Sustainable Option for Hydrogen Production: Mechanistic Study of the Interaction between Cobalt Pincer Complexes and Ammonia Borane


 ,
, 

Abstract

1. Introduction
2. Results and Discussion
2.1. When Base is Involved in the Reaction Mechanism–Formation of Cationic Iridium Species
Metal Effects
2.2. When Base is Not Involved in the Reaction Mechanism–Neutral Iridium Species
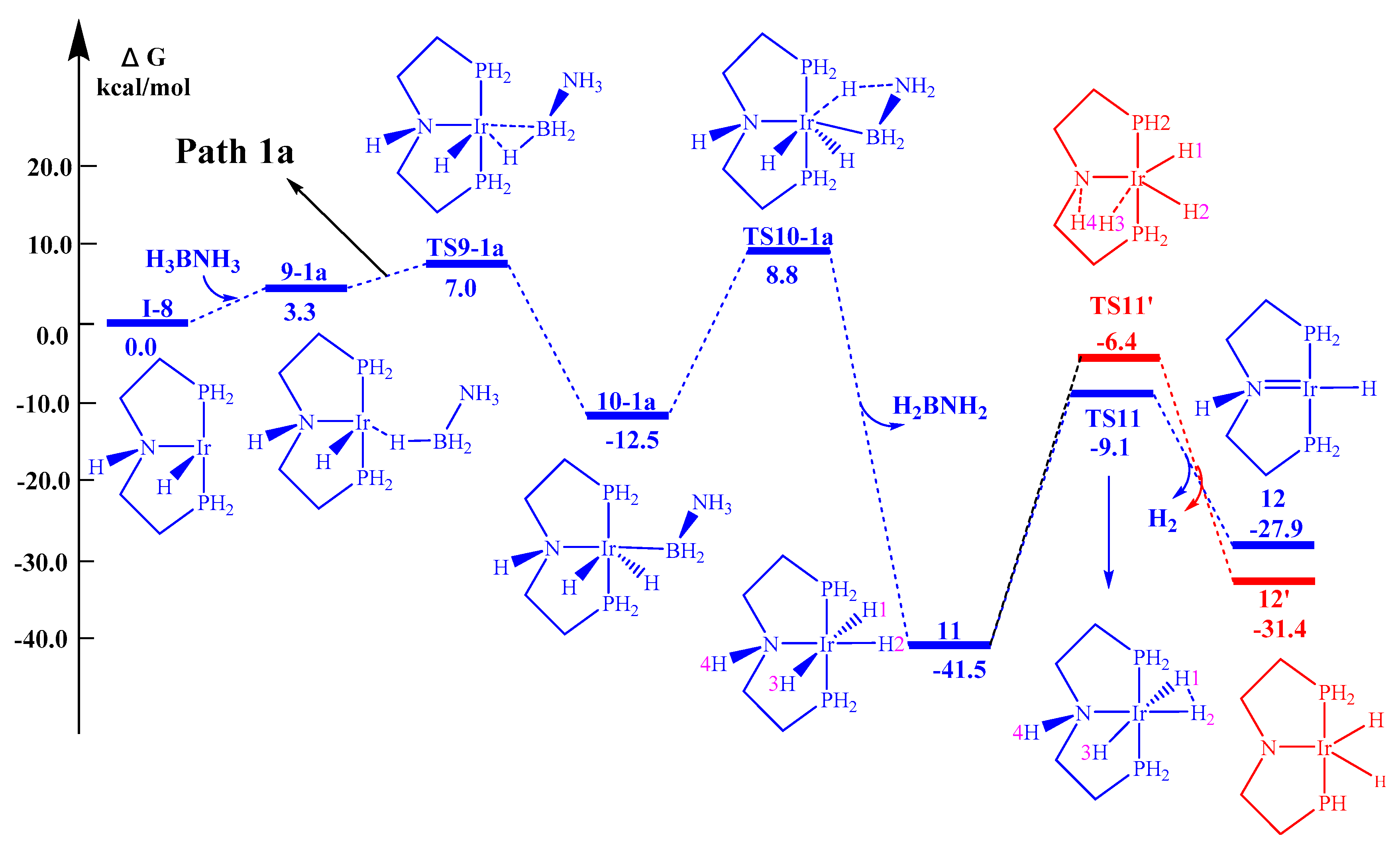
2.2.1. Stepwise Mechanism (Path 1)
2.2.2. Concerted Mechanism (Path 2)
2.2.3. Proton Transfer Mechanism
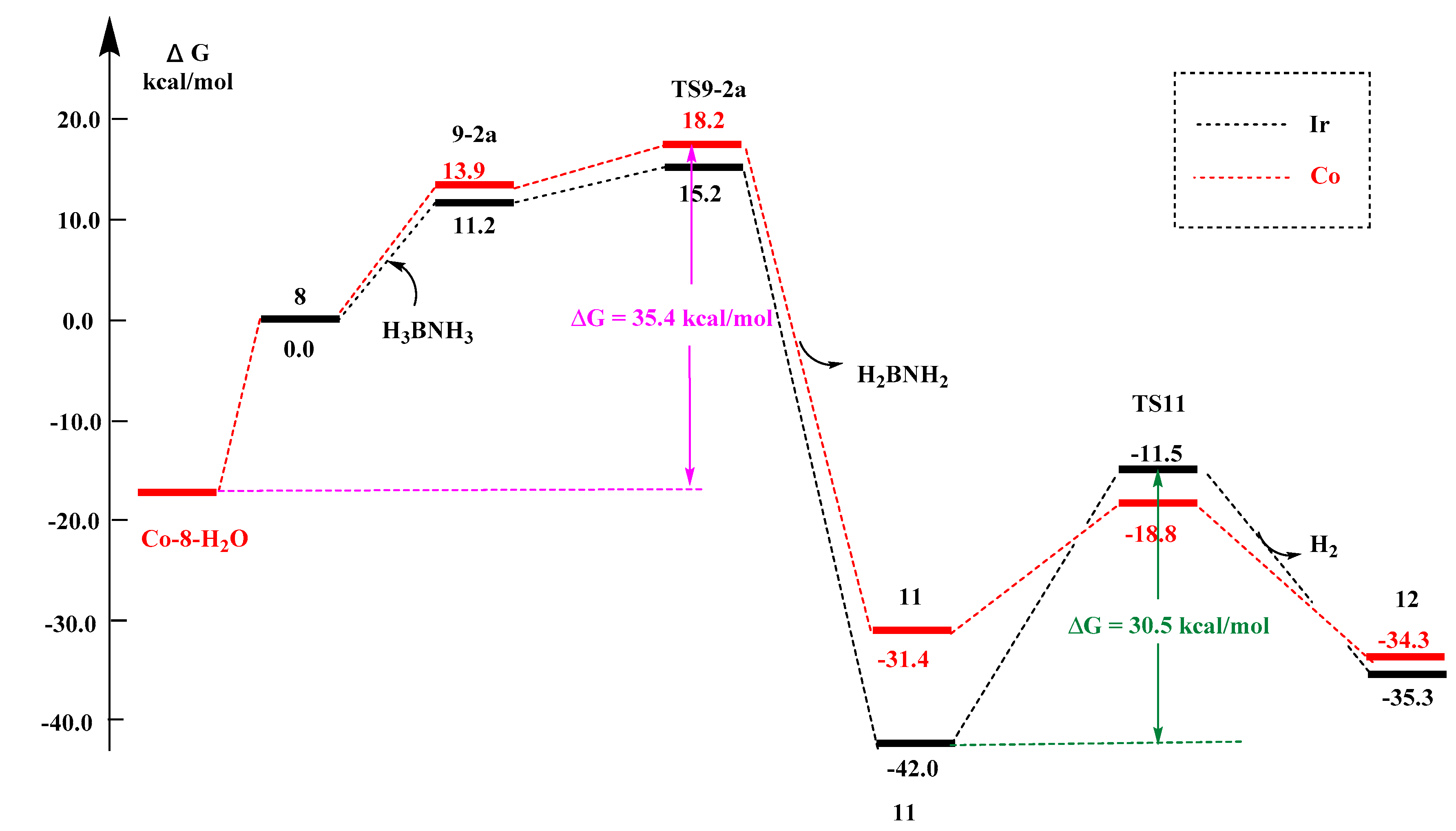
2.2.4. Metal Effects
- (1)
- except for the Ir center, the 11 → TS11′ → 12′ pathways for all the other metal centers have lower activation barriers than the 11 → TS11 → 12 pathway;
- (2)
- in Figure S6a, for the reaction of the first step (I-8 → TS9-2a) and the second step (11 → TS11), the energy barriers are calculated as follows: the energy barrier of Ir is 15.2 and 30.5 kcal/mol; the energy barrier of Co is 18.2 and 12.6 kcal/mol; the energy barrier of Fe is 44.7 and 1.8 kcal/mol; the energy barrier of Ru is 43.7 and 2.4 kcal/mol. The order of energy barrier is Co < Ir < Fe < Ru, so the reaction rate is Co > Ir > Fe > Ru;
- (3)
- in Figure S6b, for the reaction of the first step (8 → TS9-2a) and the second step (11′ → TS11′), the energy barriers are calculated as follows: the energy barrier of Ir is 15.2 and 28.6 kcal/mol; the energy barrier of Co is 18.2 and 21.0 kcal/mol; the energy barrier of Fe is 44.7 and 19.6 kcal/mol; and the energy barrier of Ru energy barrier is 43.7 and 25.4 kcal/mol. The order of energy barrier is Co < Ir < Fe < Ru, so the reaction rate is Co > Ir > Fe > Ru. It contradicts with the experimental results that the Co complex with tBu2P-substitutents did have lower reactivity towards ammonia borane; this can be explained by the finding of a much lower water molecules chelated Co-I-8 complex as a resting form (vide infra Section 2.2.6).
2.2.5. Ligand Effects
2.2.6. Effects of NH3 and H2O Adduct
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Energy Department Announces $15.8 Million Investment for Innovation in Hydrgoe and Fuel Cell Technologies; Office of Energy Efficiency & Renewable Energy, Department of Energy: Washington, DC, USA, 2017.
- Marco-Lozar, J.; Juan-Juan, J.; Suárez-García, F.; Cazorla-Amorós, D.; Linares-Solano, A. MOF-5, and activated carbons as adsorbents for gas storage. Int. J. Hydrogen Energy 2012, 37, 2370–2381. [Google Scholar] [CrossRef]
- Luo, W.; Campbell, P.G.; Zakharov, L.N.; Liu, S.-Y. A Single-component liquid-phase hydrogen storage material. J. Am. Chem. Soc. 2011, 133, 19326–19329. [Google Scholar] [CrossRef] [PubMed]
- Bogdanović, B.; Schwickardi, M. Ti-doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials. J. Alloys Compd. 1997, 253, 1–9. [Google Scholar] [CrossRef]
- Demirci, U.B. Ammonia borane, a material with exceptional properties for chemical hydrogen storage. Int. J. Hydrogen Energy 2017, 42, 9978–10013. [Google Scholar] [CrossRef]
- Demirci, U.B.; Miele, P. Sodium borohydride versus ammonia borane, in hydrogen storage and direct fuel cell applications. Energy Environ. Sci. 2009, 2, 627. [Google Scholar] [CrossRef]
- Rossin, A.; Peruzzini, M. Ammonia–borane and amine–borane dehydrogenation mediated by complex metal hydrides. Chem. Rev. 2016, 116, 8848–8872. [Google Scholar] [CrossRef]
- Moury, R.; Demirci, U.B. Hydrazine borane and hydrazinidoboranes as chemical hydrogen storage materials. Energies 2015, 8, 3118–3141. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, J.; Hamilton, E.J.; Chen, X. The continuing story of the diammoniate of diborane. J. Organomet. Chem. 2015, 798, 24–29. [Google Scholar] [CrossRef]
- Bhunya, S.; Malakar, T.; Ganguly, G.; Paul, A. Combining protons and hydrides by homogeneous catalysis for controlling the release of hydrogen from ammonia–borane: Present status and challenges. ACS Catal. 2016, 6, 7907–7934. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.; Manners, I. Ammonia-borane and related compounds as dihydrogen sources. Chem. Rev. 2010, 110, 4079–4124. [Google Scholar] [CrossRef]
- Summerscales, O.T.; Gordon, J.C. Regeneration of ammonia borane from spent fuel materials. Dalton Trans. 2013, 42, 10075. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, S.; English, N.J.; Phillips, A.D.; MacElroy, J.M.D. Exploring promising catalysts for chemical hydrogen storage in ammonia borane: A Density functional theory study. Catalysts 2017, 7, 140. [Google Scholar] [CrossRef]
- Denney, M.C.; Pons, V.; Hebden, T.J.; Heinekey, D.M.; Goldberg, K.I. Efficient catalysis of ammonia borane dehydrogenation. J. Am. Chem. Soc. 2006, 128, 12048–12049. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.W.; Tsang, C.-W.; Chen, X.; Guo, R.; Jia, W.; Lu, S.-M.; Sui-Seng, C.; Ewart, C.B.; Lough, A.; Amoroso, D.; et al. Catalytic solvolysis of ammonia borane. Angew. Chem. Int. Ed. 2010, 49, 8708–8711. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.J.; Dallanegra, R.; Chaplin, A.B.; Weller, A.S.; MacGregor, S.A.; Ward, B.; McKay, D.; Alcaraz, G.; Sabo-Etienne, S. Ir(PCy3)2(H)2(H2B‒NMe2)+ as a latent source of aminoborane: Probing the role of metal in the dehydrocoupling of H3B⋅NMe2H and retrodimerisation of [H2BNMe2]2. Chem. A Eur. J. 2011, 17, 3011–3020. [Google Scholar] [CrossRef]
- Johnson, H.C.; Robertson, A.; Chaplin, A.B.; Sewell, L.J.; Thompson, A.L.; Haddow, M.; Manners, I.; Weller, A.S. Catching the first oligomerization event in the catalytic formation of polyaminoboranes: H3B·NMeHBH2·NMeH2 Bound to iridium. J. Am. Chem. Soc. 2011, 133, 11076–11079. [Google Scholar] [CrossRef]
- Fortman, G.C.; Slawin, A.M.Z.; Nolan, S.P. Highly Active iridium (III)–NHC system for the catalytic B‒N bond activation and subsequent solvolysis of ammonia–borane. Organometallics 2011, 30, 5487–5492. [Google Scholar] [CrossRef]
- Ai, D.-X.; Qi, Z.-H.; Ruan, G.-Y.; Zhang, Y.; Liu, W.; Wang, Y. DFT studies of dehydrogenation of ammonia–borane catalyzed by Ir(ItBu′)2+: A proton transfer mechanism. Comput. Theor. Chem. 2014, 1048, 1–6. [Google Scholar] [CrossRef]
- Tang, C.Y.C.; Phillips, N.; Kelly, M.; Aldridge, S. Hydrogen shuttling: Synthesis and reactivity of a 14-electron iridium complex featuring a bis(alkyl) tethered N-heterocyclic carbene ligand. Chem. Commun. 2012, 48, 11999. [Google Scholar] [CrossRef]
- Wang, W.-H.; Tang, H.-P.; Lu, W.-D.; Li, Y.; Bao, M.; Himeda, Y. Mechanistic insights into the catalytic hydrolysis of ammonia borane with proton-responsive iridium complexes: An experimental and theoretical study. ChemCatChem 2017, 9, 3191–3196. [Google Scholar] [CrossRef]
- Rossin, A.; Caporali, M.; Gonsalvi, L.; Guerri, A.; Lledós, A.; Peruzzini, M.; Zanobini, F. Selective B‒H versus N‒H Bond activation in ammonia borane by Ir(dppm)2OTf. Eur. J. Inorg. Chem. 2009, 2009, 3055–3059. [Google Scholar] [CrossRef]
- von Colbe, J.B.; Ares, J.-R.; Barale, J.; Baricco, M.; Buckley, C.; Capurso, G.; Gallandat, N.; Grant, D.M.; Guzik, M.N.; Jacob, I.; et al. Application of hydrides in hydrogen storage and compression: Achievements, outlook and perspectives. Int. J. Hydrogen Energy 2019, 44, 7780–7808. [Google Scholar] [CrossRef]
- Cobalt prices continue to climb due to market uncertainty. Met. Powder Rep. 1999, 54, 4. [CrossRef]
- Iridium Prices. Available online: https://www.umicore.com/en/about/elements/iridium/ (accessed on 29 May 2020).
- Lin, T.-P.; Peters, J.C. Boryl–metal bonds facilitate cobalt/nickel-catalyzed olefin hydrogenation. J. Am. Chem. Soc. 2014, 136, 13672–13683. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Jing, Y.; Yang, X. Computational design of cobalt catalysts for hydrogenation of carbon dioxide and dehydrogenation of formic acid. Inorg. Chem. 2016, 55, 12179–12184. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Chen, N.-Y.; Liu, X.; Shao, Z.; Luo, S.-P.; Liu, Q. Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: Stereodivergent synthesis of Z- and E-alkenes. J. Am. Chem. Soc. 2016, 138, 8588–8594. [Google Scholar] [CrossRef]
- Alpaydın, C.Y.; Gülbay, S.K.; Colpan, C.O. A review on the catalysts used for hydrogen production from ammonia borane. Int. J. Hydrogen Energy 2020, 45, 3414–3434. [Google Scholar] [CrossRef]
- Nuss, P.; Eckelman, M.J. Life Cycle assessment of metals: A scientific synthesis. PLoS ONE 2014, 9, e101298. [Google Scholar] [CrossRef]
- Ghatak, K.; Vanka, K. A computational investigation of the role of the iridium dihydrogen pincer complex in the formation of the cyclic pentamer (NH2BH2)5. Comput. Theor. Chem. 2012, 992, 18–29. [Google Scholar] [CrossRef]
- Paul, A.; Musgrave, C.B. Catalyzed dehydrogenation of ammonia–borane by iridium dihydrogen pincer complex differs from ethane dehydrogenation. Angew. Chem. Int. Ed. 2007, 46, 8153–8156. [Google Scholar] [CrossRef]
- Ciganda, R.; Garralda, M.A.; Ibarlucea, L.; Pinilla, E.; Torres, M.R. A hydridoirida-β-diketone as an efficient and robust homogeneous catalyst for the hydrolysis of ammonia–borane or amine–borane adducts in air to produce hydrogen. Dalton Trans. 2010, 39, 7226. [Google Scholar] [CrossRef] [PubMed]
- Garralda, M.A.; Mendicute-Fierro, C.; Rodríguez-Diéguez, A.; Seco, J.M.; Ubide, C.; Zumeta, I. Efficient hydridoirida? Diketone-catalyzed hydrolysis of ammonia- or amine-boranes for hydrogen generation in air. Dalton Trans. 2013, 42, 11652. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Truscott, B.; Egbert, J.D.; Nolan, S.P. Exploring the limits of catalytic ammonia–borane dehydrogenation using a bis (N-heterocyclic carbene) iridium (III) complex. Organometallics 2013, 32, 3769–3772. [Google Scholar] [CrossRef]
- Umegaki, T.; Yan, J.-M.; Zhang, X.; Shioyama, H.; Kuriyama, N.; Xu, Q. Preparation, and catalysis of poly(N-vinyl-2-pyrrolidone) (PVP) stabilized nickel catalyst for hydrolytic dehydrogenation of ammonia borane. Int. J. Hydrogen Energy 2009, 34, 3816–3822. [Google Scholar] [CrossRef]
- Abdur-Rashid, K.; Graham, T.; Tsang, C.-W.; Chen, X.; Guo, R.; Jia, W.; Amoroso, D.; Sui-Seng, C. The Generation of Hydrogen from Ammonia Borane through Catalytic Hydrolysis. International Patent WO2008141439A1, 2008. [Google Scholar]
- Abdur-Rashid, K.; Graham, T.; Tsang, C.-W.; Chen, X.; Guo, R.; Jia, W. Method to Produce Hydrogen from the Dehydrocoupling of Amine Boranes. US Patent US9115249B2, 2015. [Google Scholar]
- Freixa, Z.; Garralda, M.A. Insights into the use of [Ru(p-Cym) (bipy)Cl]Cl as precatalyst for solvolytic dehydrogenation of ammonia-borane. Inorganica Chim. Acta 2015, 431, 184–189. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, V.; et al. Gaussian 09 (Revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., Ed.; Plenum Press: New York, NY, USA, 1977; pp. 1–28. [Google Scholar]
- Fukui, K.; Fujimoto, H. A formulation of the reaction coordinate. Quant. Chem. 1997, 7, 371–373. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Aleksandr, V.M.; Christopher, J.C.; Donald, G.T. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar]
- Tamura, H.; Yamazaki, H.; Sato, H.; Sakaki, S. Iridium-catalyzed borylation of benzene with diboron. Theoretical elucidation of catalytic cycle including unusual iridium(V) intermediate. J. Am. Chem. Soc. 2003, 125, 16114–16126. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hou, C.; Jiang, J.; Zhang, Z.; Zhao, C.; Page, A.J.; Ke, Z. General H2 activation modes for Lewis acid–transition metal bifunctional catalysts. ACS Catal. 2016, 6, 1655–1662. [Google Scholar] [CrossRef]
- Wenz, K.M.; Liu, P.; Houk, K.N. Intramolecular C–H Activation reactions of ru (NHC) complexes combined with H2 transfer to alkenes: A theoretical elucidation of mechanisms and effects of ligands on reactivities. Organometallics 2017, 36, 3613–3623. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Knecht, T.; Daniliuc, C.; Houk, K.N.; Glorius, F. Unprecedented dearomatized spirocyclopropane in a sequential rhodium (III)-catalyzed C−H activation and rearrangement reaction. Angew. Chem. Int. Ed. 2018, 57, 5520–5524. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zheng, Y.; Cui, T.; Meggers, E.; Houk, K.N. Arylketone π-conjugation controls enantioselectivity in asymmetric alkynylations catalyzed by centrochiral ruthenium complexes. J. Am. Chem. Soc. 2018, 140, 5146–5152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, S.; Hong, Y.; Winterling, E.; Tan, Y.; Hemming, M.; Harms, K.; Houk, K.N.; Meggers, E. Non-C2-symmetric chiral-at-ruthenium catalyst for highly efficient enantioselective intramolecular C(sp3)–H amidation. J. Am. Chem. Soc. 2019, 141, 19048–19057. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, H.; Zhang, X.; Wang, L.; Li, S.-J.; Wei, D.; Zhu, Y.; Zhang, W. Unravelling the origins of hydroboration chemoselectivity inversion using an N,O-chelated Ir (I) complex: A computational study. J. Org. Chem. 2019, 84, 6709–6718. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
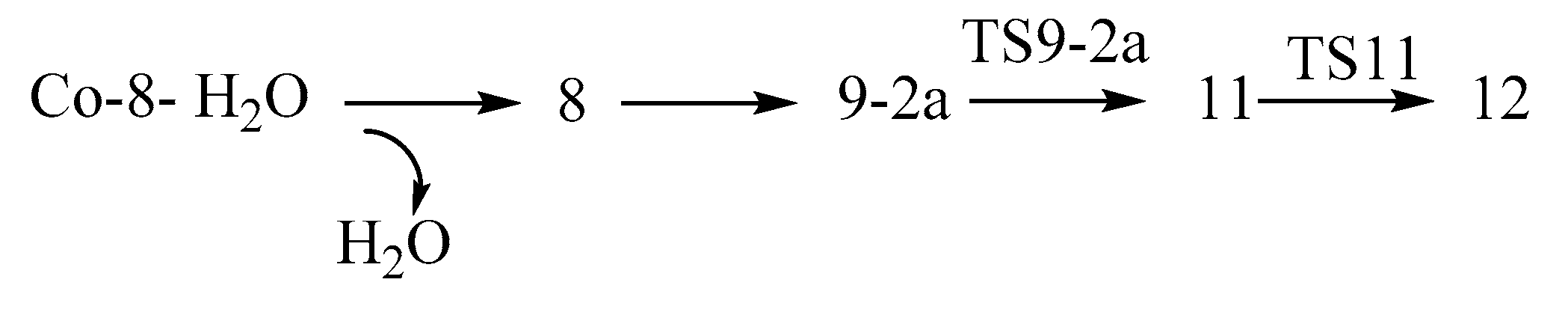{kind=link}
| Path 1a 10-1a → TS10-1a | Path 2a I-8 → TS9-2a | Path 2b I-8 → TS9-2b | Path 3 I-8 → TS9-3 | |
|---|---|---|---|---|
| Simplified model (H2P-) | 21.3 | 5.2 | 22.9 | 38.6 |
| Complete model (tBu2P-) | 19.3 | 15.2 | 34.5 | 26.8 |
| Chelation | Species | Relative Energy (kcal/mol) |
|---|---|---|
| No chelation | Co-8 | 0.0 |
| H2O | Co-8-h2o-1 | 13.6 |
| Co-8-h2o-2 | 13.7 | |
| Co-8-h2o-3 | 13.6 | |
| Co-8-h2o-4 | −17.5 | |
| Co-8-h2o-5 | 12.7 | |
| Co-8-h2o-6 | 13.3 | |
| NH3 | Co-8-nh3-1 | 7.3 |
| Co-8-nh3-2 | 7.4 | |
| Co-8-nh3-3 | 7.4 | |
| Co-8-nh3-4 | −17.2 | |
| Co-8-nh3-5 | 7.1 | |
| Co-8-nh3-6 | 7.1 | |
| THF | Co-8-thf-1 | 18.6 |
| Co-8-thf-2 | 17.5 | |
| Co-8-thf-3 | 19.1 | |
| Co-8-thf-4 | 8.3 | |
| Co-8-thf-5 | 18.4 | |
| Co-8-thf-6 | 18.4 |
| Chelation | Species | Relative Energy (kcal/mol) |
|---|---|---|
| H2O | Ir-8 | 0.0 |
| Ir-8-h2o-1 | 18.8 | |
| Ir-8-h2o-2 | 8.7 | |
| Ir-8-h2o-3 | 7.4 | |
| Ir-8-h2o-4 | 3.8 | |
| Ir-8-h2o-5 | 8.1 | |
| Ir-8-h2o-6 | 18.5 | |
| NH3 | Ir-8-nh3-1 | 13.0 |
| Ir-8-nh3-2 | 13.0 | |
| Ir-8-nh3-3 | 11.8 | |
| Ir-8-nh3-4 | 9.2 | |
| Ir-8-nh3-5 | 12.0 | |
| Ir-8-nh3-6 | 12.4 | |
| THF | Ir-8-thf-1 | 23.0 |
| Ir-8-thf-2 | 23.5 | |
| Ir-8-thf-3 | 13.0 | |
| Ir-8-thf-4 | 22.5 | |
| Ir-8-thf-5 | 22.0 |
| Chelation | Species | Relative Energy (kcal/mol) |
|---|---|---|
| H2O | Co-8 | 0.0 |
| Co-8-h2o-1 | 15.5 | |
| Co-8-h2o-2 | 14.3 | |
| Co-8-h2o-3 | 13.8 | |
| Co-8-h2o-4 | −16.6 | |
| Co-8-h2o-5 | 17.3 | |
| Co-8-h2o-6 | 16.4 | |
| NH3 | Co-8-nh3-1 | 8.6 |
| Co-8-nh3-2 | 8.4 | |
| Co-8-nh3-3 | −16.5 | |
| Co-8-nh3-4 | 18.5 | |
| Co-8-nh3-5 | 9.0 | |
| THF | Co-8-thf-1 | 19.3 |
| Co-8-thf-2 | 19.3 | |
| Co-8-thf-3 | 11.1 | |
| Co-8-thf-4 | 11.7 | |
| Co-8-thf-5 | 19.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Tsang, C.-W.; Chan, E.M.H.; Wong, E.Y.C.; Ho, D.C.K.; Lu, X.-Y.; Liang, C. Sustainable Option for Hydrogen Production: Mechanistic Study of the Interaction between Cobalt Pincer Complexes and Ammonia Borane. Catalysts 2020, 10, 723. https://doi.org/10.3390/catal10070723
Li Y, Tsang C-W, Chan EMH, Wong EYC, Ho DCK, Lu X-Y, Liang C. Sustainable Option for Hydrogen Production: Mechanistic Study of the Interaction between Cobalt Pincer Complexes and Ammonia Borane. Catalysts. 2020; 10(7):723. https://doi.org/10.3390/catal10070723
Chicago/Turabian StyleLi, Yan, Chi-Wing Tsang, Eve Man Hin Chan, Eugene Yin Cheung Wong, Danny Chi Kuen Ho, Xiao-Ying Lu, and Changhai Liang. 2020. "Sustainable Option for Hydrogen Production: Mechanistic Study of the Interaction between Cobalt Pincer Complexes and Ammonia Borane" Catalysts 10, no. 7: 723. https://doi.org/10.3390/catal10070723
APA StyleLi, Y., Tsang, C.-W., Chan, E. M. H., Wong, E. Y. C., Ho, D. C. K., Lu, X.-Y., & Liang, C. (2020). Sustainable Option for Hydrogen Production: Mechanistic Study of the Interaction between Cobalt Pincer Complexes and Ammonia Borane. Catalysts, 10(7), 723. https://doi.org/10.3390/catal10070723