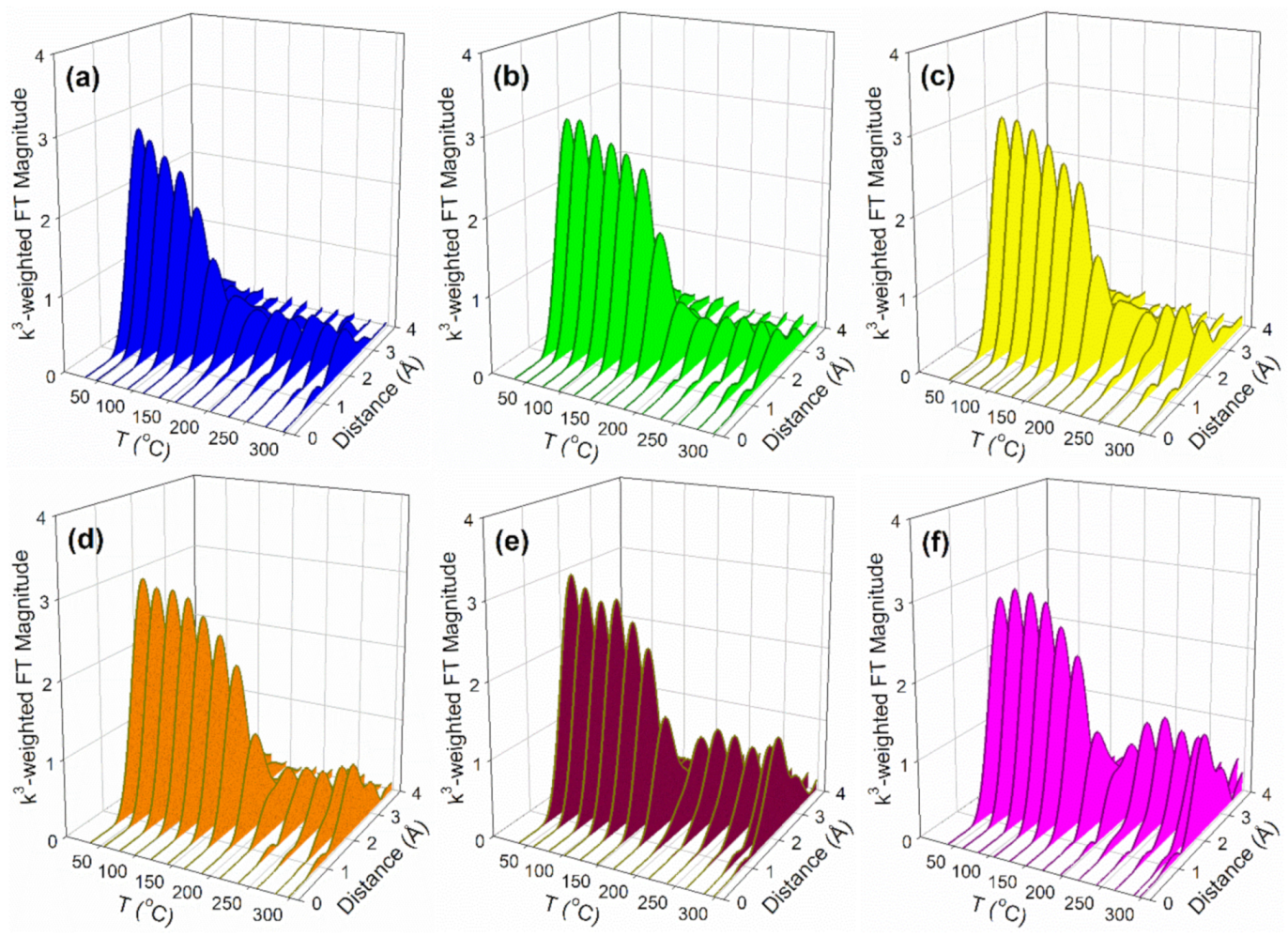
2.2. X-ray Absorption Spectroscopy
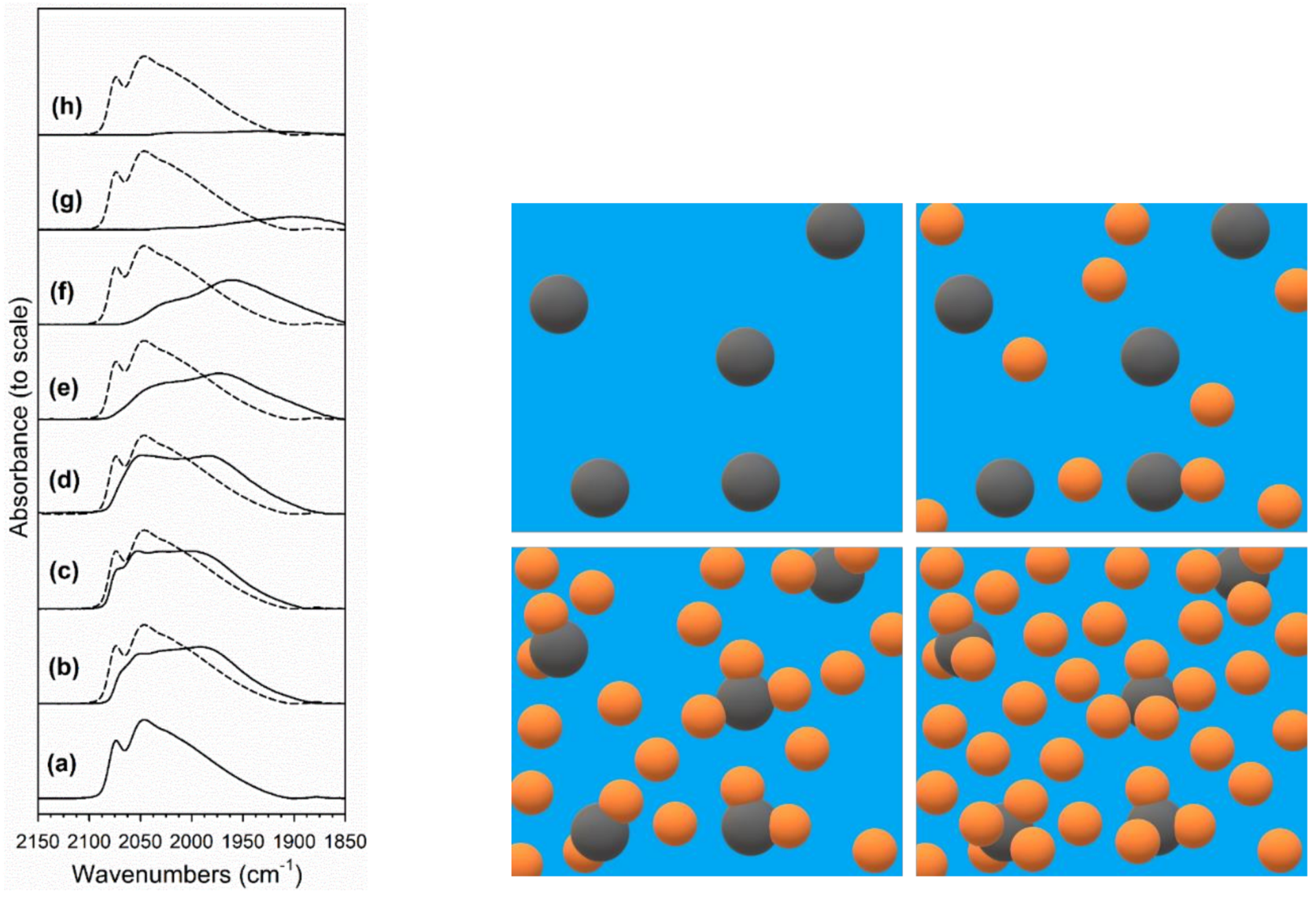
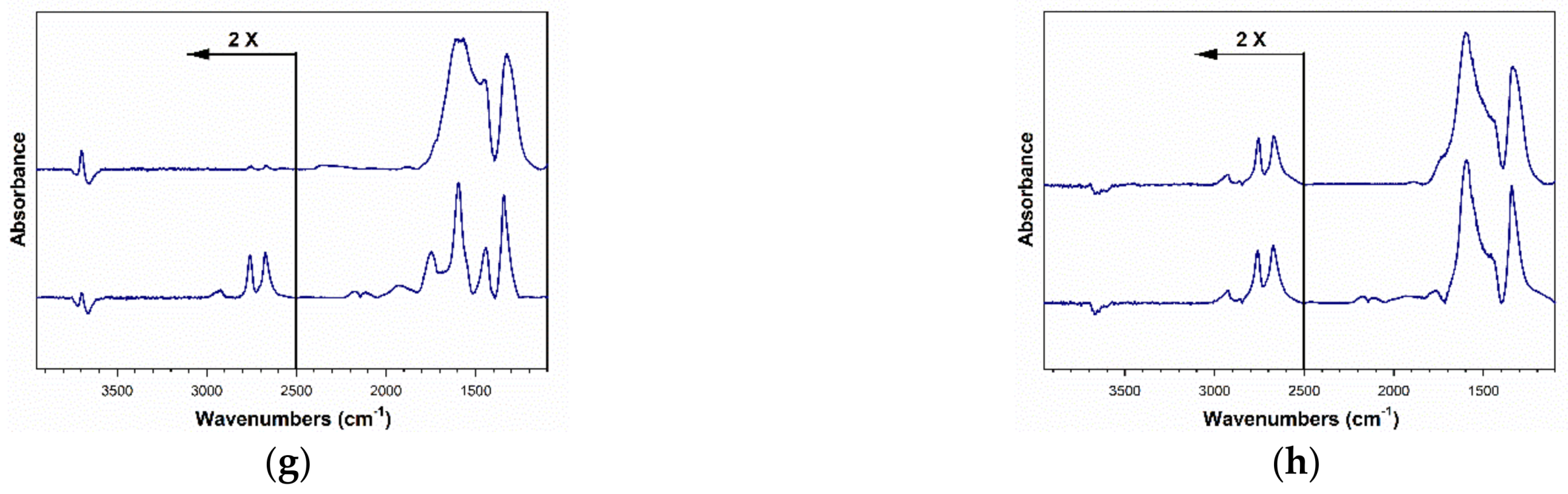
2.3. DRIFTS Studies
2.4. Reactor Testing
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Characterization
3.2.1. BET Surface Area
3.2.2. Temperature-Programmed Reduction/Mass Spectrometry
3.2.3. Temperature-Programmed Reaction and Desorption
3.2.4. EXAFS
3.2.5. DRIFTS
3.2.6. Reaction Testing
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Midilli, A.; Ay, M.; Dincer, I.; Rosen, M.A. On hydrogen and hydrogen energy strategies: I: Current status and needs. Renew. Sustain. Energy Rev. 2005, 9, 255–271. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Bethoux, O. Hydrogen Fuel Cell Road Vehicles and Their Infrastructure: An Option towards an Environmentally Friendly Energy Transition. Energies 2020, 13, 6132. [Google Scholar] [CrossRef]
- Bhat, S.A.; Sadhukhan, J. Process intensification aspects for steam methane reforming: An overview. AIChE J. 2009, 55, 408–422. [Google Scholar] [CrossRef]
- Assabumrungrat, S.; Phromprasit, J. Fuel processing technologies for hydrogen production from methane. Eng. J. 2012, 16, 1–4. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. Reactant-promoted reaction mechanism for water-gas shift reaction on Rh-doped CeO2. J. Catal. 1993, 141, 71–78. [Google Scholar] [CrossRef]
- Vignatti, C.; Avila, M.S.; Apesteguia, C.R.; Garetto, T.F. Catalytic and DRIFTS study of the WGS reaction on Pt-based catalysts. Int. J. Hydrogen Energy 2010, 35, 7302–7312. [Google Scholar] [CrossRef]
- Jacobs, G.; Ricote, S.; Graham, U.M.; Patterson, P.M.; Davis, B.H. Low temperature water gas shift: Type and loading of metal impacts forward decomposition of pseudo-stabilized formate over metal/ceria catalysts. Catal. Today 2005, 106, 259–264. [Google Scholar] [CrossRef]
- Kauppinen, M.M.; Melander, M.M.; Marko, M.; Bazhenov, A.S.; Honkala, K. Unraveling the Role of the Rh–ZrO2 Interface in the Water–Gas-Shift Reaction via a First-Principles Microkinetic Study. ACS Catal. 2018, 8, 11633–11647. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water gas shift catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I. Effect of the nature of the support on the catalytic performance of noble metal catalysts for the water–gas shift reaction. Catal. Today 2006, 112, 49–52. [Google Scholar] [CrossRef]
- Wang, Y.X.; Wang, G.C. A systematic theoretical study of the water gas shift reaction on the Pt/ZrO2 interface and Pt (111) face: Key role of a potassium additive. Catal. Sci. Technol. 2020, 10, 876–892. [Google Scholar] [CrossRef]
- Wang, X.; Gorte, R.J. The effect of Fe and other promoters on the activity of Pd/ceria for the water-gas shift reaction. Appl. Catal. A Gen. 2003, 247, 157–162. [Google Scholar] [CrossRef]
- Song, W.; Hensen, E.J. Mechanistic aspects of the water–gas shift reaction on isolated and clustered Au atoms on CeO2 (110): A density functional theory study. ACS Catal. 2014, 4, 1885–1892. [Google Scholar] [CrossRef]
- Sun, K.; Kohyama, M.; Tanaka, S.; Takeda, S. Reaction mechanism of the low-temperature water–gas shift reaction on Au/TiO2 catalysts. J. Phys. Chem. C 2017, 121, 12178–12187. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Panagiotopoulou, P.; Kondarides, D.I.; Efstathiou, A.M. Kinetic and mechanistic studies of the water–gas shift reaction on Pt/TiO2 catalyst. J. Catal. 2009, 264, 117–129. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Polychronopoulou, K.; Boghosian, S.; Garcia-Rodriguez, S.; Efstathiou, A.M. Water–Gas shift reaction on Pt/Ce1–xTix O2− δ: The effect of Ce/Ti ratio. J. Phys. Chem. C 2013, 117, 25467–25477. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Santos, M.S.; Andrade, H.M.C.; Fierro, J.L.G. Effect of alkali cations on the CuZnOAl2O3 low temperature water gas-shift catalyst. Catal. Today 2011, 172, 166–170. [Google Scholar] [CrossRef]
- Komarov, Y.M.; Il’in, A.A.; Smirnov, N.N.; Il’in, A.P.; Babaikin, D.B. Effect of alkali metal oxides on the selectivity of carbon monoxide conversion to give hydrogen on copper-containing catalysts. Russ. J. Appl. Chem. 2013, 86, 27–31. [Google Scholar] [CrossRef]
- Gao, P.; Graham, U.M.; Shafer, W.D.; Linganiso, L.Z.; Jacobs, G.; Davis, B.H. Nanostructure and kinetic isotope effect of alkali-doped Pt/silica catalysts for water-gas shift and steam-assisted formic acid decomposition. Catal. Today 2016, 272, 42–48. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water-gas shift: Characterization of Pt-based ZrO2 catalyst promoted with Na discovered by combinatorial methods. Appl. Catal. A Gen. 2007, 319, 47–57. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water–gas shift: The effect of alkali doping on the CH bond of formate over Pt/ZrO2 catalysts. Appl. Catal. A Gen. 2007, 328, 14–26. [Google Scholar] [CrossRef]
- Evin, H.N.; Jacobs, G.; Ruiz-Martinez, J.; Graham, U.M.; Dozier, A.; Thomas, G.; Davis, B.H. Low temperature water–gas shift/methanol steam reforming: Alkali doping to facilitate the scission of formate and methoxy C–H bonds over Pt/ceria catalyst. Catal. Lett. 2008, 122, 9–19. [Google Scholar] [CrossRef]
- González-Cobos, J.; Valverde, J.L.; de Lucas-Consuegra, A. Electrochemical vs. chemical promotion in the H2 production catalytic reactions. Int. J. Hydrogen Energy 2017, 42, 13712–13723. [Google Scholar] [CrossRef]
- Kusche, M.; Bustillo, K.; Agel, F.; Wasserscheid, P. Highly Effective Pt-Based Water–Gas Shift Catalysts by Surface Modification with Alkali Hydroxide Salts. ChemCatChem 2015, 7, 766–775. [Google Scholar] [CrossRef]
- Jacobs, G.; Williams, L.; Graham, U.; Thomas, G.A.; Sparks, D.E.; Davis, B.H. Low temperature water–gas shift: In situ DRIFTS-reaction study of ceria surface area on the evolution of formates on Pt/CeO2 fuel processing catalysts for fuel cell applications. Appl. Catal. A Gen. 2003, 252, 107–118. [Google Scholar] [CrossRef]
- Chenu, E.; Jacobs, G.; Crawford, A.C.; Keogh, R.A.; Patterson, P.M.; Sparks, D.E.; Davis, B.H. Water-gas shift: An examination of Pt promoted MgO and tetragonal and monoclinic ZrO2 by in situ drifts. Appl. Catal. B Environ. 2005, 59, 45–56. [Google Scholar] [CrossRef]
- Jacobs, G.; Graham, U.M.; Chenu, E.; Patterson, P.M.; Dozier, A.; Davis, B.H. Low-temperature water–gas shift: Impact of Pt promoter loading on the partial reduction of ceria and consequences for catalyst design. J. Catal. 2005, 229, 499–512. [Google Scholar] [CrossRef]
- Jentys, A. Estimation of mean size and shape of small metal particles by EXAFS. Phys. Chem. Chem. Phys. 1999, 1, 4059–4063. [Google Scholar] [CrossRef]
- Marinković, N.S.; Sasaki, K.; Adzic, R.R. Nanoparticle size evaluation of catalysts by EXAFS: Advantages and limitations. Zaštita Mater. 2016, 57, 101–109. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temprature water-gas shift: Assessing formates as potential intermediates over Pt/ZrO2 and Na doped Pt/ZrO2 catalysts employing the SSITKA-DRIFTS technique. In Advances in Fischer Tropsch Synthesis: Catalysts and Catalysis; Davis, B.H., Occelli, M., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2010; pp. 365–394. [Google Scholar]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Effect of alkali on C-H bond scission over Pt/YSZ catalyst during water-gas shift, steam-assisted formic acid decomposition and methanol steam reforming. Catal. Today 2017, 291, 29–35. [Google Scholar] [CrossRef]
- Martinelli, M.; Castro, J.D.; Alhraki, N.; Matamoros, M.E.; Kropf, A.J.; Cronauer, D.C.; Jacobs, G. Effect of sodium loading on Pt/ZrO2 during ethanol steam reforming. Appl. Catal. 2021, 610, 117947. [Google Scholar] [CrossRef]
- Iwasawa, Y. Surface Catalytic Reactions Assisted by Gas Phase Molecules. Accts. Chem. Res. 1997, 30, 103–109. [Google Scholar] [CrossRef]
- Martinelli, M.; Watson, C.D.; Jacobs, G. Sodium doping of Pt/m-ZrO2 promotes C-C scission and decarboxylation during ethanol steam reforming. Int. J. Hydrogen Energy 2020, 45, 18490–18501. [Google Scholar] [CrossRef]
- Martinelli, M.; Alhraki, N.; Castro, J.D.; Matamoros, M.E.; Jacobs, G. Water-Gas Shift: Effect of Na Loading on Pt/m-zirconia Catalysts for Low-Temperature Shift for the Production and Purification of Hydrogen. New Dimensions in Production and Utilization of Hydrogen; Elsevier: Amsterdam, The Netherlands, 2020; pp. 143–160. [Google Scholar]
- Watson, C.D.; Martinelli, M.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L.; Jacobs, G. Low temperature water-gas shift: Optimi zation of K loading on Pt/m-ZrO2 for enhancing CO conversion. Appl. Catal. A Gen. 2020, 598, 117572. [Google Scholar] [CrossRef]
- Jacoby, M. X-ray absorption spectroscopy. Chem. Eng. News 2001, 79, 33. [Google Scholar] [CrossRef]
- Ressler, T. WinXAS: A program for X-ray absorption spectroscopy data analysis under MS-Windows. J. Synchrotron Radiat. 1998, 5, 118. [Google Scholar] [CrossRef]
- Ravel, B. ATOMS: Crystallography for the X-ray absorption spectroscopist. J. Synchrotron Radiat. 2001, 8, 314–316. [Google Scholar] [CrossRef]
- Newville, M.; Ravel, B.; Haskel, D.; Rehr, J.J.; Stern, E.A.; Yacoby, Y. Analysis of multiple-scattering XAFS data using theoretical standards. Phys. B Condens. Matter 1995, 208, 154–156. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Expected AS Assuming No Pore Blocking [m2/g] | Measured AS (BET) [m2/g] | Difference between Expected & Measured [m2/g] | VP (BJH Des) [cm3/g] | DP (BJH Des) [Å] |
|---|---|---|---|---|---|
| ZrO2 | 95.4 | 0.289 | 95 | ||
| 2%Pt/ZrO2 | 93.2 | 89.6 | 3.6 | 0.272 | 96 |
| 0.72%Cs-2%Pt/ZrO2 | 92.5 | 88.5 | 4.0 | 0.259 | 93 |
| 1.45%Cs-2%Pt/ZrO2 | 91.7 | 84.6 | 7.1 | 0.249 | 92 |
| 2.17%Cs-2%Pt/ZrO2 | 91.0 | 85.3 | 5.7 | 0.246 | 92 |
| 2.89%Cs-2%Pt/ZrO2 | 90.3 | 86.6 | 3.7 | 0.251 | 91 |
| 3.87%Cs-2%Pt/ZrO2 | 89.3 | 86.8 | 2.5 | 0.256 | 89 |
| 4.80%Cs-2%Pt/ZrO2 | 88.3 | 78.7 | 9.6 | 0.238 | 89 |
| 5.78%Cs-2%Pt/ZrO2 | 87.3 | 74.1 | 13.2 | 0.229 | 91 |
| 7.22%Cs-2%Pt/ZrO2 | 85.9 | 69.1 | 16.8 | 0.211 | 93 |
| 10.41%Cs-2%Pt/ZrO2 | 82.7 | 62.1 | 20.6 | 0.197 | 94 |
| 14.45%Cs-2%Pt/ZrO2 | 78.6 | 54.9 | 23.7 | 0.177 | 97 |
| Sample Description | N Pt-Pt Metal | R Pt-Pt (Å) Metal | e0 (eV) | σ2 (Å2) | r-Factor | Est. Number of Atoms * | Est. Diam. (nm) */** | Est. % Disp. (%) |
|---|---|---|---|---|---|---|---|---|
| Pt0 foil | 12 | 2.766 | 8.99 | 0.00537 | 0.0094 | - | - | - |
| (fixed) | (0.0061) | (0.657) | (0.00045) | |||||
| 2%Pt/m-ZrO2 | 5.2 | 2.681 | 4.70 | 0.010 | 0.0062 | 13 | 0.86 0.78 | 92 |
| (0.44) | (0.0090) | (0.612) | (0.00143) | |||||
| 2.89%Cs-2%Pt/m-ZrO2 | 5.3 | 2.709 | 6.15 | 0.00905 | 0.0041 | 13 | 0.87 0.79 | 92 |
| (0.36) | (0.0069) | (0.471) | (0.00110) | |||||
| 4.80%Cs-2%Pt/m-ZrO2 | 6.3 | 2.725 | 6.55 | 0.00797 | 0.0031 | 22 | 1.0 0.93 | 87 |
| (0.36) | (0.0057) | (0.397) | (0.00090) | |||||
| 5.78%Cs-2%Pt/m-ZrO2 | 6.9 | 2.742 | 7.11 | 0.00660 | 0.0054 | 31 | 1.1 1.0 | 84 |
| (0.51) | (0.0070) | (0.507) | (0.00110) | |||||
| 10.41%Cs-2%Pt/m-ZrO2 | 8.2 | 2.755 | 8.32 | 0.00474 | 0.00089 | 82 | 1.5 1.4 | 73 |
| (0.24) | (0.0026) | (0.200) | (0.00041) | |||||
| 14.45%Cs-2%Pt/m-ZrO2 | 8.0 | 2.754 | 7.38 | 0.00594 | 0.0067 | 68 | 1.4 1.3 | 77 |
| (0.67) | (0.0076) | (0.560) | (0.00119) |
| Catalyst | Band Position (cm−1) | |||||
|---|---|---|---|---|---|---|
| ν(CH) | δ(CH) + νs(OCO) | 2δ(CH) | ν(OCO) Formate | ν(OCO) Carbonate | ν(OH) | |
| 2%Pt/ZrO2 (reference) | 2868 | 2973, (2961), 2931 | (2752) 2742 2732 | sy 1386, 1360 asy 1577 (1550) | (1613), 1562, 1471, 1437; (1387), 1350 | (3758), 3727, 3674, (3650) |
| 2.89%Cs-2%Pt/ZrO2 | 2849 2816 | 2972 2935, (2921) | 2763 2720 2686 | sy 1372, 1341, 1330, 1286 asy (1640, 1611) 1590 | 1642, (1620, 1567, 1525, 1475); (1331), 1321, (1285) | (3718), 3669 |
| 3.87%Cs-2%Pt/ZrO2 | 2829 2839 2848 | 2973, 2934 | 2762 2724 2683 | sy (1374) 1341, (1328, 1278) asy 1641, (1612, 1595) | (1677), 1643, (1587, 1565), (1512–1476); (1378) 1337, 1278 | (3755), 3725, (3678), 3665 |
| 4.80%Cs-2%Pt/ZrO2 | 2811 (2837) | 2930 | 2748 2704 2663 | sy (1377, 1363) 1345 (1331) asy (1658, 1640) 1634, 1592 | (1681, 1661), 1648, (1639), (1580), 1573, (1566–1538), (1498–1436); 1351, 1342 (1330, 1318), (1284) 1276, (1265) | (3722), 3673, 3655 |
| 5.78%Cs-2%Pt/ZrO2 | 2748 | 2927 | 2702 2664 | sy (1359) 1343, (1308, 1297) 1287, (1254) asy 1630, (1619, 1604, 1562) | (1679) 1638, (1572, 1477) (1347) 1334, (1316), 1287 | (3714), 3664 |
| 7.22%Cs-2%Pt/ZrO2 | 2758 | 2947, 2925 | 2706 2670 | sy (1358), 1344, (1321–1296) asy (1621), 1596, (1567–1540) | 1631, 1572, 1553, (1538, 1514–1441); (1344, 1335), 1330, (1313, 1303, 1293, 1286, 1280) | (3760), 3730, 3660, (3630) |
| 10.41%Cs-2%Pt/ZrO2 | 2758 | 2925 | 2706 2673 | sy (1347), 1342, (1320–1280) asy (1607, 1599), 1594, (1548) | 1613–1565, 1450, (1341, 1331), 1325, (1312, 1304, 1291) | (3735), 3716, 3663, (3635) |
| 14.45%Cs-2%Pt/ZrO2 | (2858) (2829) 2760 | 2925, (2907) | (2709) 2673 | sy 1337 (1291) asy 1596, (1550–1450) | - | (3681), 3665, (3602) |
| Sample ID | Dispersion Relative to 2%Pt/ZrO2 Calculated from EXAFS Fittings | Initial Pt-CO Magnitude Relative to 2%Pt/ZrO2 |
|---|---|---|
| 2%Pt/ZrO2 | 1 | 1 |
| 2.89%Cs-2%Pt/ZrO2 | 1 | 0.96 |
| 3.87%Cs-2%Pt/ZrO2 | - | 1.00 |
| 4.80%Cs-2%Pt/ZrO2 | 0.95 | 1.11 |
| 5.78%Cs-2%Pt/ZrO2 | 0.91 | 0.82 |
| 7.22%Cs-2%Pt/ZrO2 | - | 0.71 |
| 10.41%Cs-2%Pt/ZrO2 | 0.79 | 0.19 |
| 14.45%Cs-2%Pt/ZrO2 | 0.84 | 0.06 |
| Sample ID | T.o.S. (h) | Temperature (°C) | CO Conversion (%) | CO2 Selectivity (%) |
|---|---|---|---|---|
| 2%Pt/ZrO2 | 6.3 | 300 | 83.1 | 100 |
| 12.6 | 285 | 56.6 | 100 | |
| 19.6 | 260 | 32.0 | 100 | |
| 0.72%Cs-2%Pt/ZrO2 | 5.9 | 300 | 63.9 | 100 |
| 12.2 | 285 | 47.2 | 100 | |
| 19.2 | 260 | 25.3 | 100 | |
| 1.45%Cs-2%Pt/ZrO2 | 5.9 | 300 | 62.7 | 100 |
| 12.6 | 285 | 44.4 | 100 | |
| 19.2 | 260 | 24.1 | 100 | |
| 2.17%Cs-2%Pt/ZrO2 | 5.9 | 300 | 51.1 | 100 |
| 12.2 | 285 | 35.5 | 100 | |
| 18.9 | 260 | 18.5 | 100 | |
| 2.89%Cs-2%Pt/ZrO2 | 5.95 | 300 | 35.7 | 100 |
| 11.9 | 285 | 23.7 | 100 | |
| 18.2 | 260 | 12.7 | 100 | |
| 3.87%Cs-2%Pt/ZrO2 | 6.3 | 300 | 30.4 | 100 |
| 12.6 | 285 | 22.2 | 100 | |
| 19.9 | 260 | 12.6 | 100 | |
| 5.78%Cs-2%Pt/ZrO2 | 5.6 | 300 | 21.8 | 100 |
| 12.6 | 285 | 15.8 | 100 | |
| 17.9 | 260 | 8.7 | 100 | |
| 10.41%Cs-2%Pt/ZrO2 | 5.95 | 300 | 19.6 | 100 |
| 12.2 | 285 | 17.8 | 100 | |
| 18.9 | 260 | 9.1 | 100 | |
| 14.45%Cs-2%Pt/ZrO2 | 5.9 | 300 | 15.6 | 100 |
| 12.6 | 285 | 12.6 | 100 | |
| 18.9 | 260 | 6.0 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajabi, Z.; Martinelli, M.; Watson, C.D.; Cronauer, D.C.; Kropf, A.J.; Jacobs, G. Influence of Cs Loading on Pt/m-ZrO2 Water–Gas Shift Catalysts. Catalysts 2021, 11, 570. https://doi.org/10.3390/catal11050570
Rajabi Z, Martinelli M, Watson CD, Cronauer DC, Kropf AJ, Jacobs G. Influence of Cs Loading on Pt/m-ZrO2 Water–Gas Shift Catalysts. Catalysts. 2021; 11(5):570. https://doi.org/10.3390/catal11050570
Chicago/Turabian StyleRajabi, Zahra, Michela Martinelli, Caleb D. Watson, Donald C. Cronauer, A. Jeremy Kropf, and Gary Jacobs. 2021. "Influence of Cs Loading on Pt/m-ZrO2 Water–Gas Shift Catalysts" Catalysts 11, no. 5: 570. https://doi.org/10.3390/catal11050570
APA StyleRajabi, Z., Martinelli, M., Watson, C. D., Cronauer, D. C., Kropf, A. J., & Jacobs, G. (2021). Influence of Cs Loading on Pt/m-ZrO2 Water–Gas Shift Catalysts. Catalysts, 11(5), 570. https://doi.org/10.3390/catal11050570