
Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation

 , ,
, ,  ,
,
Abstract
:
1. Introduction
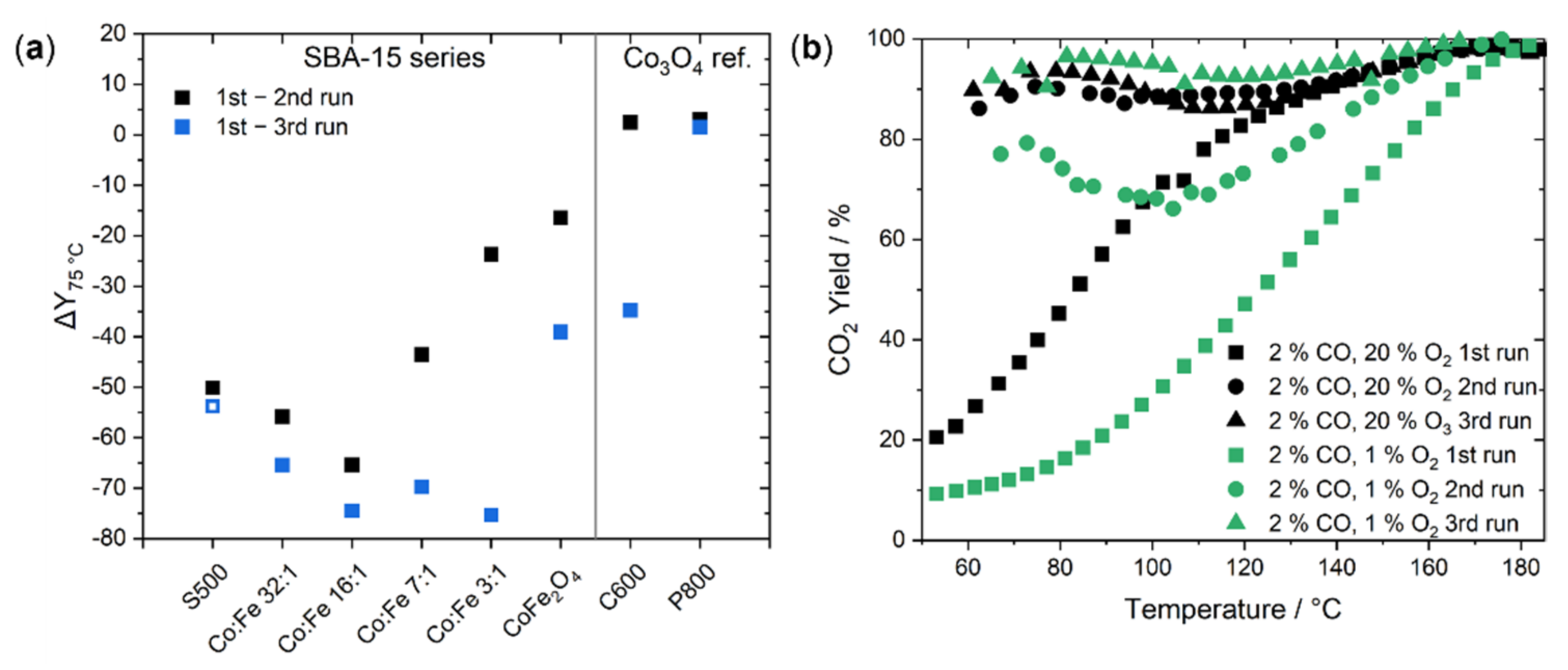
2. Results and Discussion
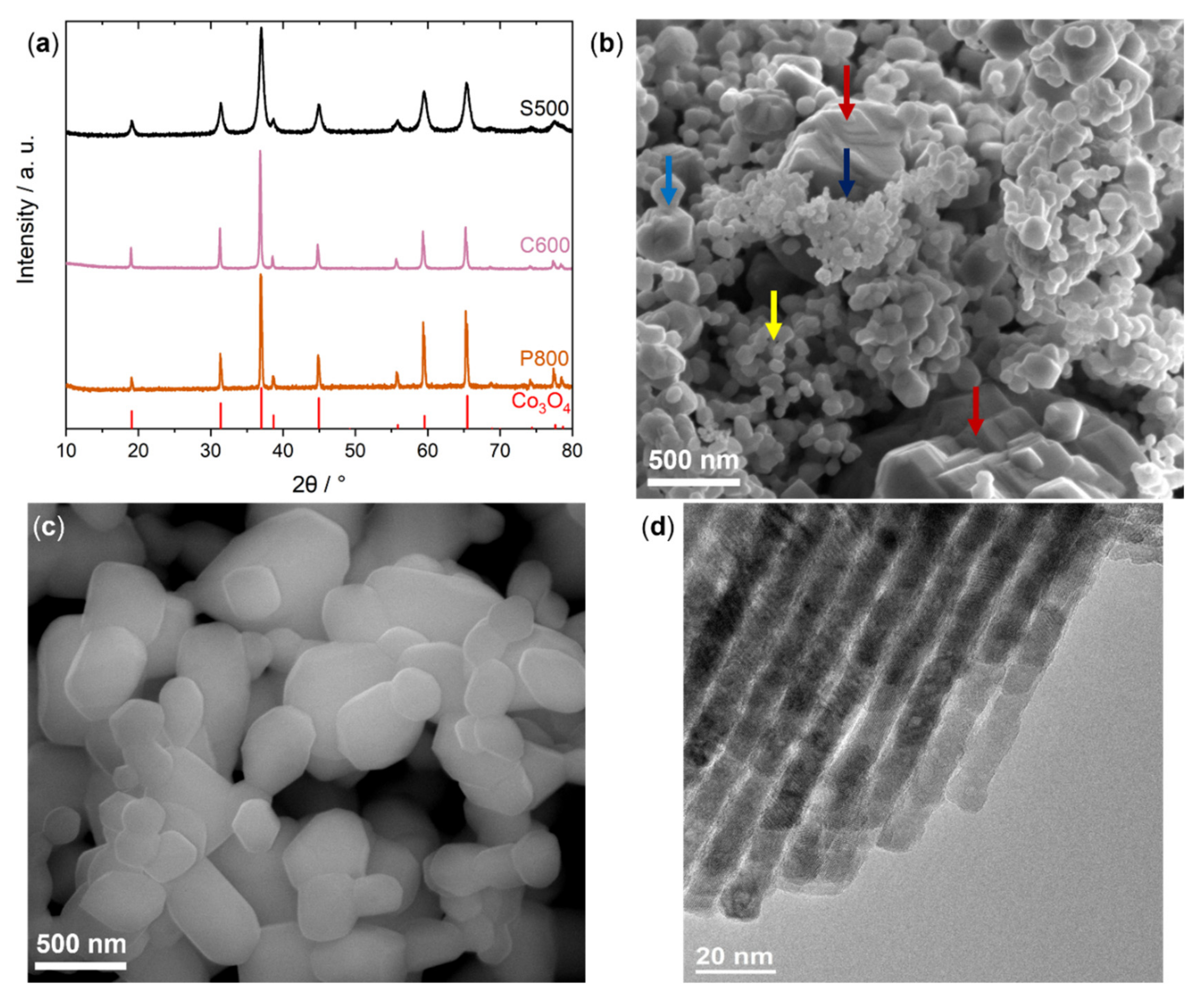
2.1. Characterization
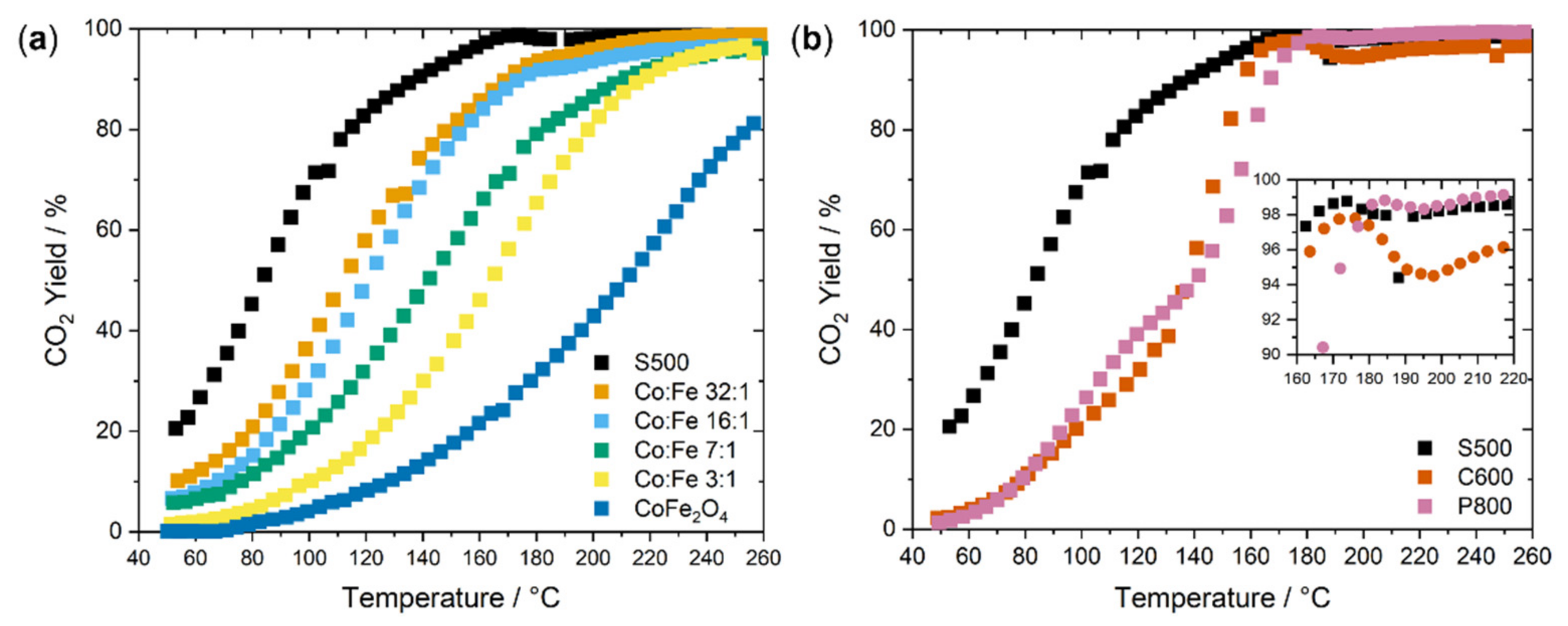
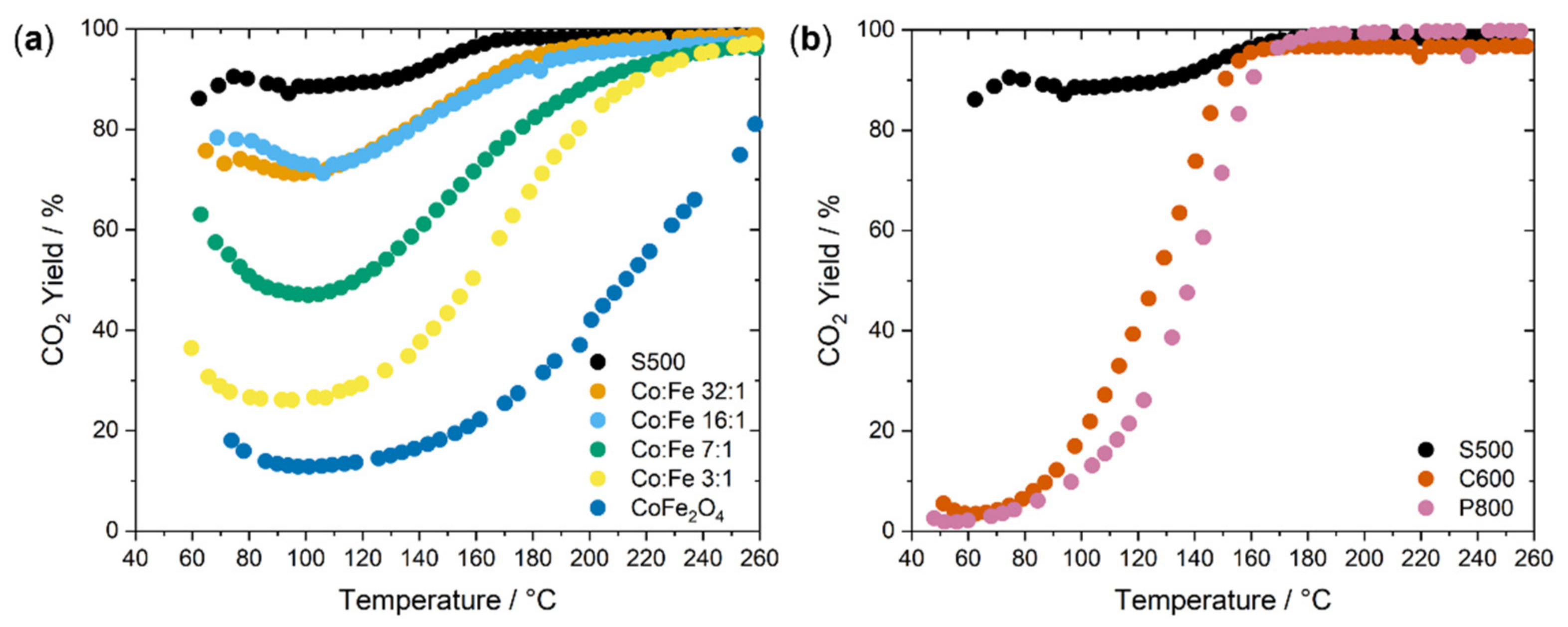
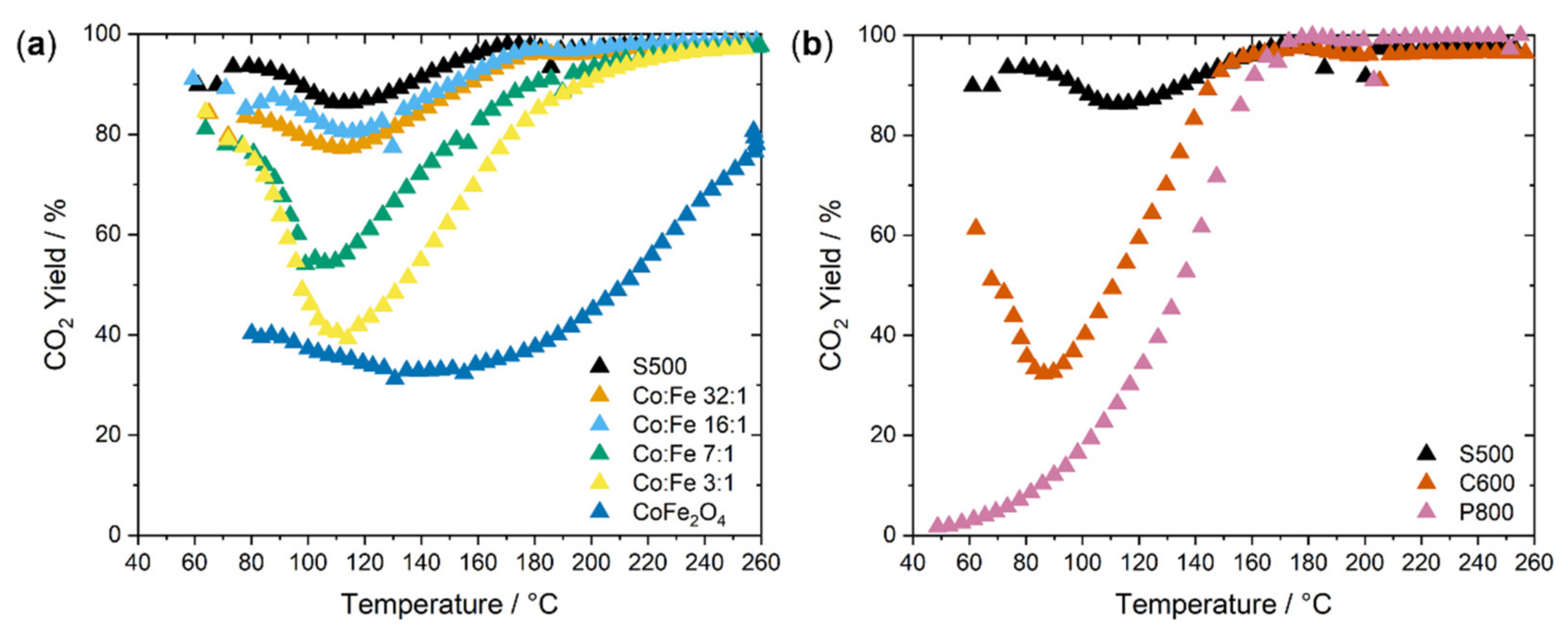
2.2. Catalysis
3. Materials and Methods
3.1. Synthesis and Sample Preparation
3.2. Catalyst Characterization
3.3. Catalytic CO Oxidation at Ambient Pressure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem. Int. Ed. Engl. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co(3)O(4) nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef]
- Yu, Y.B.; Takei, T.; Ohashi, H.; He, H.; Zhang, X.L.; Haruta, M. Pretreatments of Co3O4 at moderate temperature for CO oxidation at −80 °C. J. Catal. 2009, 267, 121–128. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Thomas, J.; Thomas, N.; Girgsdies, F.; Behrens, M.; Huang, X.; Sudheesh, V.D.; Sebastian, V. Synthesis of cobalt ferrite nanoparticles by constant pH co-precipitation and their high catalytic activity in CO oxidation. N. J. Chem. 2017, 41, 7356–7363. [Google Scholar] [CrossRef] [Green Version]
- Busca, G.; Guidetti, R.; Lorenzelli, V. Fourier-transform infrared study of the surface properties of cobalt oxides. J. Chem. Soc. Faraday Trans. 1990, 86. [Google Scholar] [CrossRef]
- Finocchio, E.; Willey, R.J.; Busca, G.; Lorenzelli, V. FTIR studies on the selective oxidation and combustion of light hydrocarbons at metal oxide surfaces Part 3.—Comparison of the oxidation of C3 organic compounds over Co3O4, MgCr2O4 and CuO. J. Chem. Soc. Faraday Trans. 1997, 93, 175–180. [Google Scholar] [CrossRef]
- Finocchio, E.; Busca, G.; Lorenzelli, V.; Escribano, V.S. FTIR studies on the selective oxidation and combustion of light hydrocarbons at metal oxide surfaces. Part 2.—Propane and propene oxidation on Co3O4. J. Chem. Soc. Faraday Trans. 1996, 92, 1587–1593. [Google Scholar] [CrossRef]
- Busca, G.; Daturi, M.; Finocchio, E.; Lorenzelli, V.; Ramis, G.; Willey, R.J. Transition metal mixed oxides as combustion catalysts: Preparation, characterization and activity mechanisms. Catal. Today 1997, 33, 239–249. [Google Scholar] [CrossRef]
- Zasada, F.; Janas, J.; Piskorz, W.; Sojka, Z. Surface oxygen dynamics and H2 oxidation on cobalt spinel surface probed by 18O/16O isotopic exchange and accounted for by DFT molecular modeling: Facile interfacial oxygen atoms flipping through transient peroxy intermediate. Res. Chem. Intermed. 2016, 43, 2865–2880. [Google Scholar] [CrossRef] [Green Version]
- Zasada, F.; Janas, J.; Piskorz, W.; Gorczynska, M.; Sojka, Z. Total Oxidation of Lean Methane over Cobalt Spinel Nanocubes Controlled by the Self-Adjusted Redox State of the Catalyst: Experimental and Theoretical Account for Interplay between the Langmuir-Hinshelwood and Mars-Van Krevelen Mechanisms. ACS Catal. 2017, 7, 2853–2867. [Google Scholar] [CrossRef]
- Zasada, F.; Grybos, J.; Budiyanto, E.; Janas, J.; Sojka, Z. Oxygen species stabilized on the cobalt spinel nano-octahedra at various reaction conditions and their role in catalytic CO and CH4 oxidation, N2O decomposition and oxygen isotopic exchange. J. Catal. 2019, 371, 224–235. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, Y.; Naschitzki, M.; Gewinner, S.; Schollkopf, W.; Kuhlenbeck, H.; Pentcheva, R.; Roldan Cuenya, B. Surface oxygen Vacancies on Reduced Co3 O4 (100): Superoxide Formation and Ultra-Low-Temperature CO Oxidation. Angew. Chem. Int. Ed. 2021, 60, 16514–16520. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Kusano, Y.; Azuma, M.; Haruta, M.; Shimakawa, Y. Morphology effects of Co3O4 nanocrystals catalyzing CO oxidation in a dry reactant gas stream. Catal. Sci. Technol. 2011, 1, 920–922. [Google Scholar] [CrossRef]
- Gu, D.; Jia, C.J.; Weidenthaler, C.; Bongard, H.J.; Spliethoff, B.; Schmidt, W.; Schüth, F. Highly Ordered Mesoporous Cobalt-Containing Oxides: Structure, Catalytic Properties, and Active Sites in Oxidation of Carbon Monoxide. J. Am. Chem. Soc. 2015, 137, 11407–11418. [Google Scholar] [CrossRef]
- Anke, S.; Bendt, G.; Sinev, I.; Hajiyani, H.; Antoni, H.; Zegkinoglou, I.; Jeon, H.; Pentcheva, R.; Roldan Cuenya, B.; Schulz, S.; et al. Selective 2-Propanol Oxidation over Unsupported Co3O4 Spinel Nanoparticles: Mechanistic Insights into Aerobic Oxidation of Alcohols. ACS Catal. 2019, 9, 5974–5985. [Google Scholar] [CrossRef]
- Lukashuk, L.; Yigit, N.; Rameshan, R.; Kolar, E.; Teschner, D.; Havecker, M.; Knop-Gericke, A.; Schlögl, R.; Föttinger, K.; Rupprechter, G. Operando Insights into CO Oxidation on Cobalt Oxide Catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 2018, 8, 8630–8641. [Google Scholar] [CrossRef]
- Dreyer, M.; Krebs, M.; Najafishirtari, S.; Rabe, A.; Friedel Ortega, K.; Behrens, M. The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites. Catalysts 2021, 11, 550. [Google Scholar] [CrossRef]
- Hu, L.H.; Sun, K.Q.; Peng, Q.; Xu, B.Q.; Li, Y.D. Surface Active Sites on Co3O4 Nanobelt and Nanocube Model Catalysts for CO Oxidation. Nano Res. 2010, 3, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Guo, J.; Wang, Y.B.; Wang, T.; Gu, J.; Peng, L.M.; Xue, N.H.; Zhu, Y.; Guo, X.F.; Ding, W.P. Reduction-oxidation pretreatment enhanced catalytic performance of Co3O4/Al2O3 over CO oxidation. Appl. Surf. Sci. 2018, 453, 330–335. [Google Scholar] [CrossRef]
- Jansson, J.; Palmqvist, A.E.C.; Fridell, E.; Skoglundh, M.; Osterlund, L.; Thormahlen, P.; Langer, V. On the catalytic activity of Co3O4 in low-temperature CO oxidation. J. Catal. 2002, 211, 387–397. [Google Scholar] [CrossRef]
- Chakrapani, K.; Bendt, G.; Hajiyani, H.; Lunkenbein, T.; Greiner, M.T.; Masliuk, L.; Salamon, S.; Landers, J.; Schlögl, R.; Wende, H.; et al. The Role of Composition of Uniform and Highly Dispersed Cobalt Vanadium Iron Spinel Nanocrystals for Oxygen Electrocatalysis. ACS Catal. 2018, 8, 1259–1267. [Google Scholar] [CrossRef]
- Büker, J.; Alkan, B.; Fu, Q.; Xia, W.; Schulwitz, J.; Waffel, D.; Falk, T.; Schulz, C.; Wiggers, H.; Muhler, M.; et al. Selective cyclohexene oxidation with O2, H2O2 and tert-butyl hydroperoxide over spray-flame synthesized LaCo1−xFexO3 nanoparticles. Catal. Sci. Technol. 2020, 10, 5196–5206. [Google Scholar] [CrossRef]
- Waffel, D.; Alkan, B.; Fu, Q.; Chen, Y.T.; Schmidt, S.; Schulz, C.; Wiggers, H.; Muhler, M.; Peng, B. Towards Mechanistic Understanding of Liquid-Phase Cinnamyl Alcohol Oxidation with tert-Butyl Hydroperoxide over Noble-Metal-Free LaCo1-x Fex O3 Perovskites. ChemPlusChem 2019, 84, 1155–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waag, F.; Gökce, B.; Kalapu, C.; Bendt, G.; Salamon, S.; Landers, J.; Hagemann, U.; Heidelmann, M.; Schulz, S.; Wende, H.; et al. Adjusting the catalytic properties of cobalt ferrite nanoparticles by pulsed laser fragmentation in water with defined energy dose. Sci. Rep. 2017, 7, 13161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrapani, K.; Bendt, G.; Hajiyani, H.; Schwarzrock, I.; Lunkenbein, T.; Salamon, S.; Landers, J.; Wende, H.; Schlögl, R.; Pentcheva, R.; et al. Role of Composition and Size of Cobalt Ferrite Nanocrystals in the Oxygen Evolution Reaction. ChemCatChem 2017, 9, 2988–2995. [Google Scholar] [CrossRef]
- Anke, S.; Falk, T.; Bendt, G.; Sinev, I.; Havecker, M.; Antoni, H.; Zegkinoglou, I.; Jeon, H.; Knop-Gericke, A.; Schlögl, R.; et al. On the reversible deactivation of cobalt ferrite spinel nanoparticles applied in selective 2-propanol oxidation. J. Catal. 2020, 382, 57–68. [Google Scholar] [CrossRef]
- Friedel Ortega, K.; Anke, S.; Salamon, S.; Özcan, F.; Heese, J.; Andronescu, C.; Landers, J.; Wende, H.; Schuhmann, W.; Muhler, M.; et al. Topotactic Synthesis of Porous Cobalt Ferrite Platelets from a Layered Double Hydroxide Precursor and Their Application in Oxidation Catalysis. Chemistry 2017, 23, 12443–12449. [Google Scholar] [CrossRef] [Green Version]
- Budiyanto, E.; Zerebecki, S.; Weidenthaler, C.; Kox, T.; Kenmoe, S.; Spohr, E.; DeBeer, S.; Rüdiger, O.; Reichenberger, S.; Barcikowski, S.; et al. Impact of Single-Pulse, Low-Intensity Laser Post-Processing on Structure and Activity of Mesostructured Cobalt Oxide for the Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2021. [Google Scholar] [CrossRef]
- Alkan, B.; Medina, D.; Landers, J.; Heidelmann, M.; Hagemann, U.; Salamon, S.; Andronescu, C.; Wende, H.; Schulz, C.; Schuhmann, W.; et al. Spray-Flame-Prepared LaCo1-xFexO3 Perovskite Nanoparticles as Active OER Catalysts: Influence of Fe Content and Low-Temperature Heating. ChemElectroChem 2020, 7, 2564–2574. [Google Scholar] [CrossRef]
- Alkan, B.; Cychy, S.; Varhade, S.; Muhler, M.; Schulz, C.; Schuhmann, W.; Wiggers, H.; Andronescu, C. Spray-Flame-Synthesized LaCo1-xFexO3 Perovskite Nanoparticles as Electrocatalysts for Water and Ethanol Oxidation. ChemElectroChem 2019, 6, 4266–4274. [Google Scholar] [CrossRef] [Green Version]
- Angel, S.; Neises, J.; Dreyer, M.; Friedel Ortega, K.; Behrens, M.; Wang, Y.; Arandiyan, H.; Schulz, C.; Wiggers, H. Spray-flame synthesis of La(Fe, Co)O3 nano-perovskites from metal nitrates. AlChE J. 2019, 66, 441. [Google Scholar] [CrossRef] [Green Version]
- Bahlawane, N.; Ngamou, P.H.; Vannier, V.; Kottke, T.; Heberle, J.; Kohse-Hoinghaus, K. Tailoring the properties and the reactivity of the spinel cobalt oxide. Phys. Chem. Chem. Phys. 2009, 11, 9224–9232. [Google Scholar] [CrossRef] [PubMed]
- Budiyanto, E.; Yu, M.Q.; Chen, M.M.; DeBeer, S.; Rudiger, O.; Tüysüz, H. Tailoring Morphology and Electronic Structure of Cobalt Iron Oxide Nanowires for Electrochemical Oxygen Evolution Reaction. ACS Appl. Energy Mater. 2020, 3, 8583–8594. [Google Scholar] [CrossRef]
- Falk, T.; Budiyanto, E.; Dreyer, M.; Pflieger, C.; Waffel, D.; Büker, J.; Weidenthaler, C.; Ortega, K.F.; Behrens, M.; Tüysüz, H.; et al. Identification of Active Sites in the Catalytic Oxidation of 2-Propanol over Co1+xFe2–xO4 Spinel Oxides at Solid/Liquid and Solid/Gas Interfaces. ChemCatChem 2021, 13, 2942–2951. [Google Scholar] [CrossRef]
- Waffel, D.; Budiyanto, E.; Porske, T.; Büker, J.; Falk, T.; Fu, Q.; Schmidt, S.; Tüysüz, H.; Muhler, M.; Peng, B.X. Investigation of Synergistic Effects between Co and Fe in Co3-xFexO4 Spinel Catalysts for the Liquid-Phase Oxidation of Aromatic Alcohols and Styrene. Mol. Catal. 2020, 498, 111251. [Google Scholar] [CrossRef]
- Wang, X.T.; Ouyang, T.; Wang, L.; Zhong, J.H.; Ma, T.; Liu, Z.Q. Redox-Inert Fe(3+) Ions in Octahedral Sites of Co-Fe Spinel Oxides with Enhanced Oxygen Catalytic Activity for Rechargeable Zinc-Air Batteries. Angew. Chem. Int. Ed. 2019, 58, 13291–13296. [Google Scholar] [CrossRef] [PubMed]
- Thommes, M. Physical Adsorption Characterization of Nanoporous Materials. Chem. Ing. Tech. 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Neimark, A.V.; Sing, K.S.W.; Thommes, M. Surface Area and Porosity. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 721–738. [Google Scholar]
- Rabe, A.; Büker, J.; Salamon, S.; Koul, A.; Hagemann, U.; Landers, J.; Ortega, K.F.; Peng, B.; Muhler, M.; Wende, H.; et al. The Roles of Composition and Mesostructure of Cobalt-based Spinel Catalysts in Oxygen Evolution Reactions. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Kotousova, I.; Polyakov, S. Electron-diffraction study of Co3O4. Kristallografiya 1972, 17, 661–663. [Google Scholar]
- Rietveld, H.M. Line Profiles of Neutron Powder-Diffraction Peaks for Structure Refinement. Acta Crystallogr. 1967, 22, 151. [Google Scholar] [CrossRef]
- Wang, H.F.; Kavanagh, R.; Guo, Y.L.; Guo, Y.; Lu, G.Z.; Hu, P. Origin of extraordinarily high catalytic activity of Co3O4 and its morphological chemistry for CO oxidation at low temperature. J. Catal. 2012, 296, 110–119. [Google Scholar] [CrossRef]
- Tüysuz, H.; Comotti, M.; Schüth, F. Ordered mesoporous Co3O4 as highly active catalyst for low temperature CO-oxidation. Chem. Commun. 2008, 34, 4022–4024. [Google Scholar] [CrossRef] [PubMed]
- Zasada, F.; Piskorz, W.; Janas, J.; Budiyanto, E.; Sojka, Z. Dioxygen Activation Pathways over Cobalt Spinel Nanocubes—From Molecular Mechanism into Ab Initio Thermodynamics and 16O2/18O2 Exchange Microkinetics. J. Phys. Chem. C 2017, 121, 24128–24143. [Google Scholar] [CrossRef]
- Jia, C.J.; Schwickardi, M.; Weidenthaler, C.; Schmidt, W.; Korhonen, S.; Weckhuysen, B.M.; Schüth, F. Co3O4-SiO2 nanocomposite: A very active catalyst for CO oxidation with unusual catalytic behavior. J. Am. Chem. Soc. 2011, 133, 11279–11288. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SABET/m2 g−1 |
|---|---|
| Co3O4 S500 (SBA-15 templated) | 107 |
| Co:Fe 32:1 (SBA-15 templated) | 118 |
| Co:Fe 16:1 (SBA-15 templated) | 103 |
| Co:Fe 7:1 (SBA-15 templated) | 113 |
| Co:Fe 3:1 (SBA-15 templated) | 125 |
| CoFe2O4 (SBA-15 templated) | 178 |
| Co3O4 C600 (commercial) | 9 |
| Co3O4 P800 (precipitated) | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreyer, M.; Rabe, A.; Budiyanto, E.; Friedel Ortega, K.; Najafishirtari, S.; Tüysüz, H.; Behrens, M. Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation. Catalysts 2021, 11, 1312. https://doi.org/10.3390/catal11111312
Dreyer M, Rabe A, Budiyanto E, Friedel Ortega K, Najafishirtari S, Tüysüz H, Behrens M. Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation. Catalysts. 2021; 11(11):1312. https://doi.org/10.3390/catal11111312
Chicago/Turabian StyleDreyer, Maik, Anna Rabe, Eko Budiyanto, Klaus Friedel Ortega, Sharif Najafishirtari, Harun Tüysüz, and Malte Behrens. 2021. "Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation" Catalysts 11, no. 11: 1312. https://doi.org/10.3390/catal11111312
APA StyleDreyer, M., Rabe, A., Budiyanto, E., Friedel Ortega, K., Najafishirtari, S., Tüysüz, H., & Behrens, M. (2021). Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation. Catalysts, 11(11), 1312. https://doi.org/10.3390/catal11111312