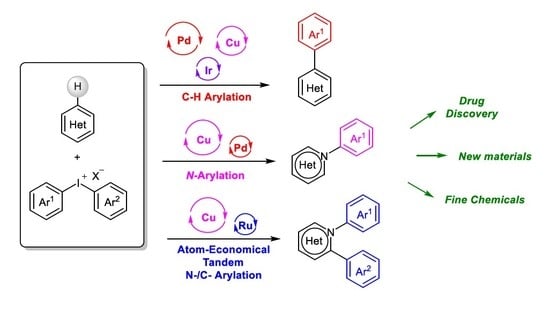
Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes




Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
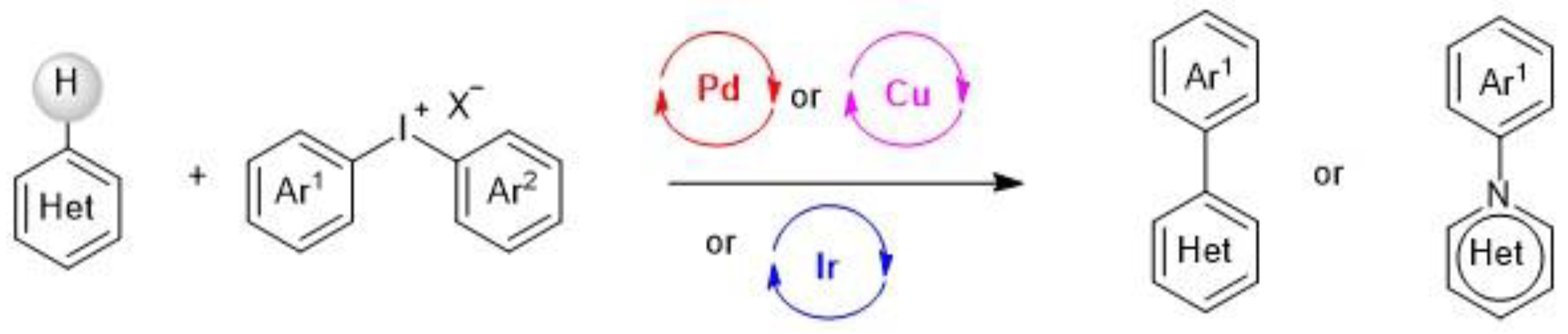
2. Metal-Catalyzed sp2 C–H Arylation of Heteroarenes
2.1. Palladium-Catalyzed C-H Arylation
2.2. Copper-Catalyzed C–H Arylation
2.3. Iridium-Catalyzed C–H Arylation and Visible-light mediated photoredox catalysis
3. Metal-Catalyzed N–H Arylation of Heteroarenes
4. Atom-Economical Tandem N–H/CH Arylation of Heteroarenes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stang, P.J. Polyvalent Iodine in Organic Chemistry. J. Org. Chem. 2003, 68, 2997–3008. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.A.; Pike, V.W.; Widdowson, D.A. New synthesis of diaryliodonium sulfonates from arylboronic acids. Tetrahedron Lett. 2000, 41, 5393–5396. [Google Scholar] [CrossRef]
- Hossain, M.D.; Kitamura, T. Reaction of iodoarenes with potassium peroxodisulfate/ trifluoroacetic acid in the presence of aromatics. Direct preparation of diaryliodonium triflates from iodoarenes. Tetrahedron 2006, 62, 6955–6960. [Google Scholar] [CrossRef]
- Bielawski, M.; Zhu, M.; Olofsson, B. Efficient and General One-Pot Synthesis of Diaryliodonium Triflates: Optimization, Scope and Limitations. Adv. Synth. Catal. 2007, 349, 2610–2618. [Google Scholar] [CrossRef]
- Bielawski, M.; Olofsson, B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or aryl iodides. Chem. Commun. 2007, 2521–2523. [Google Scholar] [CrossRef]
- Dohi, T.; Ito, M.; Morimoto, K.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Versatile direct dehydrative approach for diaryliodonium(iii) salts in fluoroalcohol media. Chem. Commun. 2007, 4152–4154. [Google Scholar] [CrossRef]
- Hossain, M.D.; Kitamura, T. New and Direct Approach to Hypervalent Iodine Compounds from Arenes and Iodine. Straightforward Synthesis of (Diacetoxyiodo)Arenes and Diaryliodonium Salts Using Potassium μ-Peroxo-Hexaoxodisulfate. Bull. Chem. Soc. Jpn. 2007, 80, 2213–2219. [Google Scholar] [CrossRef]
- Bielawski, M.; Aili, D.; Olofsson, B. Regiospecific One-Pot Synthesis of Diaryliodonium Tetrafluoroborates from Arylboronic Acids and Aryl Iodides. J. Org. Chem. 2008, 73, 4602–4607. [Google Scholar] [CrossRef]
- Dohi, T.; Yamaoka, N.; Kita, Y. Fluoroalcohols: versatile solvents in hypervalent iodine chemistry and syntheses of diaryliodonium(III) salts. Tetrahedron 2010, 66, 5775–5785. [Google Scholar] [CrossRef]
- Cardinale, J.; Ermert, J.; Coenen, H.H. Convenient preparation of (4-iodophenyl)aryliodonium salts. Tetrahedron 2012, 68, 4112–4116. [Google Scholar] [CrossRef]
- Chun, J.-H.; Pike, V.W. Regiospecific Syntheses of Functionalized Diaryliodonium Tosylates via [Hydroxy(tosyloxy)iodo]arenes Generated in Situ from (Diacetoxyiodo)arenes. J. Org. Chem. 2012, 77, 1931–1938. [Google Scholar] [CrossRef]
- Seidl, T.L.; Sundalam, S.K.; McCullough, B.; Stuart, D.R. Unsymmetrical Aryl(2,4,6-trimethoxyphenyl)iodonium Salts: One-Pot Synthesis, Scope, Stability, and Synthetic Studies. J. Org. Chem. 2016, 81, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Carreras, V.; Sandtorv, A.H.; Stuart, D.R. Synthesis of Aryl(2,4,6-trimethoxyphenyl)iodonium Trifluoroacetate Salts. J. Org. Chem. 2017, 82, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, E.; Reitti, M.; Olofsson, B. One-Pot Synthesis of Unsymmetric Diaryliodonium Salts from Iodine and Arenes. J. Org. Chem. 2017, 82, 11909–11914. [Google Scholar] [CrossRef] [PubMed]
- Soldatova, N.; Postnikov, P.; Kukurina, O.; Zhdankin, V.V.; Yoshimura, A.; Wirth, T.; Yusubov, M.S. One-pot synthesis of diaryliodonium salts from arenes and aryl iodides with Oxone–sulfuric acid. Beilstein J. Org. Chem. 2018, 14, 849–855. [Google Scholar] [CrossRef]
- Soldatova, N.S.; Postnikov, P.S.; Yusubov, M.S.; Wirth, T. Flow Synthesis of Iodonium Trifluoroacetates through Direct Oxidation of Iodoarenes by Oxone®. Eur. J. Org. Chem. 2019, 2081–2088. [Google Scholar] [CrossRef]
- Gallagher, R.T.; Seidl, T.L.; Bader, J.; Orella, C.; Vickery, T.; Stuart, D.R. Anion Metathesis of Diaryliodonium Tosylate Salts with a Solid Phase Column Constructed from Readily Available Laboratory Consumables. Org. Process Res. Dev. 2019, 23, 1269–1274. [Google Scholar] [CrossRef]
- Seidl, T.L.; Moment, A.; Orella, C.; Vickery, T.; Stuart, D.R. Synthesis of 4-Methylbenzoate(2ʹ,4ʹ,6ʹ-trimethoxyphenyl)iodonium Tosylate. Org. Synth. 2019, 96, 137–149. [Google Scholar] [CrossRef]
- Dohi, T.; Hayashi, T.; Ueda, S.; Shoji, T.; Komiyama, K.; Takeuchi, H.; Kita, Y. Recyclable synthesis of mesityl iodonium(III) salts. Tetrahedron 2019, 75, 3617–3627. [Google Scholar] [CrossRef]
- Tóth, B.L.; Béke, F.; Egyed, O.; Bényei, A.; Stirling, A.; Novák, Z. Synthesis of Multifunctional Aryl(trifloxyalkenyl)iodonium Triflate Salts. ACS Omega 2019, 4, 9188–9197. [Google Scholar] [CrossRef]
- Gallagher, R.T.; Basu, S.; Stuart, D.R. Trimethoxyphenyl (TMP) as a Useful Auxiliary for in situ Formation and Reaction of Aryl(TMP)iodonium Salts: Synthesis of Diaryl Ethers. Adv. Synth. Catal. 2020, 362, 320–325. [Google Scholar] [CrossRef]
- Beringer, F.M.; Brierley, A.; Drexler, M.; Gindler, E.M.; Lumpkin, C.C. Diaryliodonium Salts. II. The Phenylation of Organic and Inorganic Bases. J. Am. Chem. Soc. 1953, 75, 2708–2712. [Google Scholar] [CrossRef]
- Deprez, N.R.; Sanford, M.S. Reactions of Hypervalent Iodine Reagents with Palladium: Mechanisms and Applications in Organic Synthesis. Inorg. Chem. 2007, 46, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef] [PubMed]
- Yusubov, M.S.; Maskaev, A.V.; Zhdankin, V.V. Iodonium salts in organic synthesis. ARKIVOC 2011, 370–409. [Google Scholar]
- Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Arylation with Unsymmetrical Diaryliodonium Salts: A Chemoselectivity Study. Chem.-A Eur. J. 2013, 19, 10334–10342. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure and Applications of Polyvalent Iodine Compounds; John Wiley & Sons: Chichester, UK, 2014. [Google Scholar]
- Hartrampf, F.; Toombs-Ruane, H. Diaryliodonium Salts: Aryl Transfer Reagents for Alkyne Difunctionalization. Aust. J. Chem. 2015, 68, 699–702. [Google Scholar] [CrossRef]
- Olofsson, B. Arylation with Diaryliodonium Salts. Top. Curr. Chem. 2016, 373, 135–166. [Google Scholar] [CrossRef]
- Aradi, K.; Tóth, B.L.; Tolnai, G.L.; Novák, Z. Diaryliodonium Salts in Organic Syntheses: A Useful Compound Class for Novel Arylation Strategies. Synlett 2016, 27, 1456–1485. [Google Scholar] [CrossRef]
- Kumar, D.; Arun, V.; Pilania, M.; Mehra, M.K.; Khandagale, S.B. Diaryliodonium salts: Emerging reagents for arylations and heterocycles synthesis. Chem. Biol. Interface 2016, 6, 270–281. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Goswami, A. Synthesis and Application of Cyclic Diaryliodonium Salts: A Platform for Bifunctionalization in a Single Step. Eur. J. Org. Chem. 2017, 3023–3032. [Google Scholar] [CrossRef]
- Fañanás-Mastral, M. Copper-Catalyzed Arylation with Diaryliodonium Salts. Synthesis 2017, 49, 1905–1930. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Jiang, X. Atom-Economical Applications of Diaryliodonium Salts. Chem. Asian J. 2018, 13, 2195–2207. [Google Scholar] [CrossRef]
- Boelke, A.; Finkbeiner, P.; Nachtsheim, B.J. Atom-economical group-transfer reactions with hypervalent iodine compounds. Beilstein J. Org. Chem. 2018, 14, 1263–1280. [Google Scholar] [CrossRef]
- Villo, P.; Olofsson, B. Arylations Promoted by Hypervalent Iodine Reagents. Hypervalent Halogen Compounds in PATAI’S Chemistry of Functional Groups 2018, 1–61. [Google Scholar] [CrossRef]
- Hyatt, I.D.; Dave, L.; David, N.; Kaur, K.; Medard, M.; Mowdawallla, C. Hypervalent iodine reactions utilized in carbon–carbon bond formations. Org. Biomol. Chem. 2019, 17, 7822–7848. [Google Scholar] [CrossRef]
- Pike, V.W.; Aigbirhio, F.I. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts—a novel single-step route to no-carrier-added [18F]fluoroarenes. J. Chem. Soc., Chem. Commun. 1995, 21, 2215–2216. [Google Scholar] [CrossRef]
- Ross, T.L.; Ermert, J.; Hocke, C.; Coenen, H.H. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J. Am. Chem. Soc. 2007, 129, 8018–8025. [Google Scholar] [CrossRef]
- Telu, S.; Chun, J.H.; Simeon, F.G.; Lu, S.; Pike, V.W. Syntheses of mGluR5 PET radioligands through the radiofluorination of diaryliodonium tosylates. Org. Biomol. Chem. 2011, 9, 6629–6638. [Google Scholar] [CrossRef]
- Yusubov, M.S.; Svitich, D.Y.; Larkina, M.S.; Zhdankin, V.V. Applications of iodonium salts and iodonium ylides as precursors for nucleophilic fluorination in Positron Emission Tomography. ARKIVOC 2013, 364–395. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-catalyzed [18F]fluorination of (Mesityl)(aryl)iodonium salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.F.; Ichiishi, N.; Topczewski, J.J.; Sanford, M.S.; Scott, P.J.H. Synthesis of PET Radiotracers by Copper-mediated 18F-Fluorination of (Mesityl)(Aryl)Iodonium Salts and Aryl Iodides. J. Nucl. Med. 2015, 56, 218. [Google Scholar]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. [18F]Fluorination of (Mesityl)(Aryl)Iodonium Salts. Radiochemical Syntheses. In Further Radiopharmaceuticals for Positron Emission Tomography and New Strategies for Their Production, 1st ed.; Scott, P.J.H., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 2, pp. 129–137. [Google Scholar] [CrossRef]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Mossine, A.V.; Thompson, S.; Brooks, A.F.; Sowa, A.R.; Miller, J.M.; Scott, P.J. Fluorine-18 patents (2009–2015). Part 2: new radiochemistry. Pharmaceutical Patent Analyst 2016, 5, 319–349. [Google Scholar] [CrossRef]
- Yuan, Z.; Cheng, R.; Chen, P.; Liu, G.; Liang, S.H. Efficient Pathway for the Preparation of Aryl(isoquinoline)iodonium(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem. Int. Ed. 2016, 55, 11882–11886. [Google Scholar] [CrossRef]
- Warnier, C.; Lemaire, C.; Becker, G.; Zaragoza, G.; Giacomelli, F.; Aerts, J.; Otabashi, M.; Bahri, M.A.; Mercier, J.; Plenevaux, A.; et al. Enabling Efficient Positron Emission Tomography (PET) Imaging of Synaptic Vesicle Glycoprotein 2A (SV2A) with a Robust and One-Step Radiosynthesis of a Highly Potent 18F-Labeled Ligand ([18F]UCB-H). J. Med. Chem. 2016, 59, 8955–8966. [Google Scholar] [CrossRef]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef]
- McCammant, M.S.; Thompson, S.; Brooks, A.F.; Krska, S.W.; Scott, P.J.H.; Sanford, M.S. Cu-Mediated C–H 18F-Fluorination of Electron-Rich (Hetero)arenes. Org. Lett. 2017, 19, 3939–3942. [Google Scholar] [CrossRef]
- Kim, J.; Moon, B.S.; Lee, B.C.; Lee, H.-Y.; Kim, H.-J.; Choo, H.; Pae, A.N.; Cho, Y.S.; Min, S.-J. A Potential PET Radiotracer for the 5-HT2C Receptor: Synthesis and in Vivo Evaluation of 4-(3-[18F]fluorophenethoxy)pyrimidine. ACS Chem. Neurosci. 2017, 8, 996–1003. [Google Scholar] [CrossRef]
- Pike, V.W. Hypervalent aryliodine compounds as precursors for radiofluorination. J. Labelled Compd. Radiopharm. 2018, 61, 196–227. [Google Scholar] [CrossRef]
- Kwon, Y.-D.; Son, J.; Chun, J.-H. Chemoselective Radiosyntheses of Electron-Rich [18F]Fluoroarenes from Aryl(2,4,6-trimethoxyphenyl)iodonium Tosylates. J. Org. Chem. 2019, 84, 3678–3686. [Google Scholar] [CrossRef] [PubMed]
- Pauton, M.; Aubert, C.; Bluet, G.; Gruss-Leleu, F.; Roy, S.; Perrio, C. Development, Optimization, and Scope of the Radiosynthesis of 3/5-[18F]Fluoropyridines from Readily Prepared Aryl(pyridinyl) Iodonium Salts: The Importance of TEMPO and K2CO3. Org. Process Res. Dev. 2019, 23, 900–911. [Google Scholar] [CrossRef]
- Jang, K.S.; Lee, S.-S.; Oh, Y.-H.; Lee, S.H.; Kim, S.E.; Kim, D.W.; Lee, B.C.; Lee, S.; Raffel, D.M. Control of Reactivity and Selectivity of Guanidinyliodonium Salts Toward 18F-Labeling by Monitoring of Protecting Groups: Experiment and Theory. J. Fluor. Chem. 2019, 227, 109387–109410. [Google Scholar] [CrossRef]
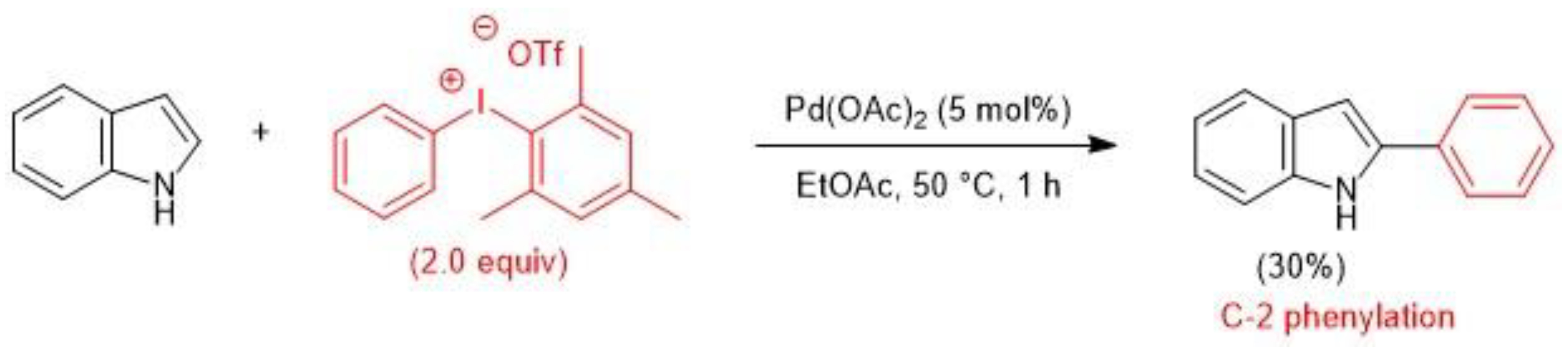
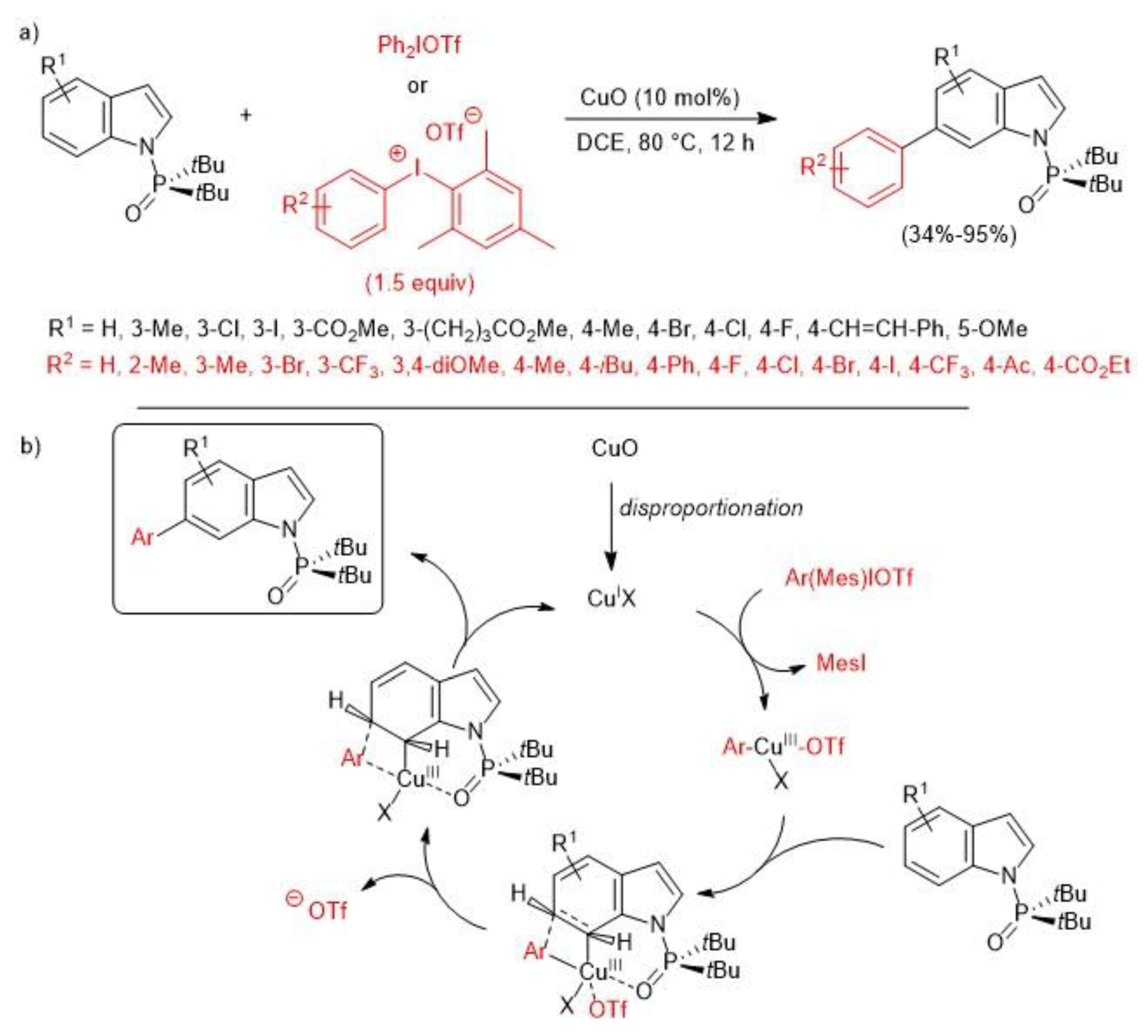
- Deprez, N.R.; Kalyani, D.; Sanford, M.S. Room Temperature Palladium-Catalyzed 2-Arylation of Indoles. J. Am. Chem. Soc. 2006, 128, 4972–4973. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Grimster, N.P.; Gaunt, M.J. Cu(II)-Catalyzed Direct and Site-Selective Arylation of Indoles Under Mild Conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174. [Google Scholar] [CrossRef] [PubMed]
- Budhwan, R.; Yadav, S.; Murarka, S. Late stage functionalization of heterocycles using hypervalent iodine(III) reagents. Org. Biomol. Chem. 2019, 17, 6326–6341. [Google Scholar] [CrossRef]
- Dröge, T.; Notzon, A.; Fröhlich, R.; Glorius, F. Palladium-Catalyzed C-H Bond Functionalization of a Metal–Organic Framework (MOF): Mild, Selective, and Efficient. Chem. Eur. J. 2011, 17, 11974–11977. [Google Scholar] [CrossRef]
- Wagner, A.M.; Sanford, M.S. Palladium-Catalyzed C-H Arylation of 2,5-Substituted Pyrroles. Org. Lett. 2011, 13, 288–291. [Google Scholar] [CrossRef]
- Bhunia, S.K.; Polley, A.; Natarajan, R.; Jana, R. Through-Space 1,4-Palladium Migration and 1,2-Aryl Shift: Direct Access to Dibenzo[a,c]carbazoles through a Triple C-H Functionalization Cascade. Chem. Eur. J. 2015, 21, 16786–16791. [Google Scholar] [CrossRef]
- Leitch, J.A.; Bhonoah, Y.; Frost, C.G. Beyond C2 and C3: Transition-Metal-Catalyzed C–H Functionalization of Indole. ACS Catal. 2017, 7, 5618–5627. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, Y. Regioselective Direct Arylation of Indoles on the Benzenoid Moiety. Chem. Commun. 2018, 54, 1676–1685. [Google Scholar] [CrossRef]
- Borah, A.J.; Shi, Z. Palladium-catalyzed regioselective C–H fluoroalkylation of indoles at the C4-position. Chem. Commun. 2017, 53, 3945–3948. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, P.; Zhao, T.; Shi, Z. Regiocontrolled Direct C-H Arylation of Indoles at the C4 and C5 Positions. Angew. Chem. Int. Ed. 2017, 56, 3966–3971. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, A.M.; Shanahan, R.; Hickey, A.; Harrington, F.; Schönbauer, D.; Byrne, P.A.; Schnürch, M.; McGlacken, G.P. Synthesis of a Diaryliodonium Salt and Its Use in the Direct Arylation of Indole: A Two-Step Experiment for the Organic Teaching Laboratory. J. Chem. Educ. 2020, 97, 200–206. [Google Scholar] [CrossRef]
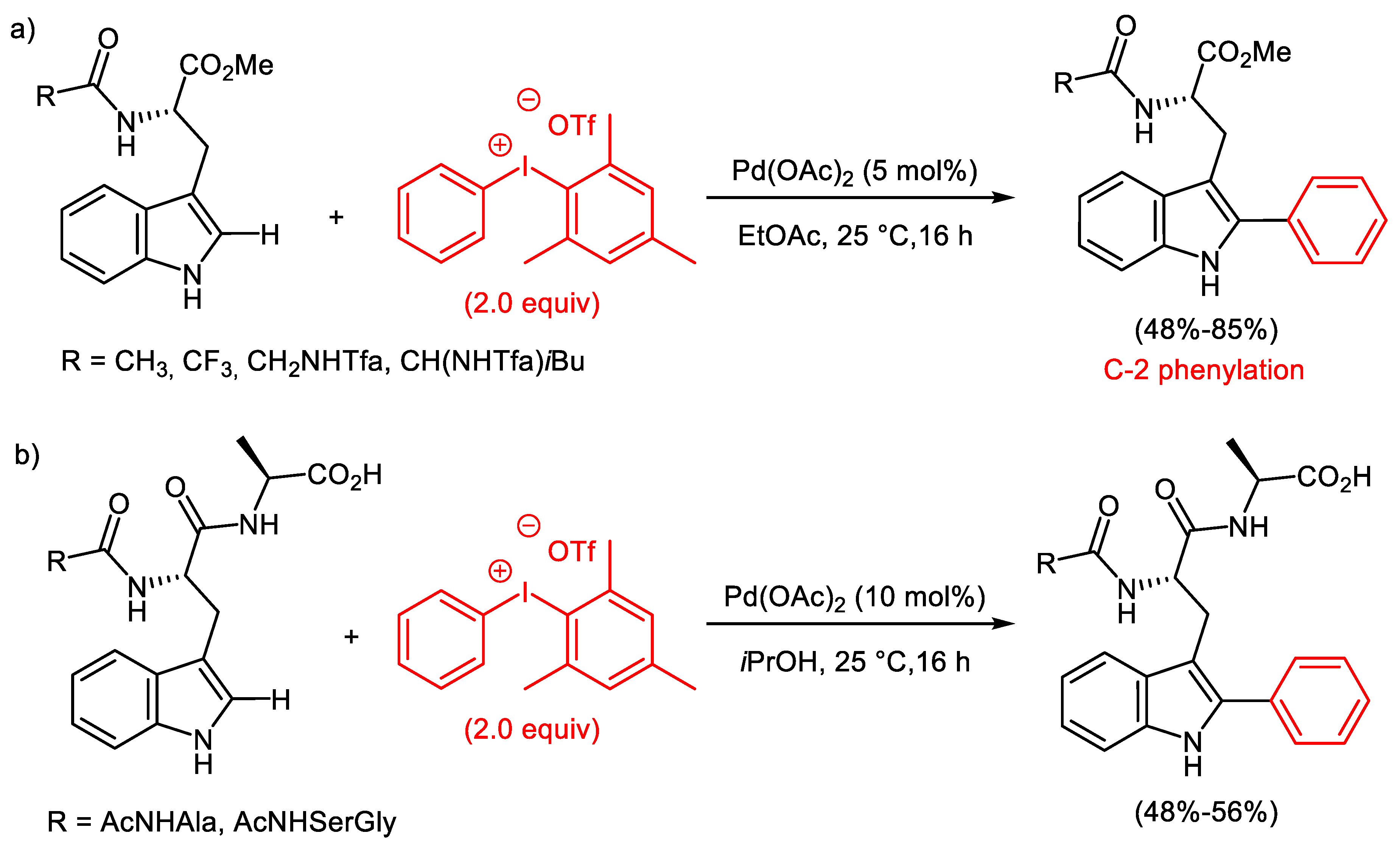
- Reay, A.J.; Williams, T.J.; Fairlamb, I.J.S. Unified mild reaction conditions for C2-selective Pd-catalysed tryptophan arylation, including tryptophan-containing peptides. Org. Biomol. Chem. 2015, 13, 8298–8309. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, J.; Pan, H.; Li, J. Additive-free Pd-catalysed C-2 arylation of tryptophan derivatives with diaryliodonium salts. J. Chem. Res. 2018, 42, 184–188. [Google Scholar] [CrossRef]
- Arun, V.; Pilania, M.; Kumar, D. Access to 2-Arylindoles via Decarboxylative C-C Coupling in Aqueous Medium and to Heteroaryl Carboxylates under Base-Free Conditions using Diaryliodonium Salts. Chem. Asian J. 2016, 11, 3345–3349. [Google Scholar] [CrossRef]
- Malmgren, J.; Nagendiran, A.; Tai, C.-W.; Bäckvall, J.-E.; Olofsson, B. C-2 Selective Arylation of Indoles with Heterogeneous Nanopalladium and Diaryliodonium Salts. Chem. Eur. J. 2014, 20, 13531–13535. [Google Scholar] [CrossRef]
- Duan, L.; Fu, R.; Zhang, B.; Shi, W.; Chen, S.; Wan, Y. An Efficient Reusable Mesoporous Solid-Based Pd Catalyst for Selective C2 Arylation of Indoles in Water. ACS Catal. 2016, 6, 1062–1074. [Google Scholar] [CrossRef]
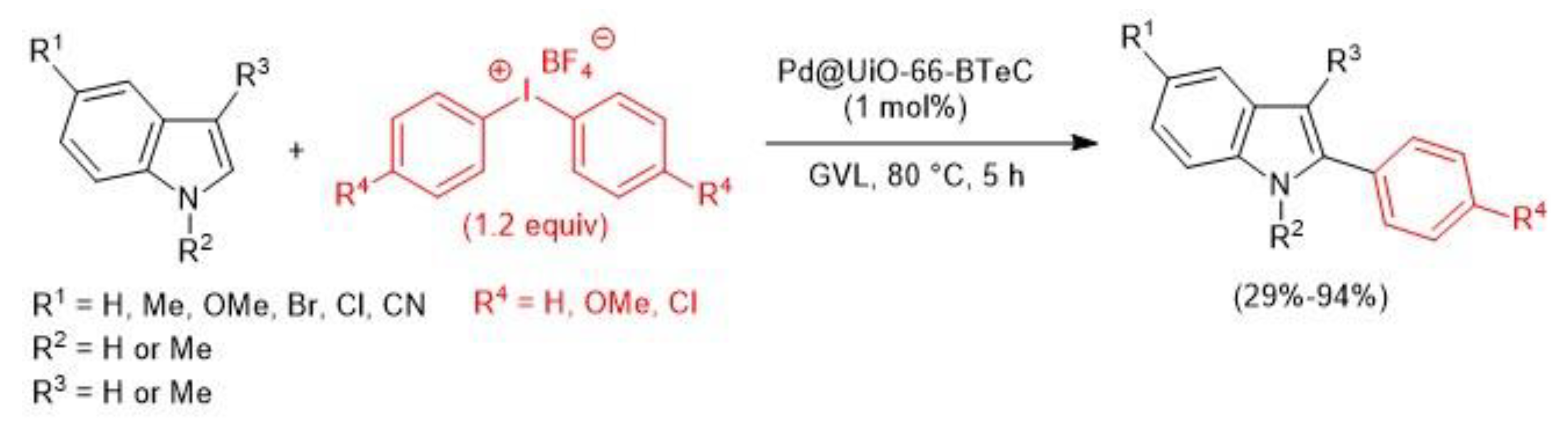
- Anastasiou, I.; Van Velthoven, N.; Tomarelli, E.; Lombi, A.; Lanari, D.; Liu, P.; Bals, S.; De Vos, D.E.; Vaccaro, L. C2–H Arylation of indoles catalyzed by Palladium containing Metal-Organic-Framework (Pd-MOF) in bioderived γ-valerolactone (GVL). ChemSusChem 2020, asap. [Google Scholar] [CrossRef]
- Tang, D.-T.D.; Collins, K.D.; Ernst, J.B.; Glorius, F. Pd/C as a Catalyst for Completely Regioselective C-H Functionalization of Thiophenes under Mild Conditions. Angew. Chem. Int. Ed. 2014, 53, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
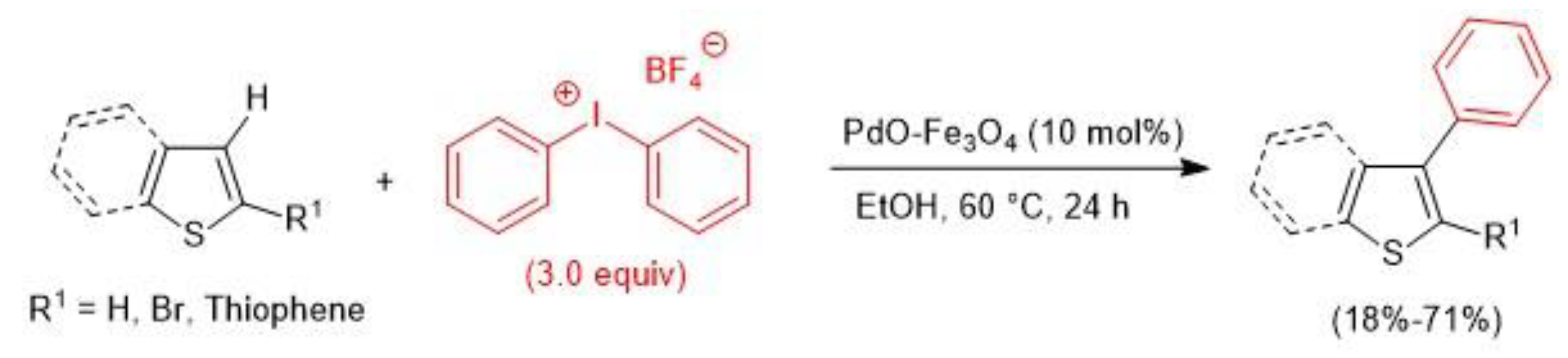
- Cano, R.; Perez, J.M.; Ramon, D.J.; McGlacken, G.P. Impregnated palladium on magnetite as catalyst for direct arylation of heterocycles. Tetrahedron 2016, 72, 1043–1050. [Google Scholar] [CrossRef]
- Yang, Q.; Chang, J.; Wu, Q.; Zhang, B. A simple phenylation of heteroaromatic compounds using diphenyliodonium triflate. Res. Chem. Intermed. 2012, 38, 1335–1340. [Google Scholar] [CrossRef]
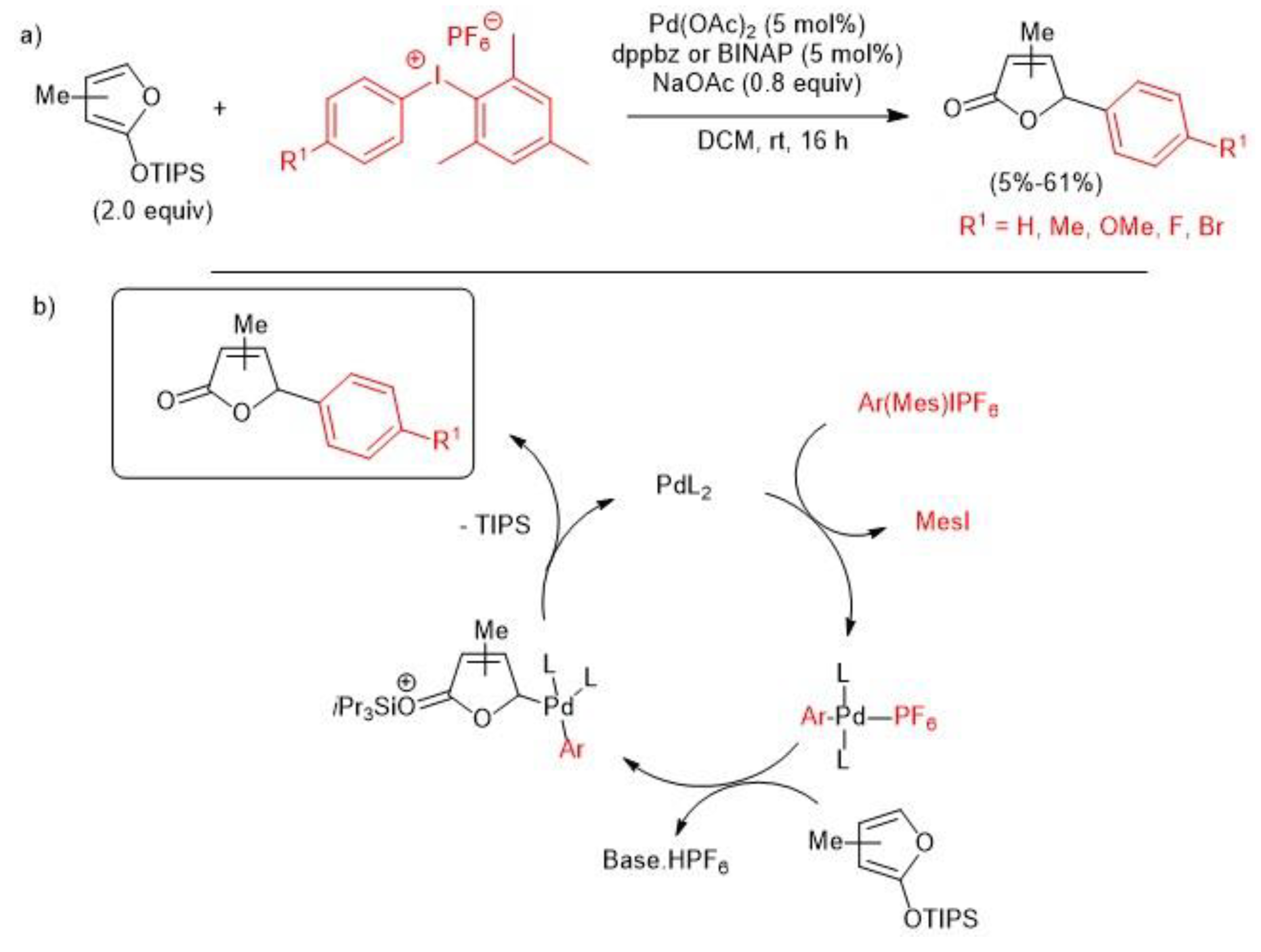
- Alexander, T.S.; Clay, T.J.; Maldonado, B.; Nguyen, J.M.; Martin, D.B.C. Comparative studies of palladium and copper-catalysed γ-arylation of silyloxy furans with diaryliodonium salts. Tetrahedron 2019, 75, 2229–2238. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Y.; Han, J.; Wang, L. Palladium Catalyzed C–I and Vicinal C–H Dual Activation of Diaryliodonium Salts for Diarylation: Synthesis of 4, 5-Benzocoumarins. Org. Lett. 2015, 17, 5654–5657. [Google Scholar] [CrossRef]
- Lee, S.; Mah, S.; Hong, S. Catalyst Controlled Divergent C4/C8 Site-Selective C−H Arylation of Isoquinolones. Org. Lett. 2015, 17, 3864–3867. [Google Scholar] [CrossRef]
- Garad, D.N.; Viveki, A.B.; Mhaske, S.B. Pd-Catalyzed Regioselective Mono-Arylation: Quinazolinone as the Inherent Directing Group for C(sp2)−H Activation. J. Org. Chem. 2017, 82, 6366–6372. [Google Scholar] [CrossRef]
- Mehra, M.K.; Sharma, S.; Rangan, K.; Kumar, D. Substrate or Solvent-Controlled PdII-Catalyzed Regioselective Arylation of Quinolin-4(1H)-ones Using Diaryliodonium Salts: Facile Access to Benzoxocine and Aaptamine Analogues. Eur. J. Org. Chem. 2020, asap. [Google Scholar] [CrossRef]
- Dey, A.; Sinha, S.K.; Achar, T.K.; Maiti, D. Accessing Remote meta- and para-C(sp2)-H Bonds with Covalently Attached Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 10820–10843. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schonbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, S.; Sharma, A. Palladium-Catalyzed Regioselective C−H Arylation of Quinoline N-Oxides at C-8 Position using Diaryliodonium Salts. Asian J. Org. Chem. 2020, 9, 660–667. [Google Scholar] [CrossRef]
- Yang, P.; Wang, R.; Wu, H.; Du, Z.; Fu, Y. Pd-Catalyzed C-H Arylation of Benzothiazoles with Diaryliodonium Salt: One-Pot Synthesis of 2-Arylbenzothiazoles. Asian J. Org. Chem. 2017, 6, 184–188. [Google Scholar] [CrossRef]
- Srivastava, S.; Thakur, N.; Singh, A.; Shukla, P.; Maikhuri, V.K.; Garg, N.; Prasad, A.; Pandey, R. Development of a fused imidazo[1–α]pyridine based fluorescent probe for Fe3+ and Hg2+ in aqueous media and HeLa cells. RSC Adv. 2019, 9, 29856–29863. [Google Scholar] [CrossRef]
- Banerji, B.; Chatterjee, S.; Chandrasekhar, K.; Bera, S.; Majumder, L.; Prodhan, C.; Chaudhuri, K. Expedient synthesis of a phenanthro-imidazo-pyridine fused heteropolynuclear framework via CDC coupling: a new class of luminophores. Org. Biomol. Chem. 2017, 15, 4130–4134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bagdi, A.K.; Hajra, A. Design, Synthesis, and Functionalization of Imidazoheterocycles. Chem. Rec. 2016, 16, 1868–1885. [Google Scholar] [CrossRef]
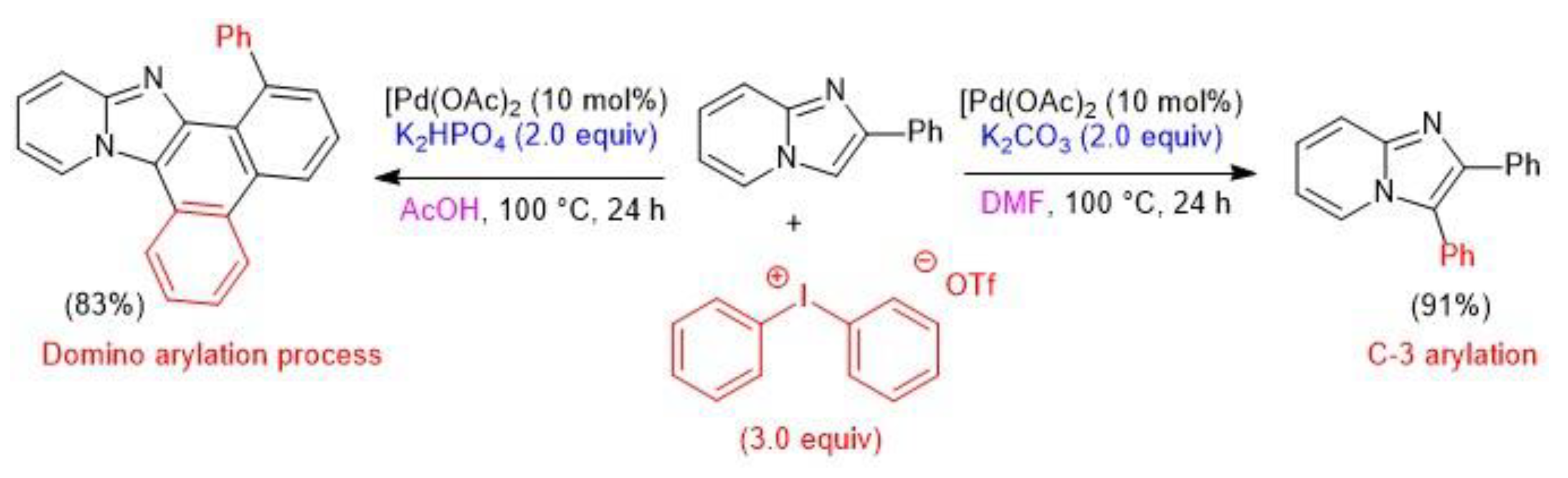
- Xue, C.; Han, J.; Zhao, M.; Wang, L. Rapid Construction of Fused Heteropolycyclic Aromatics via Palladium-Catalyzed Domino Arylations of Imidazopyridine Derivatives. Org. Lett. 2019, 21, 4402–4406. [Google Scholar] [CrossRef]
- Zhu, S.; MacMillan, D.W.C. Enantioselective Copper-Catalyzed Construction of Aryl Pyrroloindolines via an Arylation−Cyclization Cascade. J. Am. Chem. Soc. 2012, 134, 10815–10818. [Google Scholar] [CrossRef]
- Kieffer, M.E.; Chuang, K.V.; Reisman, S.E. A copper-catalyzed arylation of tryptamines for the direct synthesis of aryl pyrroloindolines. Chem. Sci. 2012, 3, 3170–3174. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, W.; Dai, L.-X.; You, S.-L. Copper-Catalyzed C-2 Arylation or Vinylation of Indole Derivatives with Iodonium Salts. Chem. Asian J. 2014, 9, 2113–2118. [Google Scholar] [CrossRef]
- Yang, Y.; Li, R.; Zhao, Y.; Zhao, D.; Shi, Z. Cu-Catalyzed Direct C6-Arylation of Indoles. J. Am. Chem. Soc. 2016, 138, 8734–8737. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhou, L.; Yang, Y.; Zhang, X.; Shi, Z.; Wu, Y.-D. Directing Effects on the Copper-Catalyzed Site-Selective Arylation of Indoles. Org. Lett. 2018, 20, 6502–6505. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Pilania, M.; Arun, V.; Pooniya, S. C–H arylation of azaheterocycles: a direct ligand-free and Cu-catalyzed approach using diaryliodonium salts. Org. Biomol. Chem. 2014, 12, 6340–6344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, Q. Arylation, Vinylation, and Alkynylation of Electron-Deficient (Hetero)arenes Using Iodonium Salts. Org. Lett. 2016, 18, 5118–5121. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Liu, L.; Shi, Z.; Yuan, Y. Iridium(III)-catalyzed regioselective direct arylation of sp2 C–H bonds with diaryliodonium salts. Org. Biomol. Chem. 2016, 14, 7109–7113. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium-Catalyzed C-H Arylation with Diaryliodonium Salts. Adv. Synth. Catal. 2012, 354, 3517–3522. [Google Scholar] [CrossRef]
- Tobisu, M.; Furukawa, T.; Chatani, N. Visible Light-mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42, 1203–1205. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Xue, D.; Wang, J.-D.; Zhao, C.-J.; Zou, Q.-Z.; Wang, C.; Xiao, J. Room-Temperature Arylation of Arenes and Heteroarenes with Diaryl iodonium Salts by Photoredox Catalysis. Synlett 2013, 24, 507–513. [Google Scholar] [CrossRef]
- Najib, A.; Tabuchi, S.; Hirano, K.; Miura, M. Highly C3-Selective Direct Alkylation and Arylation of 2-Pyridinones under Visible-Light-Promoted Photoredox Catalysis. Heterocycles 2016, 92, 1187–1203. [Google Scholar] [CrossRef]
- Li, D.; Liang, C.; Jiang, Z.; Zhang, J.; Zhuo, W.-T.; Zou, F.-Y.; Wang, W.-P.; Gao, G.-L.; Song, J. Visible-Light-Promoted C2 Selective Arylation of Quinoline and Pyridine N-Oxides with Diaryliodonium Tetrafluoroborate. J. Org. Chem. 2020, 85, 2733–2742. [Google Scholar] [CrossRef]
- Hari, D.P.; Hering, T.; Koenig, B. Chapter 8: Arene functionalization by visible light photoredox catalysis. In Visible Light Photocatalysis in Organic Chemistry; Yoon, T.P., Stephenson, C.R.J., MacMillan, D.W.C., Eds.; Whiley-VCH Verlag GmbH & Co. KGaA: Wheinheim, Germany, 2018; pp. 253–281. [Google Scholar] [CrossRef]
- Dixneuf, P.H.; Soulé, J.F. Functionalization of C(sp2)–H bonds of arenes and heteroarenes assisted by photoredox catalysts for the C–C Bond Formation. In Organometallics for Green Catalysis. Topics in Organometallic Chemistry; Dixneuf, P., Soulé, J.F., Eds.; Springer: Cham, Switzerland, 2018; Volume 63, pp. 225–265. [Google Scholar] [CrossRef]
- Rae, J.; Frey, J.; Jerhaoui, S.; Choppin, S.; Wencel-Delord, J.; Colobert, F. Synthesis of Axially Chiral C−N Scaffolds via Asymmetric Coupling with Enantiopure Sulfinyl Iodanes. ACS Catal. 2018, 8, 2805–2809. [Google Scholar] [CrossRef]
- Neha, D.; Ashish, R.; Kumar, R.; Kumar, V. Recent Synthetic Strategies for Monocyclic Azole Nucleus and Its Role in Drug Discovery and Development. Curr. Org. Synth. 2018, 15, 321–340. [Google Scholar] [CrossRef]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-K.; Lee, S.-H.; Lee, D. Copper-Catalyzed N-Arylation of Amines with Hypervalent Iodonium Salts. Synlett 2000, 7, 1022–1024. [Google Scholar] [CrossRef]
- Berzina, B.; Sokolovs, I.; Suna, E. Copper-Catalyzed para-Selective C−H Amination of Electron-Rich Arenes. ACS Catal. 2015, 5, 7008–7014. [Google Scholar] [CrossRef]
- Lv, T.; Wang, Z.; You, J.; Lan, J.; Gao, G. Copper-Catalyzed Direct Aryl Quaternization of N-Substituted Imidazoles to Form Imidazolium Salts. J. Org. Chem. 2013, 78, 5723–5730. [Google Scholar] [CrossRef]
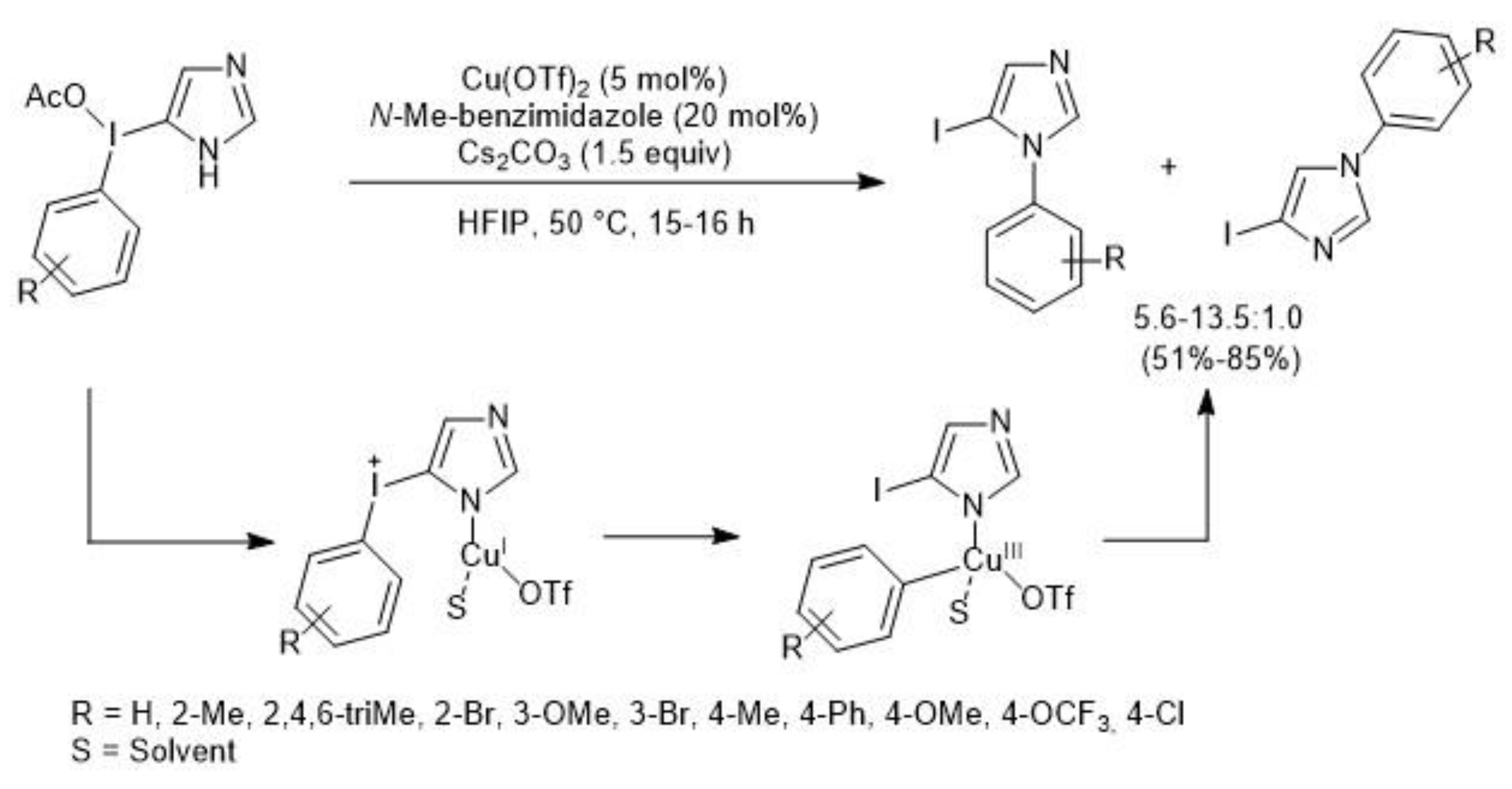
- Wu, Y.; Izquierdo, S.; Vidossich, P.; Lledýs, A.; Shafir, A. NH-Heterocyclic Aryliodonium Salts and their Selective Conversion into N1-Aryl-5-iodoimidazoles. Angew. Chem. Int. Ed. 2016, 55, 7152–7156. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Kapur, M. Catalyst-controlled positional-selectivity in C–H functionalizations. Org. Biomol. Chem. 2019, 17, 1007–1026. [Google Scholar] [CrossRef]
- Hartwig, J.F. Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res. 2017, 50, 549–555. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Davydov, D.V.; Moreno-Mafias, M. Pd- and Cu-catalyzed selective Arylation of Benzotriazole by Diaryliodonium salts in Water. Tetrahedron Lett. 1998, 39, 5621–5622. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Davydov, D.V.; Gorovo, M.S. Palladium- and copper-catalyzed selective arylation of 5-aryltetrazoles by diaryliodonium salts. Tetrahedron Lett. 2002, 43, 6221–6223. [Google Scholar] [CrossRef]
- Davydov, D.V.; Oprunenko, Y.F.; Beletskaya, I.P. Pd/Al2O3-catalysed regioselective N-1-modification of benzotriazoles using iodonium salts. Tetrahedron Lett. 2017, 58, 4465–4467. [Google Scholar] [CrossRef]
- Davydov, D.V.; Chernyshev, V.V.; Rybakov, V.B.; Oprunenko, Y.F.; Beletskaya, I.P. Regioselective N1- or N2-modification of benzotriazoles with iodonium salts in the presence of copper compounds. Mendeleev Commun. 2018, 28, 287–289. [Google Scholar] [CrossRef]
- Pankajakshan, S.; Chng, Z.G.; Gangulya, R.; Loh, T.P. Copper-catalyzed aerobic carboxygenation and N-arylation of [1,2,3]triazolo[1,5-a]pyridines towards pyridinium triazolinone ylides. Chem. Commun. 2015, 51, 5929–5931. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Jia, X.; Du, Z.; Fu, Y. Cu-catalyzed arylation of 1-acyl-1H-1,2,3-Benzotriazoles via C-N activation. J. Organomet. Chem. 2019, 895, 64–67. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Z.; Peng, Q.; Zhou, Y.; Xu, L.; Pan, X. Access to 2-substituted-2H indazoles via a copper-catalyzed regioselective cross-coupling reaction. Org. Biomol. Chem. 2018, 16, 1816–1822. [Google Scholar] [CrossRef]
- Koseki, D.; Aoto, E.; Shoji, T.; Watanabe, K.; In, Y.; Kita, Y.; Dohi, T. Efficient N-arylation of azole compounds utilizing selective aryl-transfer TMP-iodonium(III) reagents. Tetrahedron Lett. 2019, 60, 1281–1286. [Google Scholar] [CrossRef]
- Zeng, Q.; Ma, C.; Wu, X.; Zhou, L.; Huang, Y. Selective C–N coupling reaction of diaryliodonium salts and dinucleophiles. New J. Chem. 2017, 41, 2873–2877. [Google Scholar] [CrossRef]
- Jiang, J.; Li, J. Mechanically Induced N-arylation of Amines with Diaryliodonium Salts. Chemistry Select 2020, 5, 542–548. [Google Scholar] [CrossRef]
- Sheng, J.; He, R.; Xue, J.; Wu, C.; Qiao, J.; Chen, C. Cu-Catalyzed π-Core Evolution of Benzoxadiazoles with Diaryliodonium Salts for Regioselective Synthesis of Phenazine Scaffolds. Org. Lett. 2018, 20, 4458–4461. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Bonasera, A.; Hristov, L.; Garmshausen, Y.; Schmidt, B.M.; Jacquemin, D.; Hecht, S. N,N′-Disubstituted Indigos as Readily Available Red-Light Photoswitches with Tunable Thermal Half-Lives. J. Am. Chem. Soc. 2017, 139, 15205–15211. [Google Scholar] [CrossRef]
- Zhou, T.; Li, T.-C.; Chen, Z.-C. Hypervalent Iodine in Synthesis. Part 86. Selective Copper-catalyzed N-Monoarylation and N1,N3-Diarylation of Uracil and Its Derivatives with Diaryliodonium Salts. Helv. Chim. Acta 2005, 88, 290–296. [Google Scholar] [CrossRef]
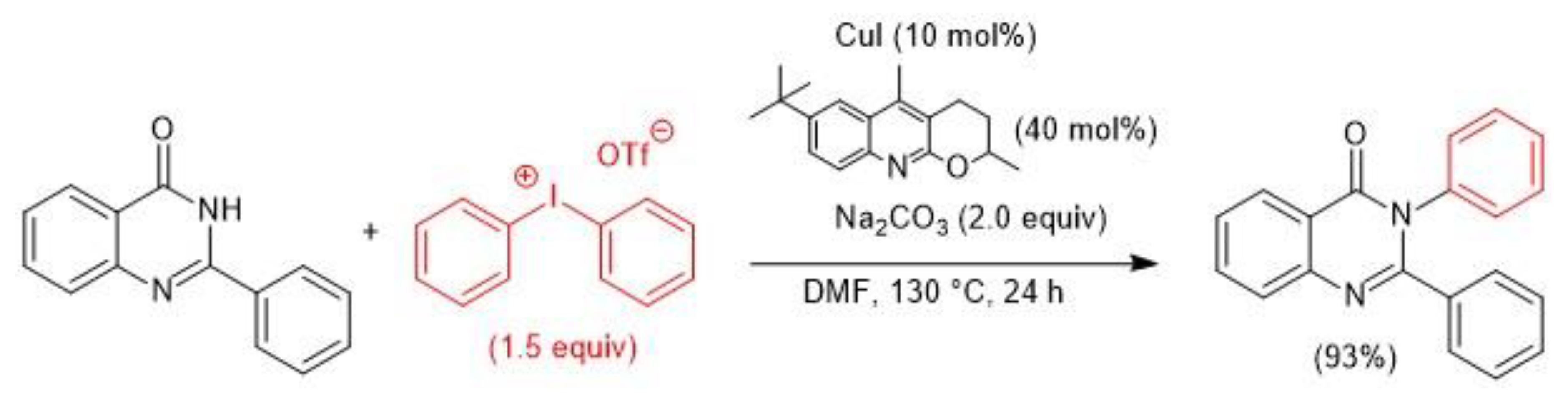
- Lee, J.B.; Kang, M.E.; Kim, J.; Lee, C.Y.; Kee, J.-M.; Myung, K.; Park, J.-U.; Hong, S.Y. Direct diversification of unmasked quinazolin-4(3H)-ones through orthogonal reactivity modulation. Chem. Commun. 2017, 53, 10394–10397. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Sung, D.-B.; Park, C.-H.; Kim, W.-S. Copper-Catalyzed N-Arylation of 2-Pyridones Employing Diaryliodonium Salts at Room Temperature. J. Org. Chem. 2016, 81, 7717–7724. [Google Scholar] [CrossRef] [PubMed]
- Reus, C.; Stolar, M.; Vanderkley, J.; Nebauer, J.; Baumgartner, T. A Convenient N Arylation Route for Electron-Deficient Pyridines: The Case of π Extended Electrochromic Phosphaviologens. J. Am. Chem. Soc. 2015, 137, 11710–11717. [Google Scholar] [CrossRef] [PubMed]
- Grelier, G.; Darses, B.; Dauban, P. Hypervalent organoiodine compounds: from reagents to valuable building blocks in synthesis. Beilstein J. Org. Chem. 2018, 14, 1508–1528. [Google Scholar] [CrossRef]
- Hu, T.; Kai, X.; Ye, Z.; Zhu, K.; Wu, Y.; Zhang, F. Two-in-One Strategy for the Pd(II)-Catalyzed Tandem C–H Arylation/Decarboxylative Annulation Involved with Cyclic Diaryliodonium Salts. Org. Lett. 2019, 21, 7233–7237. [Google Scholar] [CrossRef]
- Hu, T.; Ye, Z.; Zhu, K.; Xu, K.; Wu, T.; Zhang, F. Synthesis of Tribenzo[b,d,f]azepines via Cascade π-Extended Decarboxylative Annulation Involving Cyclic Diaryliodonium Salts. Org. Lett. 2020, 22, 505–509. [Google Scholar] [CrossRef]
- Modha, S.G.; Popescu, M.V.; Greaney, M.F. Synthesis of Triarylamines via Sequential C−N Bond Formation. J. Org. Chem. 2017, 82, 11933–11938. [Google Scholar] [CrossRef]
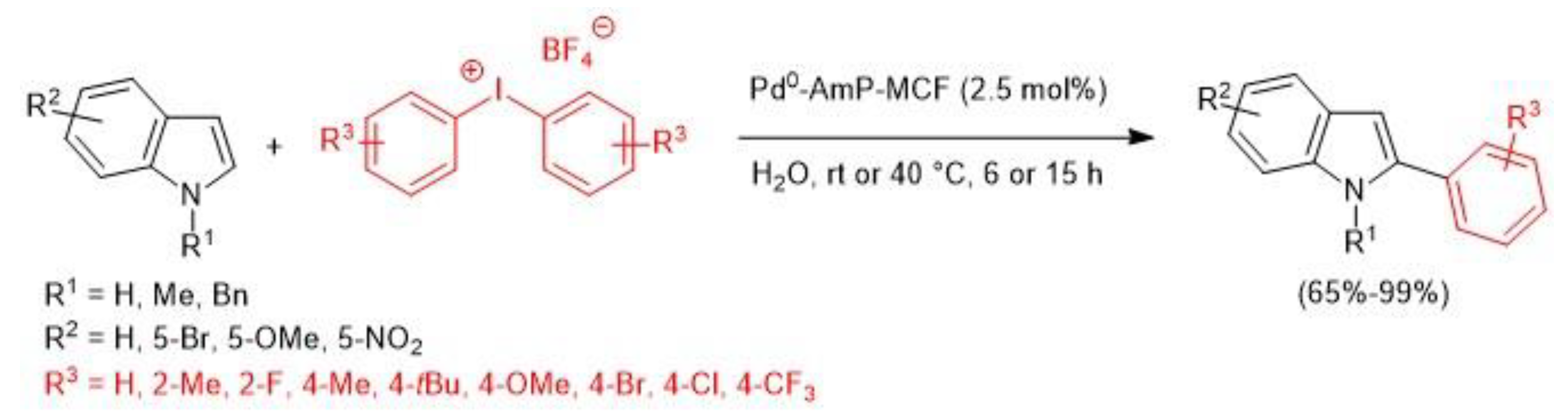
- Modha, S.G.; Greaney, M.F. Atom-Economical Transformation of Diaryliodonium Salts: Tandem C−H and N−H Arylation of Indoles. J. Am. Chem. Soc. 2015, 137, 1416–1419. [Google Scholar] [CrossRef]
- Teskey, C.J.; Sohel, S.M.A.; Bunting, D.L.; Modha, S.G.; Greaney, M.F. Domino N-/C-Arylation via In Situ Generation of a Directing Group: Atom-Efficient Arylation Using Diaryliodonium Salts. Angew. Chem., Int. Ed. 2017, 56, 5263–5266. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Li, S.; Lv, H.; Yu, Y.; Ye, X.; Li, B.; Yang, S.; Mo, Y.; Kong, X. Domino N-/C- or N-/N-/C-arylation of imidazoles to yield polyaryl imidazolium salts via atom-economical use of diaryliodonium salts. Chem. Commun. 2019, 55, 11267–11270. [Google Scholar] [CrossRef] [PubMed]









































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco-Benichou, A.; Besson, T.; Fruit, C. Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes. Catalysts 2020, 10, 483. https://doi.org/10.3390/catal10050483
Pacheco-Benichou A, Besson T, Fruit C. Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes. Catalysts. 2020; 10(5):483. https://doi.org/10.3390/catal10050483
Chicago/Turabian StylePacheco-Benichou, Alexandra, Thierry Besson, and Corinne Fruit. 2020. "Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes" Catalysts 10, no. 5: 483. https://doi.org/10.3390/catal10050483
APA StylePacheco-Benichou, A., Besson, T., & Fruit, C. (2020). Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes. Catalysts, 10(5), 483. https://doi.org/10.3390/catal10050483