3.1. Chemicals
The chemicals used for this investigation include sodium trisilicate (Na2Si3O7, Si as SiO2 ≥ 60% and Na as Na2O ≥ 18%, Fluka Honeywell, Charlotte, North Carolina, USA), sodium aluminate (NaAlO2 technical, Al as Al2O3 50–56% and Na as Na2O 37–45%, Sigma-Aldrich, St. Louis, Missouri, USA), tetrapropylammonium bromide (TPABr, C12H28BrN > 98.0%, TCI, Tokio, Japan), sulfuric acid (H2SO4 95–97%, VWR International, Radnor, Pennsylvania, USA), ammonium nitrate (NH4NO3 99%, Grüssing GmbH, Filsum, Germany), hydrofluoric acid (HF 40%, Merck Millipore, Burlington, Massachusetts, USA), nitric acid (HNO3 69%, Supra-quality, Carl Roth GmbH & Co. KG, Karlsruhe, Germany), hydrochloric acid (HCl 37%, VWR International) and boric acid (H3BO3 99.9995%, Alfa Aesar, Haverhill, Massachusetts, USA). Feed for catalytic tests consists of methanol (CH3OH 99.9%, TCI), ethanol (C2H5OH abs., Sigma-Aldrich), n-propanol (C3H7OH 99.7%, Sigma-Aldrich), n-butanol (C4H9OH 99.8%, Sigma-Aldrich), n-pentanol (C5H11OH > 99%, Sigma-Aldrich), n-hexanol (C6H13OH > 99%, Sigma-Aldrich), n-heptanol (C7H15OH > 98%, Sigma-Aldrich), ethyl octanoate (C7H15COOC2H5 > 98%, Sigma-Aldrich), octanal (C7H15CHO 99%, Sigma-Aldrich), octanoic acid/caprylic acid (C7H15COOH > 98%, Sigma-Aldrich).
3.5. Temperature-Programmed Ammonia Desorption (TPAD)
The surface acidity of H-ZSM-5 zeolite samples was investigated using a TPDRO 1100 Series from Thermo Fisher Scientific (Waltham, Massachusetts, USA). The premeasurement preparation consisted of drying the calcined sample for 20 min in a muffle furnace. Then, 250 mg of the dried sample was calcined at 550 °C for 6 h using argon and cooled down to 100 °C. The ammonia loading was performed at 100 °C for 10 min, with consecutive conditioning at 100 °C with helium for 3 h. The measurement was performed, increasing the temperature from 100 °C to 550 °C or 750 °C with 10 K/min. The deconvolution of the TPAD profile was accomplished by applying asymmetric Gaussian distributions. The area was estimated by intern calibration of the device. The ammonia desorption profile of H-ZSM-5 consisted of an l-peak (“low-temperature peak”) and an h-peak (“high-temperature peak”). The signal maximum of the l-peak was approximately 270 °C and the signal maximum of the h-peak was approximately 490 °C. The h-peak was closely related to the chemisorption of NH
3 on the Brønsted acid sites and partly also the Lewis acid sites of the aluminosilicate material. The l-peak, on the other hand, was primarily caused by the adsorption of NH
3 by weaker physisorption interactions. These included the interactions between NH
3 and (1) silanol groups [
44,
45], (2) Na
+-ions [
44,
46], (3) non-zeolitic material [
44], (4) extra framework aluminum [
47] and (5) already chemisorbed NH
3 [
48]. The total acidity was given by the specific amount of adsorbed ammonia (mmol NH
3/g) and calculated by calibration of the signal area of the TPAD profile. Finally, the amount of desorbed ammonia and the surface acidity were determined (
Table 1). For further details, see
Table A3.
3.10. Catalytic Test Devices and Procedure
The catalytic tests were performed using a plug flow tube reactor (inner diameter 10 mm) connected to an electric oven, gas supply and a liquid pump. The reactor was filled with 500 mg of dried, calcined and fractionated (315–400 µm) catalyst sample between two layers of quartz glass wool. The reactor was heated using nitrogen at 500 °C for 60 min. The excess pressure inside the reactor was adjusted to 5 bar using hydrogen. After starting the feed pump, the reaction was equilibrated for 20 min. In turns of every 30 min, a gas and a liquid phase were abstracted. The gas phase was measured immediately using an HP 6890 Series GC System from Hewlett-Packard (Palo Alto, California, CA, USA) (HP-1 (100 m)) by a shortened DHA method (detailed hydrocarbon analysis). A qualitative check for CO and CO
2 was possible using gas chromatography and mass spectrometry (GC/MS) with a Clarus 680 GC and a Clarus SQ 8 S MS from PerkinElmer Instruments (Walham, Massachusatts, MA, USA) by means of the same DHA method. For this reason, hydrogen atmosphere was used as carrier gas, which was necessary to efficiently detect these small gases and which did not influence the catalytic results in this case, as already reported by Rao et al. [
52]. The liquid phase was stored at 10 °C and measured after the reaction using the same gas chromatograph (GC/FID) by means of an extended DHA method (based on ASTM D6730-01 [
53]). The calculation used is explained below. Since the oxygenated feeds differed in the wt.-% of carbon, all catalytic results were presented as carbon yield, which gives the yield normalized to the amount of hydrocarbon C
nH
m in the oxygenate feed after initial deoxygenation.
3.11. Analysis and Evaluation of Catalytic Test Reactions
The catalytic testing of the zeolite samples at standard parameters (
Table A1) provided the gas chromatographic analysis of the gaseous, organic and aqueous phase as a function of the reaction time (ToS), the mass flow of feed (
) and the mass flow of aqueous (
) and organic phase (
). Coke analysis of the catalyst samples after the reaction was performed using thermogravimetric analysis and revealed no significant amount of coke that had to be taken into account during this calculation.
The application of the DHA method for the separation of the products in the individual phases provided the area fractions of hydrocarbons and oxygenates but did not provide any information on the area fraction of H
2O, CO and CO
2, since these products cannot be detected by a flame ionization detector [
54]. In order to be able to conclude the mass balance of the reaction, the proportions of these products were determined indirectly by calculation. The calculation was based on the assumption that the deoxygenation of the oxygenates treated here takes place exclusively via decarbonylation and/or dehydration [
55], decarboxylation [
56,
57,
58] being excluded by definition as a possible reaction path. The product yield (
) was calculated for a defined reaction period (ToS) from the mass flow of a product in relation to the mass flow of the feed supplied. The feed conversion (x) was calculated analogously for a defined reaction period (ToS) taking into account the possible presence of the feed in all three product phases (aqueous, organic, gaseous).
The mass flow (
) and, indirectly, the flow rate of the amount of substance (
) of a product were calculated from the mass fraction of the product related to the mass flow of the analysed phase (
). Since the gas phase contained water and CO in non-analyzable amounts, the mass fraction of a product in the gas phase could only be based on the mass flow of analyzable products in the gas phase (hydrocarbons in gaseous phase) (
).
The mass fraction of a product (
) was obtained by dividing the area of the peak (
) and the resulting total area of all peaks of the chromatogram.
The feed types converted in this work differed in the presence and type of the functional group. The above deoxygenation assumption has different effects on the calculation of the mass flow of CO and H2O depending on the feed type. These two quantities have a direct effect on the calculation of the mass flow of analyzable products in the gas phase () and thus, according to (1), also to the product yield ().
Feed type I: “Non-oxygenates (pure hydrocarbons)”. Since no water or CO can be produced from the feed, the following applies:
Feed type II: “Alcohol oxygenates (primary alcohols)”. Deoxygenation for this type of feed takes place exclusively via dehydration. The mass flow of formed water thus corresponds to the mass flow of converted feed, which can be determined by gas chromatographic calibration of the mass flow of the feed used in the aqueous and gaseous product phase.
Feed type III: “Carbonyl oxygenates (carboxylic acid, aldehyde, ketone, ester)”. Deoxygenation of the carbonyl oxygenates takes place via decarbonylation and dehydration, whereby these two reactions, by definition, take place in a 1:1 ratio. By summing up the mass flow of all oxygenated products in the organic product phase, a mass flow rate of “converted oxygen” is defined. This mass flow rate is compared to the mass flow rate of oxygen supplied by the feed. The difference provides the mass flow of CO and water.
3.12. Experimental Series
For the catalytic tests, several different oxygenate feeds and four prepared H-ZSM-5 catalyst samples were used. The investigation of these feeds can be divided into two experimental series.
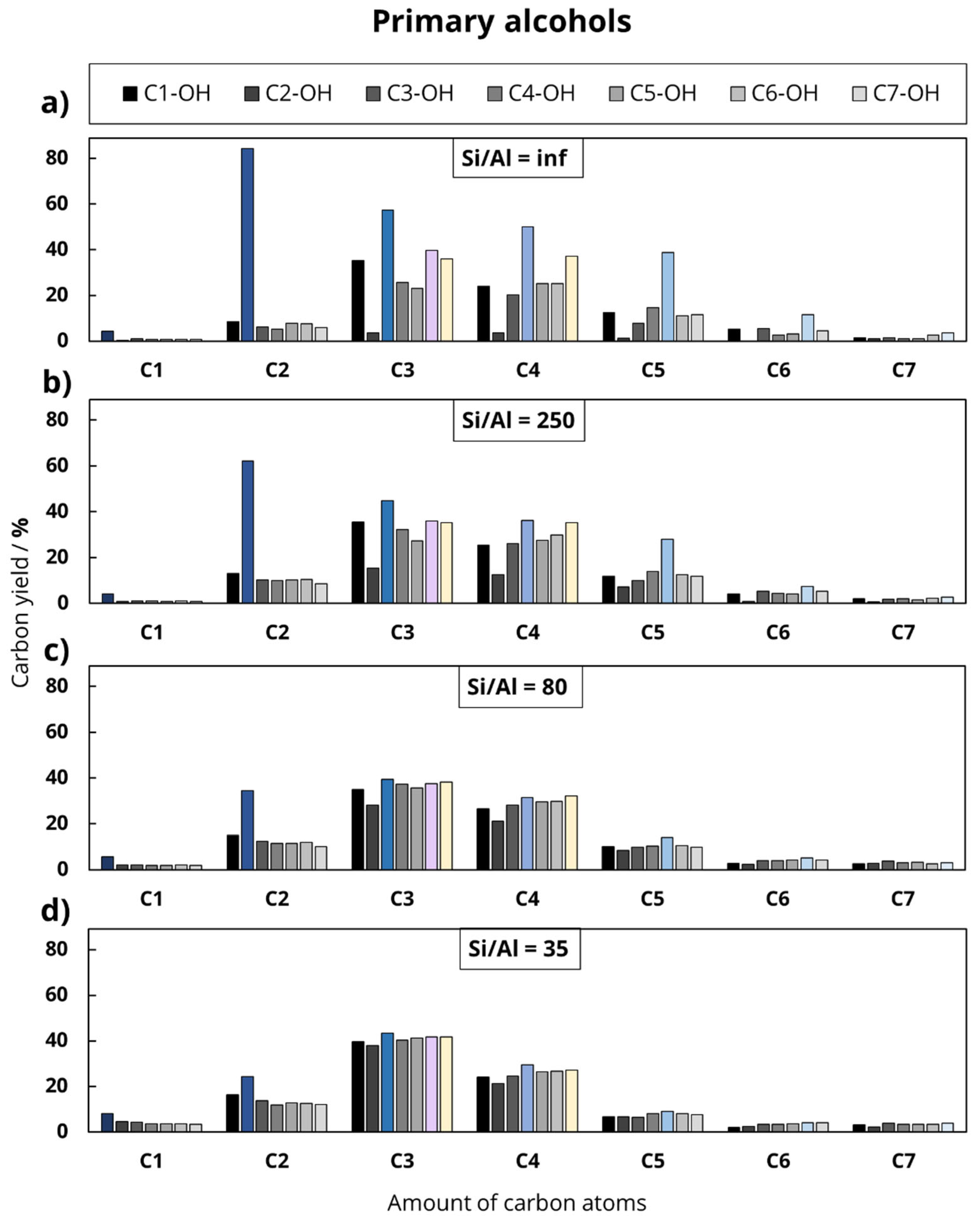
In the first experimental series, seven primary alcohols were investigated, which differed in the length of the hydrocarbon chain, starting with methanol (C1-OH) up to 1-heptanol (C7-OH).
In the second experimental series, the length of the main hydrocarbon chain remained unchanged, while the functional group varied (octanoic acid, octanal, ethyllaurate, 1-heptanol, equimolar solution of 1-heptanol and ethanol). This division was chosen to investigate the influence of the chain length and the deoxygenation mechanism on the product distribution during the catalytic conversion of oxygenates at 500 °C. In addition to the feed variation, the Al content of the used H-ZSM-5 zeolites was varied to investigate the influence of Al-based catalytic active sites.


 ,
, 
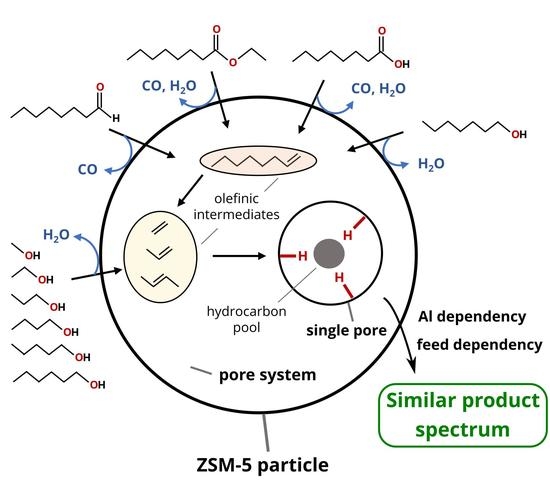


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







