Investigation of the Synergistic Effect of Sonolysis and Photocatalysis of Titanium Dioxide for Organic Dye Degradation

 ,
, 
Abstract
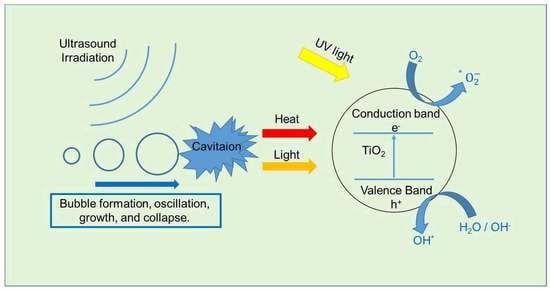
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Analysis of Sonoluminescence
2.2. Sonolysis of Eosin B
2.3. Influence of Sonolysis on Photocatalytic Activity of TiO2
3. Methods
3.1. Chemicals and Instruments
3.2. Sonoluminescence
3.3. Sonolysis, Photocatalysis and Sonophotocatalysis of Eosin B
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oturan, M.A.; Aaron, J.J. Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications. A Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 1. Combinative and hybrid sonophotochemical oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, J.; Kumar, P.S.S.; Anandan, S.; Zhou, M.; Grieser, F.; Ashokkumar, M. Ultrasound assisted photocatalytic degradation of diclofenac in an aqueous environment. Chemosphere 2010, 80, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Chakma, S.; Moholkar, V.S. Physical mechanism of sono-Fenton process. AIChE J. 2013, 59, 4303–4313. [Google Scholar] [CrossRef]
- Asghar, A.; Raman, A.A.A.; Daud, W.M.A.W. Advanced oxidation processes for in-situ production of hydrogen peroxide/hydroxyl radical for textile wastewater treatment: A review. J. Clean. Prod. 2015, 87, 826–838. [Google Scholar] [CrossRef]
- Reuter, F.; Mettin, R. Mechanisms of single bubble cleaning. Ultrason. Sonochem. 2016, 29, 550–562. [Google Scholar] [CrossRef]
- Suslick, K.S.; Didenko, Y.; Fang, M.M.; Hyeon, T.; Kolbeck, K.J.; McNamara, W.B.; Mdleleni, M.M.; Wong, M. Acoustic cavitation and its chemical consequences. Philos. Trans. R. Soc. A 1999, 357, 335–353. [Google Scholar] [CrossRef]
- Skorb, E.V.; Fix, D.; Shchukin, D.G.; Möhwald, H.; Sviridov, D.V.; Mousa, R.; Wanderka, N.; Schäferhans, J.; Pazos-Pérez, N.; Fery, A. Sonochemical formation of metal sponges. Nanoscale 2011, 3, 985–993. [Google Scholar] [CrossRef]
- Cravotto, G.; Gaudino, E.C.; Cintas, P. On the mechanochemical activation by ultrasound. Chem. Soc. Rev. 2013, 42, 7521–7534. [Google Scholar] [CrossRef]
- Qiu, P.; Park, B.; Choi, J.; Thokchom, B.; Pandit, A.B.; Khim, J. A review on heterogeneous sonocatalyst for treatment of organic pollutants in aqueous phase based on catalytic mechanism. Ultrason. Sonochem. 2018, 45, 29–49. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Mahamuni, N.N.; Adewuyi, Y.G. Advanced oxidation processes (AOPs) involving ultrasound for waste water treatment: A review with emphasis on cost estimation. Ultrason. Sonochem. 2010, 17, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 2. Heterogeneous sonophotocatalytic oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 8557–8570. [Google Scholar] [CrossRef] [PubMed]
- Lops, C.; Ancona, A.; Di Cesare, K.; Dumontel, B.; Garino, N.; Canavese, G.; Hérnandez, S.; Cauda, V. Sonophotocatalytic degradation mechanisms of Rhodamine B dye via radicals generation by micro-and nano-particles of ZnO. Appl. Catal. B Environ. 2019, 243, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Sun, M.; Yuan, X.; Zhu, Y.; Lin, X.; Anandan, S. One-step hydrothermal synthesis of N/Ti3+ co-doping multiphasic TiO2/BiOBr heterojunctions towards enhanced sonocatalytic performance. Ultrason. Sonochem. 2018, 49, 69–78. [Google Scholar] [CrossRef]
- Soltani, R.D.C.; Jorfi, S.; Ramezani, H.; Purfadakari, S. Ultrasonically induced ZnO-biosilica nanocomposite for degradation of a textile dye in aqueous phase. Ultrason. Sonochem. 2016, 28, 69–78. [Google Scholar] [CrossRef]
- Balaji, C.; Moholkar, V.S.; Pandit, A.B.; Ashokkumar, M. Mechanistic investigations on sonophotocatalytic degradation of textile dyes with surface active solutes. Ind. Eng. Chem. Res. 2011, 50, 11485–11494. [Google Scholar] [CrossRef]
- Chakma, S.; Moholkar, V.S. Investigation in mechanistic issues of sonocatalysis and sonophotocatalysis using pure and doped photocatalysts. Ultrason. Sonochem. 2015, 22, 287–299. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. Sonophotocatalytic reactors for wastewater treatment: A critical review. AIChE J. 2004, 50, 1051–1079. [Google Scholar] [CrossRef]
- Mahdavi, R.; Talesh, S.S.A. Enhancement of ultrasound-assisted degradation of Eosin B in the presence of nanoparticles of ZnO as sonocatalyst. Ultrason. Sonochem. 2019, 51, 230–240. [Google Scholar] [CrossRef]
- Rooze, J.; Rebrov, E.V.; Schouten, J.C.; Keurentjes, J.T. Dissolved gas and ultrasonic cavitation—A review. Ultrason. Sonochemistry 2013, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Didenko, Y.T.; Pugach, S.P. Spectra of Water Sonoluminescence. J. Phys. Chem. 1994, 98, 9742–9749. [Google Scholar] [CrossRef]
- Ji, R.; Pflieger, R.; Virot, M.; Nikitenko, S.I. Multibubble Sonochemistry and Sonoluminescence at 100 kHz: The Missing Link between Low- and High-Frequency Ultrasound. J. Phys. Chem. B 2018, 122, 6989–6994. [Google Scholar] [CrossRef] [PubMed]
- Luminol. Available online: https://en.wikipedia.org/wiki/Luminol (accessed on 8 January 2020).
- McMurray, H.N.; Wilson, B.P. Mechanistic and spatial study of ultrasonically induced luminol chemiluminescence. J. Phys. Chem. A 1999, 103, 3955–3962. [Google Scholar] [CrossRef]
- Crum, L.A.; Mason, T.J.; Reisse, J., Suslick. Sonochemistry and Sonoluminescence; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999; pp. 225–246. [Google Scholar]
- Janzen, E.G.; Kotake, Y.; Hinton, R.D. Stabilities of Hydroxyl Radical Spin Adducts of Pbn-Type Spin Traps. Free Radic. Biol. Med. 1992, 12, 169–173. [Google Scholar] [CrossRef]
- Khan, P.; Idrees, D.; Moxley, M.A.; Corbett, J.A.; Ahmad, F.; von Figura, G.; Sly, W.S.; Waheed, A.; Hassan, M.I. Luminol-based chemiluminescent signals: Clinical and non-clinical application and future uses. Appl. Biochem. Biotechnol. 2014, 173, 333–355. [Google Scholar] [CrossRef]
- Electromagnetic Absorption by Water. Available online: https://en.wikipedia.org/wiki/Electromagnetic_absorption_by_water (accessed on 23 February 2020).
- Ashokkumar, M.; Grieser, F. The effect of surface active solutes on bubbles in an acoustic field. Phys. Chem. Chem. Phys. 2007, 9, 5631–5643. [Google Scholar] [CrossRef]
- Rayaroth, M.P.; Aravind, U.K.; Aravindakumar, C.T. Sonochemical degradation of Coomassie Brilliant Blue: Effect of frequency, power density, pH and various additives. Chemosphere 2015, 119, 848–855. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.; Lee, D.; Hong, S.; Khan, S.; Darya, B.; Lee, J.-Y.; Chung, J.; Cho, S.-H. Investigation of the Synergistic Effect of Sonolysis and Photocatalysis of Titanium Dioxide for Organic Dye Degradation. Catalysts 2020, 10, 500. https://doi.org/10.3390/catal10050500
Choi Y, Lee D, Hong S, Khan S, Darya B, Lee J-Y, Chung J, Cho S-H. Investigation of the Synergistic Effect of Sonolysis and Photocatalysis of Titanium Dioxide for Organic Dye Degradation. Catalysts. 2020; 10(5):500. https://doi.org/10.3390/catal10050500
Chicago/Turabian StyleChoi, Yunseok, Daein Lee, Sungje Hong, Sovann Khan, Burak Darya, Jae-Young Lee, Jaewon Chung, and So-Hye Cho. 2020. "Investigation of the Synergistic Effect of Sonolysis and Photocatalysis of Titanium Dioxide for Organic Dye Degradation" Catalysts 10, no. 5: 500. https://doi.org/10.3390/catal10050500
APA StyleChoi, Y., Lee, D., Hong, S., Khan, S., Darya, B., Lee, J.-Y., Chung, J., & Cho, S.-H. (2020). Investigation of the Synergistic Effect of Sonolysis and Photocatalysis of Titanium Dioxide for Organic Dye Degradation. Catalysts, 10(5), 500. https://doi.org/10.3390/catal10050500