Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid

 and
and
Abstract
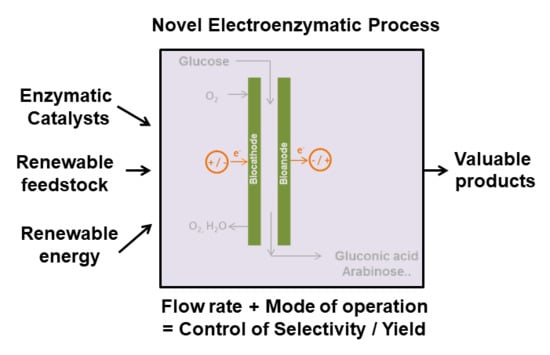
1. Introduction
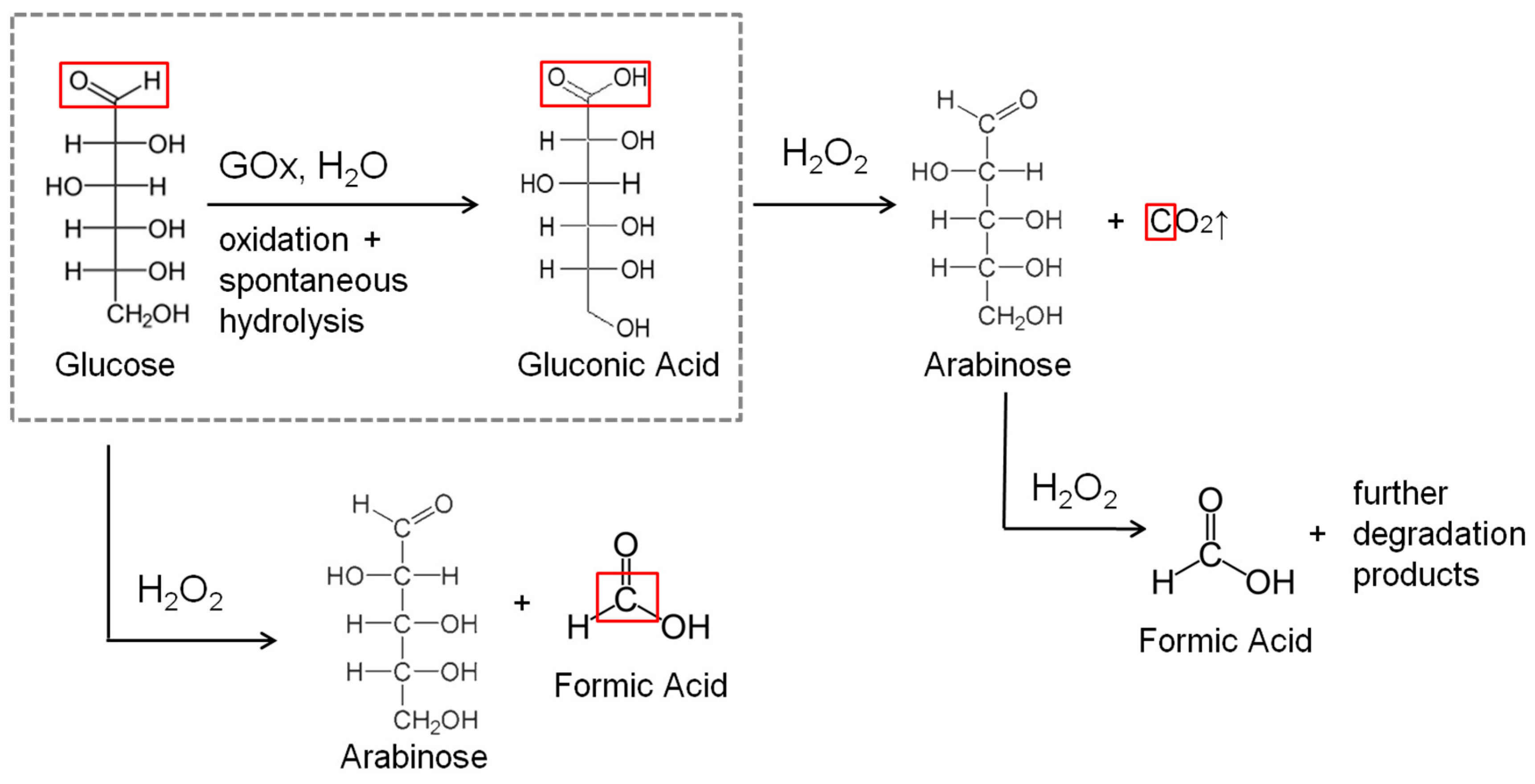
2. Results and Discussion
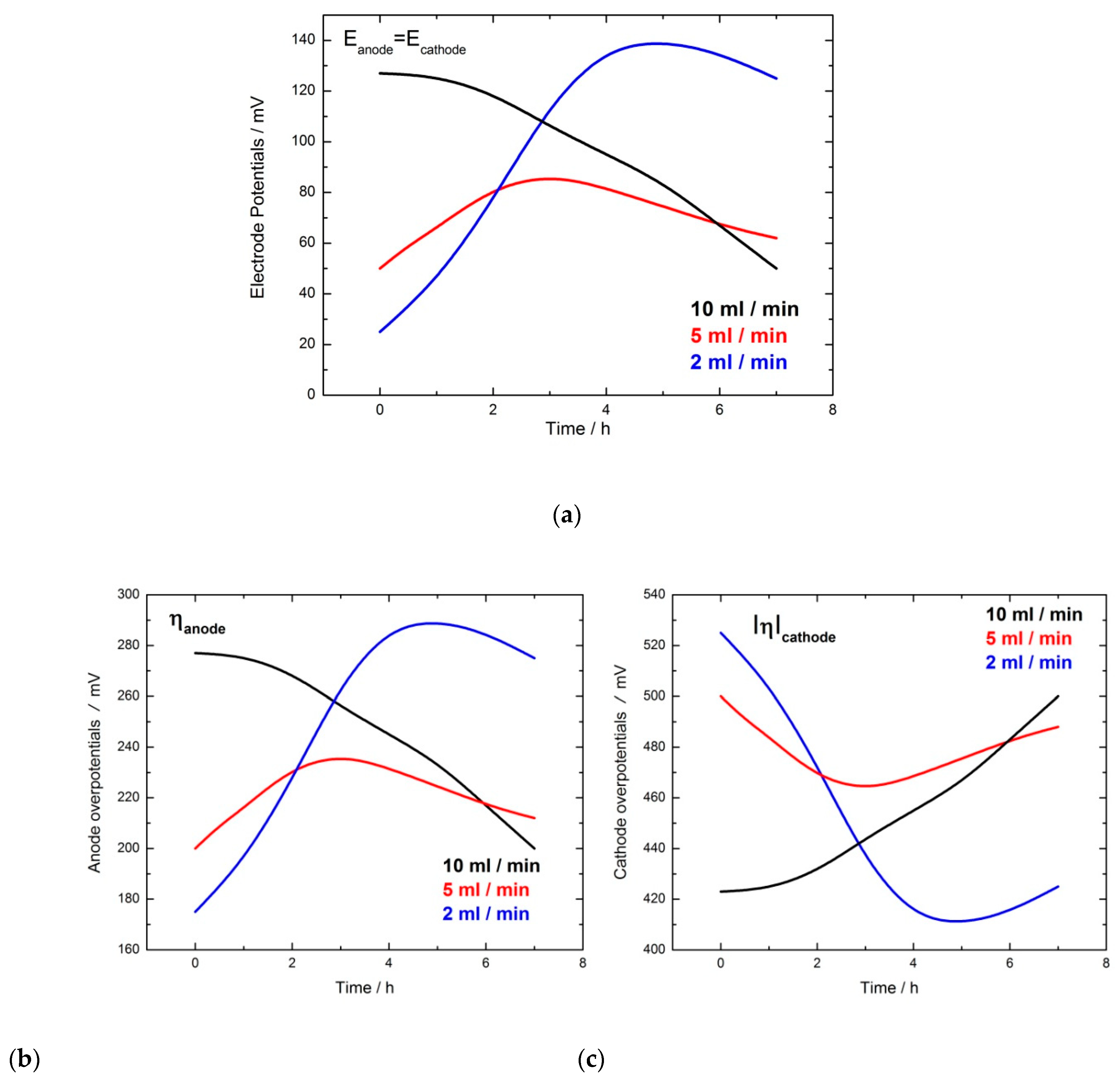
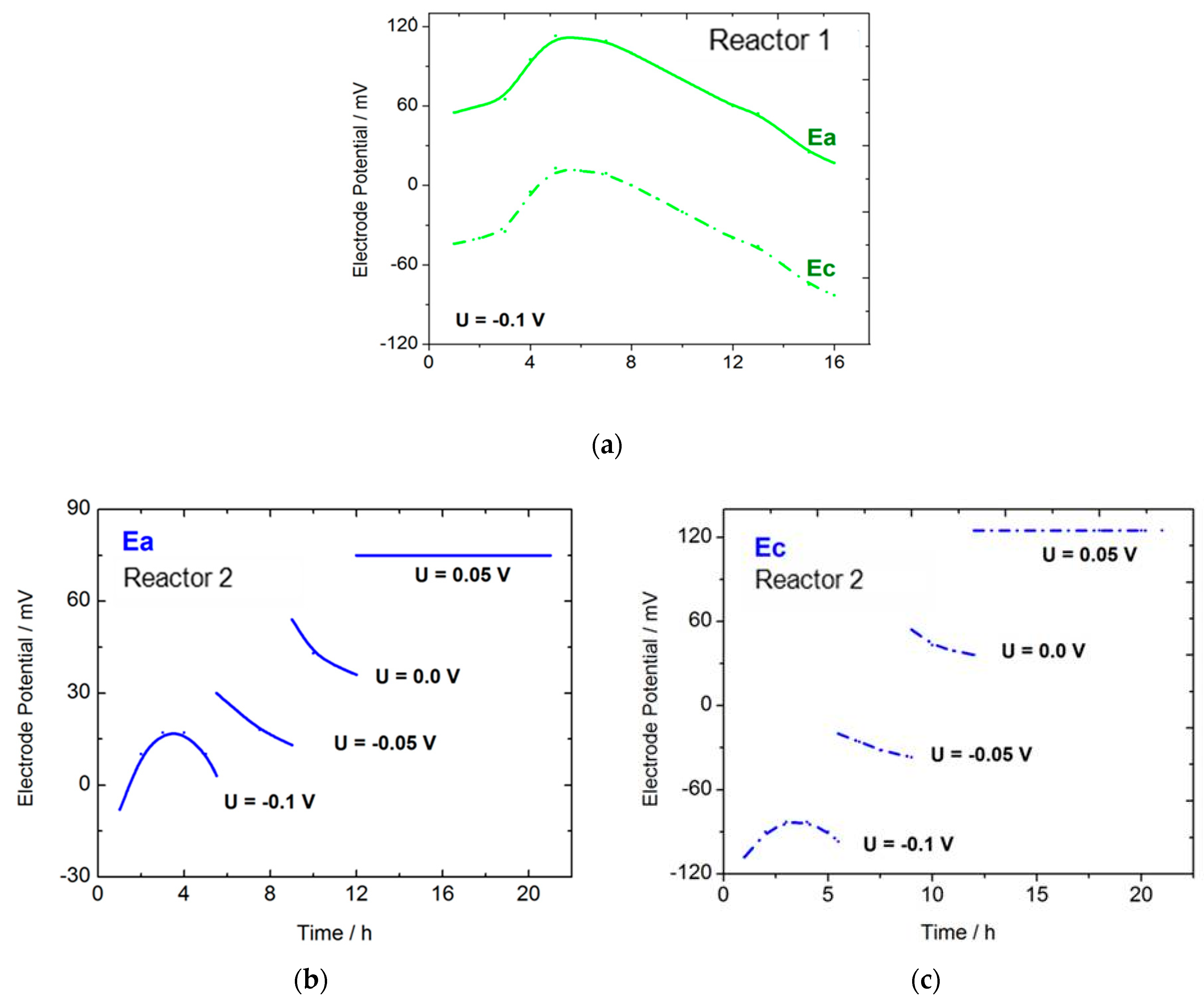
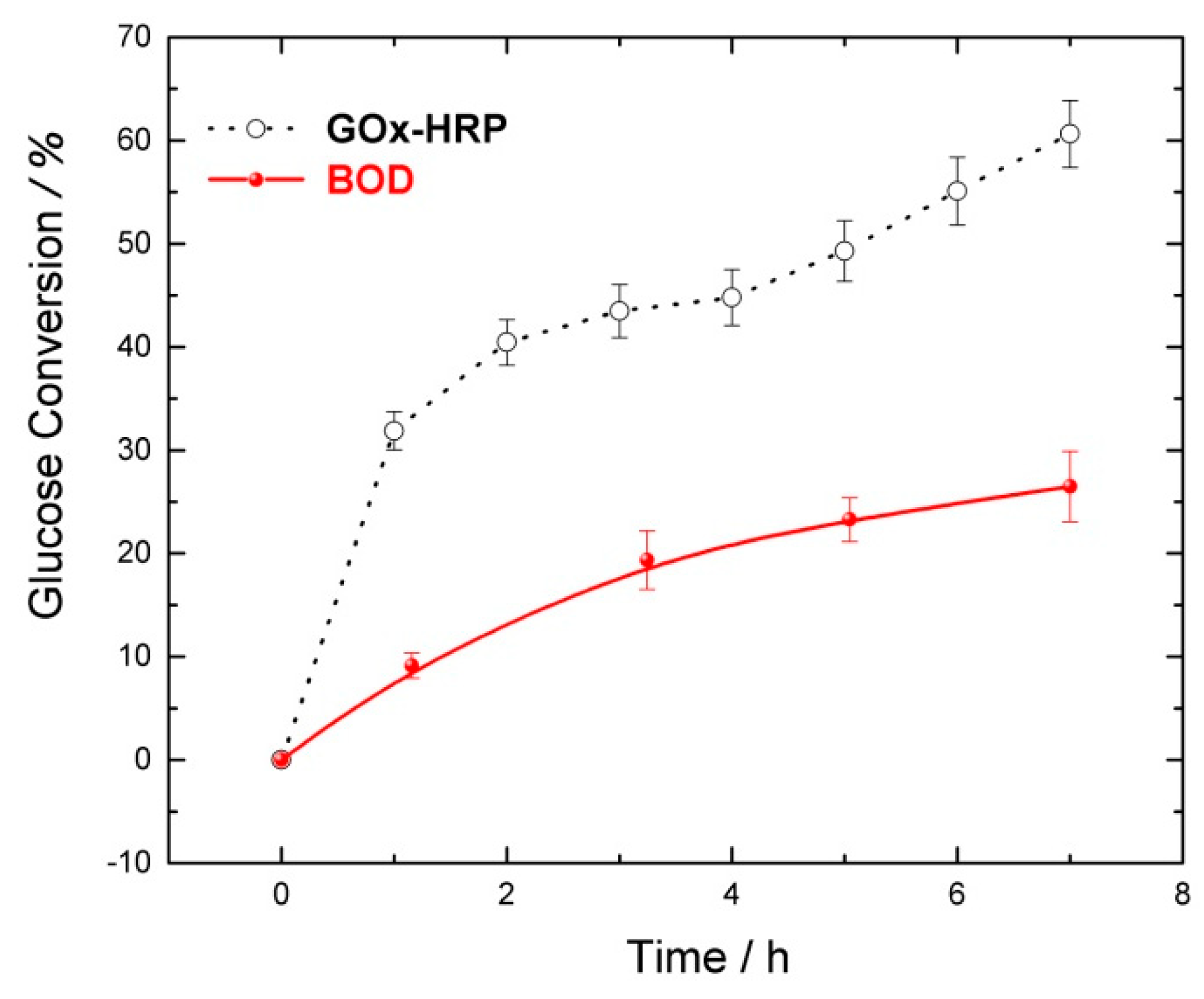
2.1. GOx/GOx-HRP Reactor
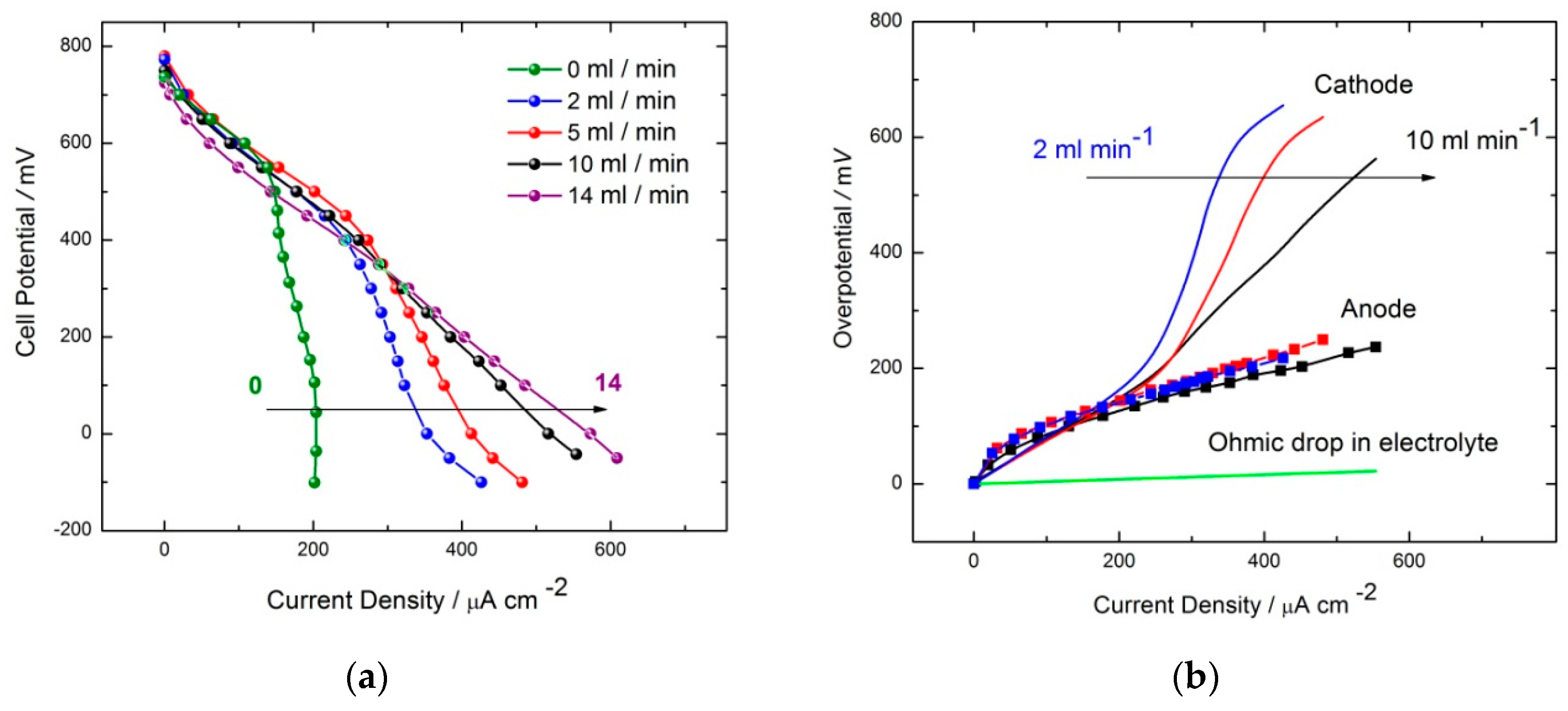
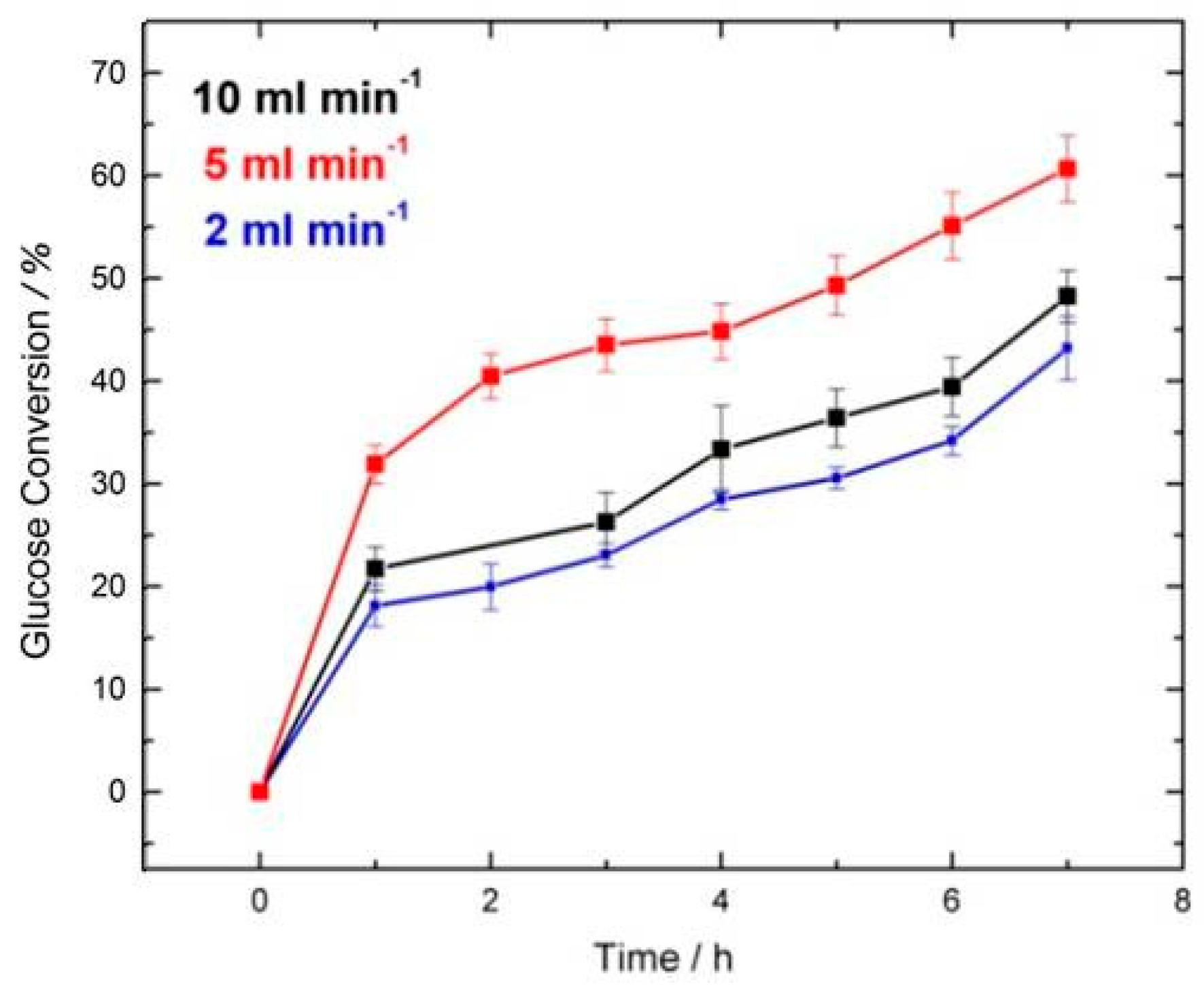
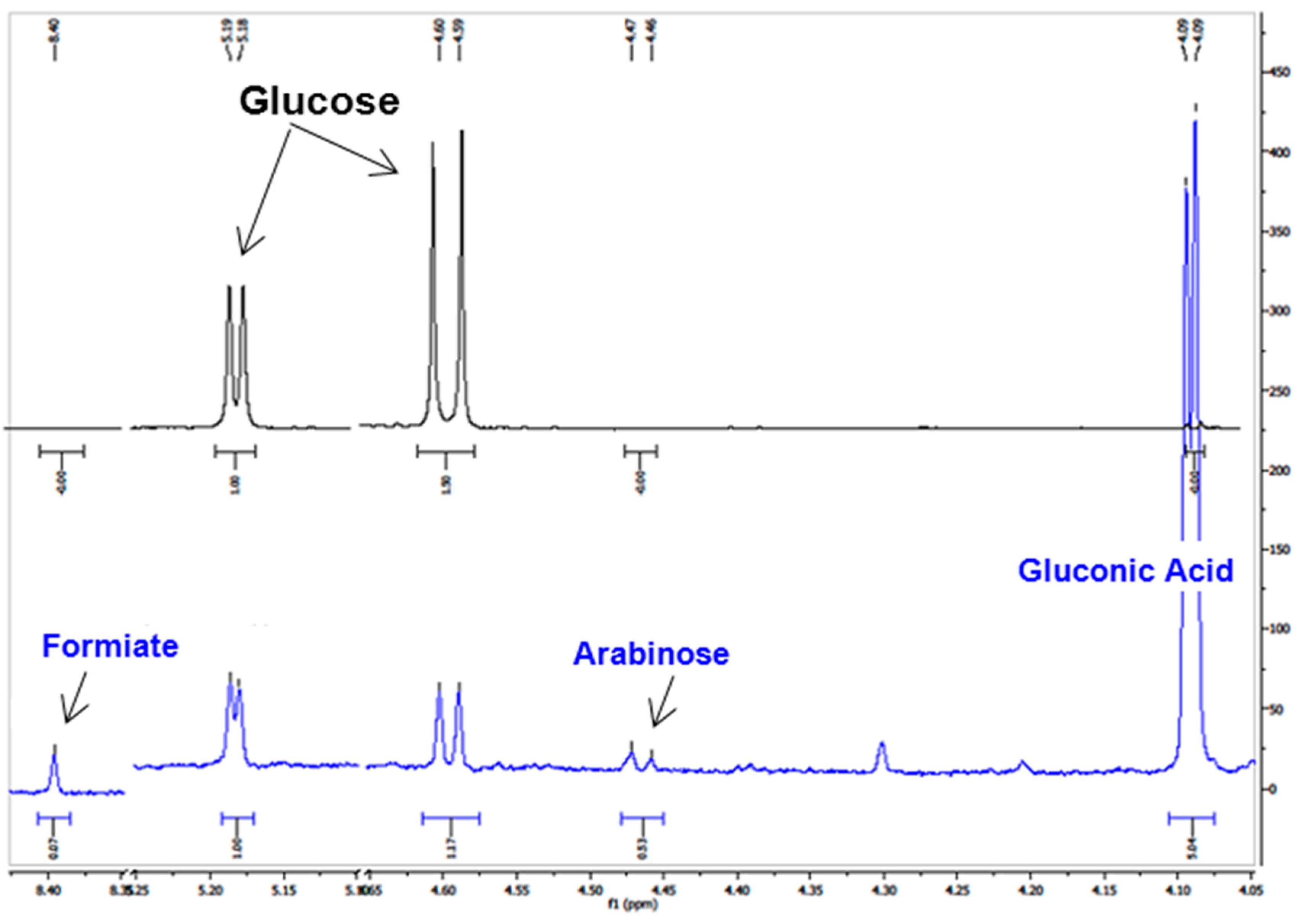
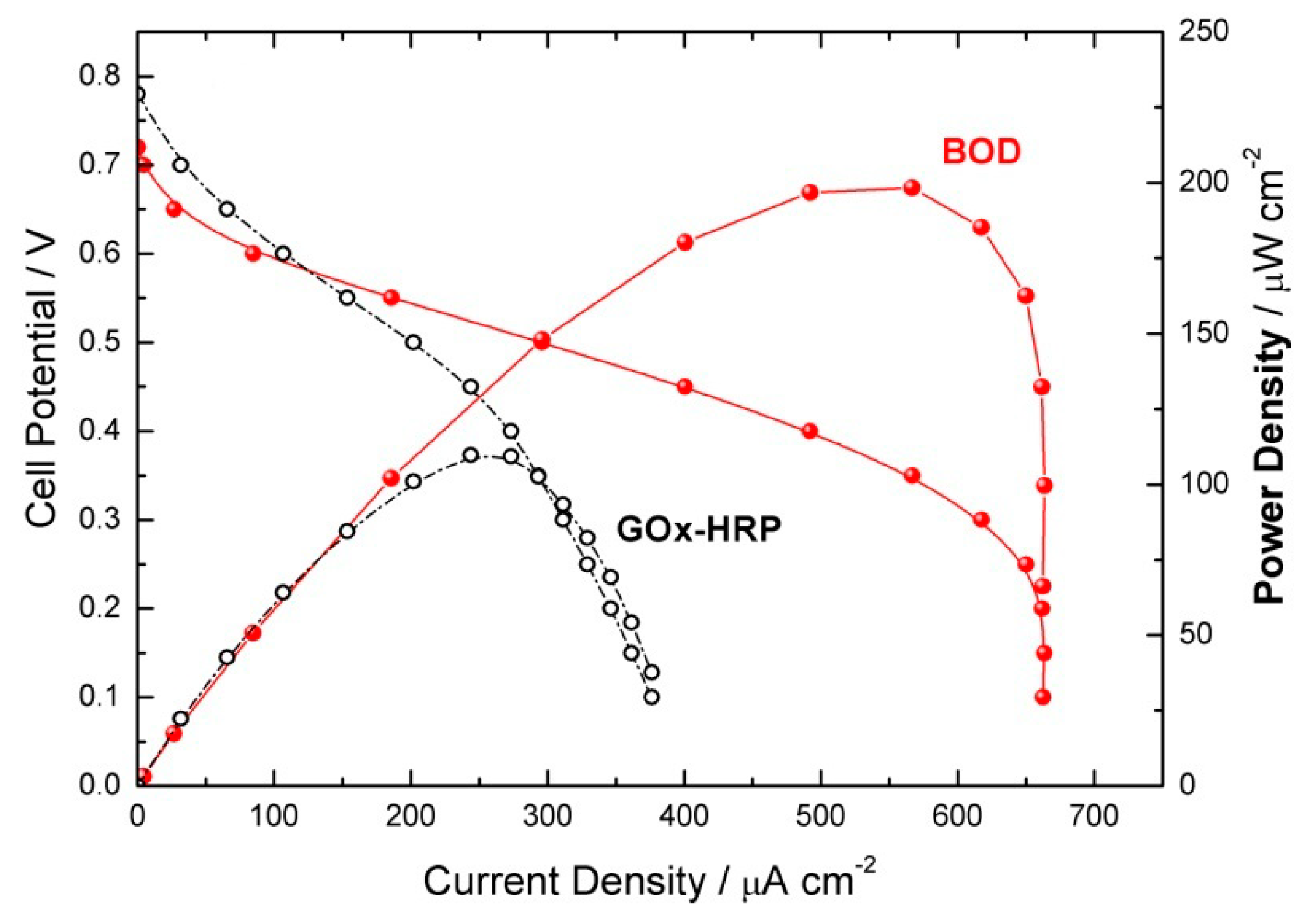
2.2. GOx/BOD Reactor
2.3. Process Sustainability
3. Materials and Methods
3.1. Chemicals and Instrumentation
3.2. Enzymatic Electrode Preparations
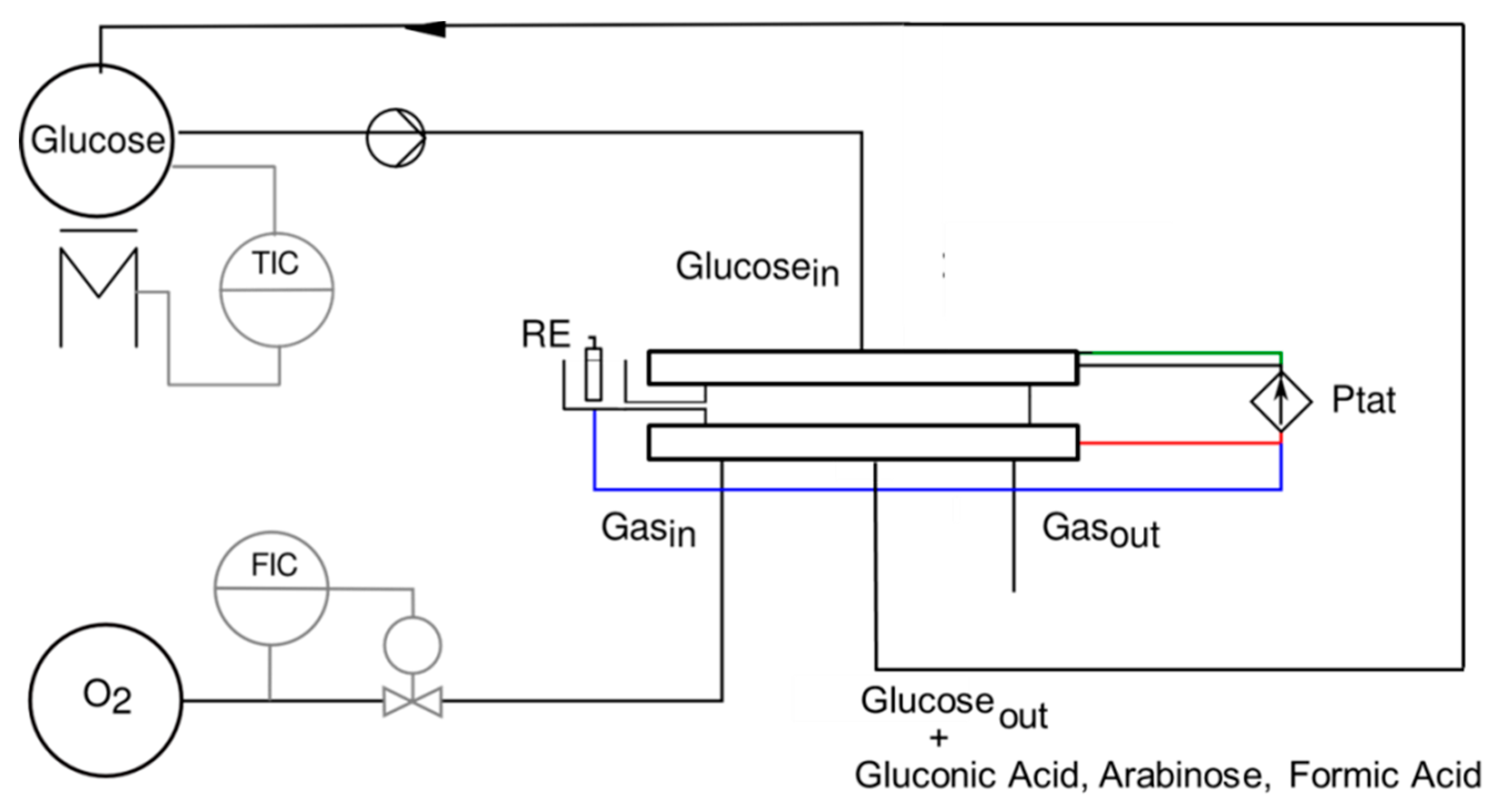
3.3. Electroenzymatic Reactor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anastassiadis, S.; Morgunov, I.G. Gluconic acid production. Recent Pat. Biotechnol. 2007, 1, 167–180. [Google Scholar] [CrossRef]
- Ramachandran, S.; Fontanille, P.; Pandey, A.; Larroche, C. Gluconic acid: Properties, applications and microbial production. Food Technol. Biotechnol. 2006, 44, 185–195. [Google Scholar]
- Tong, X.L.; Ma, Y.; Li, Y.D. Biomass into chemicals: Conversion of sugars to furan derivatives by catalytic processes. Appl. Catal. A-Gen. 2010, 385, 1–13. [Google Scholar] [CrossRef]
- Murzin, D.Y.; Leino, R. Sustainable chemical technology through catalytic multistep reactions. Chem. Eng. Res. Des. 2008, 86, 1002–1010. [Google Scholar] [CrossRef]
- Bin, D.; Wang, H.; Li, J.; Wang, H.; Yin, Z.; Kang, J.; He, B.; Li, Z. Controllable oxidation of glucose to gluconic acid and glucaric acid using an electrocatalytic reactor. Electrochim. Acta 2014, 130, 170–178. [Google Scholar] [CrossRef]
- Biella, S.; Prati, L.; Rossi, M. Selective oxidation of D-glucose on gold catalyst. J. Catal. 2002, 206, 242–247. [Google Scholar] [CrossRef]
- Zhang, H.; Watanabe, T.; Okumura, M.; Haruta, M.; Toshima, N. Catalytically highly active top gold atom on palladium nanocluster. Nat. Mater. 2012, 11, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Blom, R.H.; Pfeifer, V.F.; Moyer, A.J.; TraufRer, D.H.; Conway, H.F.; Crocker, C.K.; Farison, R.E.; Hannibal, D.V. Sodium gluconate production-fermentation with Asppergillus niger. Ind. Eng. Chem. 1952, 44, 435–440. [Google Scholar] [CrossRef]
- Sankpal, N.V.; Kulkarni, B.D. Optimization of fermentation conditions for gluconic acid production using Aspergillus niger immobilized on cellulose microfibrils. Process Biochem. 2002, 37, 1343–1350. [Google Scholar] [CrossRef]
- Velizarov, S.; Beschkov, V. Biotransformation of glucose to free gluconic acid by Gluconobacter oxydans: Substrate and product inhibition situations. Process Biochem. 1998, 33, 527–534. [Google Scholar] [CrossRef]
- Anastassiadis, S.; Aivasidis, A.; Wandrey, C. Continuous gluconic acid production by isolated yeast-like mould strains of Aureobasidium pullulans. Appl. Microbiol. Biotechnol. 2003, 61, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Doneva, T.; Vassilieff, C.; Donev, R. Catalytic and biocatalytic oxidation of glucose to gluconic acid in a modified three-phase reactor. Biotechnol. Lett. 1999, 21, 1107–1111. [Google Scholar] [CrossRef]
- Krieg, T.; Sydow, A.; Schröder, U.; Schrader, J.; Holtmann, D. Reactor concepts for bioelectrochemical syntheses and energy conversion. Trends Biotechnol. 2014, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Dencic, I.; Meuldijk, J.; Croon, M.; Hessel, V. From a review of noble metal versus enzyme catalysts for glucose oxidation under conventional conditions towards a process design analysis for continuous-flow operation. J. Flow Chem. 2011, 1, 13–23. [Google Scholar] [CrossRef]
- DevauxBasseguy, R.; Gros, P.; Bergel, A. Electroenzymatic processes: A clean technology alternative for highly selective synthesis? J. Chem. Technol. Biotechnol. 1997, 68, 389–396. [Google Scholar] [CrossRef]
- Varničić, M.; Vidaković-Koch, T.; Sundmacher, K. Gluconic acid synthesis in an electroenzymatic reactor. Electrochim. Acta 2015, 174, 480–487. [Google Scholar] [CrossRef]
- Manjon, A.; Obón, J.M.; Casanova, P.; Fernández, V.M.; Iborra, J.L. Increased activity of glucose dehydrogenase co-immobilized with a redox mediator in a bioreactor with electrochemical NAD(+) regeneration. Biotechnol. Lett. 2002, 24, 1227–1232. [Google Scholar] [CrossRef]
- Obon, J.M.; Casanova, P.; Manjon, A.; Fernández, V.M.; Iborra, J.L. Stabilization of glucose dehydrogenase with polyethyleneimine in an electrochemical reactor with NAD(P)(+) regeneration. Biotechnol. Prog. 1997, 13, 557–561. [Google Scholar] [CrossRef]
- Gros, P.; Bergel, A. Electrochemically enhanced biosynthesis of gluconic acid. AIChE J. 2005, 51, 989–997. [Google Scholar] [CrossRef]
- Miyawaki, O.; Yano, T. Electrochemical bioreactor with immobilized glucose-6-phosphate-dehydrogenase on the rotating graphite disc electrode modified with phenazine methosulfate. Enzym. Microb. Technol. 1993, 15, 525–529. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Biocatalysis-key to sustainable industrial chemistry. Curr. Opin. Biotechnol. 2010, 21, 713–724. [Google Scholar] [CrossRef]
- Varnicic, M.; Bettenbrock, K.; Hermsdorf, D.; Vidaković-Koch, T.; Sundmacher, K. Combined electrochemical and microscopic study of porous enzymatic electrodes with direct electron transfer mechanism. RSC Adv. 2014, 4, 36471–36479. [Google Scholar] [CrossRef]
- Wendt, H.; Kreysa, G. Electrochemical Engineering Science and Technology in Chemical and Other Industries; Springer: Berlin, Germany, 1999; p. 199. ISBN 978-3-642-08406-5. [Google Scholar]
- Reid, R.C.; Giroud, F.; Minteer Shelley, D.; Bruce, K.G. Enzymatic biofuel cell with a flow-through toray paper bioanode for improved fuel utilization. J. Electrochem. Soc. 2013, 160, H612–H619. [Google Scholar] [CrossRef]
- Agnes, C.; Reuillard, B.; Goff, A.; Holzinger, M.; Cosnier, S. A double-walled carbon nanotube-based glucose/H2O2 biofuel cell operating under physiological conditions. Electrochem. Commun. 2013, 34, 105–108. [Google Scholar] [CrossRef]
- Ramanavicius, A.; Kausaite-Minkstimiene, A.; Morkvenaite-Vilkonciene, I.; Genys, P.; Mikhailovar, R.; Semashko, T.; Voronovic, J.; Ramanaviciene, A. Biofuel cell based on glucose oxidase from Penicillium funiculosum 46.1 and horseradish peroxidase. Chem. Eng. J. 2015, 264, 165–173. [Google Scholar] [CrossRef]
- Krikstolaityte, V.; Oztekin, Y.; Kuliesius, J.; Ramanaviciene, A.; Yazicigil, Z.; Ersoz, M.; Okumus, A.; Kausaite-Minkstimiene, A.; Kilic, Z.; Solak, A.O.; et al. Biofuel cell based on anode and cathode modified by glucose oxidase. Electroanalysis 2013, 25, 2677–2683. [Google Scholar] [CrossRef]
- Gomez, C.; Shipovskov, S.; Ferapontova, E.E. Peroxidase biocathodes for a biofuel cell development. J. Renew. Sustain. Energy 2010, 2, 013103. [Google Scholar] [CrossRef]
- Holade, Y.; Servat, K.; Napporn, T.W.; Morais, C.; Berjeaud, J.M.; Kokoh, K.B. Highly selective oxidation of carbohydrates in an efficient electrochemical energy converter: Cogenerating organic electrosynthesis. Chemsuschem 2016, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.F.; Chou, T.C. Paired electrooxidation IV. Decarboxylation of sodium gluconate to D-arabinose. J. Appl. Electrochem. 2003, 33, 741–745. [Google Scholar] [CrossRef]
- Zargari, N.; Kim, Y.; Jung, K.W. Conversion of saccharides into formic acid using hydrogen peroxide and a recyclable palladium(II) catalyst in aqueous alkaline media at ambient temperatures. Green Chem. 2015, 17, 2736–2740. [Google Scholar] [CrossRef]
- Zhang, H.; Li, N. Oxidative conversion of glucose to gluconic acid by iron(III) chloride in water under mild conditions. Green Chem. 2016, 16, 2558. [Google Scholar] [CrossRef]
- Do, T.Q.N. Model-Based Analysis of an Electroenzymatic System for Glucose Oxidation; Otto-von-Guericke University Magdeburg: Magdeburg, Germany, 2017; p. 136. [Google Scholar]
- Vandenberg, R.; Peters, J.A.; Vanbekkum, H. Selective alkaline oxidative-degradation of monosaccharides and disaccharides by hydrogen-peroxide using borate as catalyst and protecting group. Carbohydr. Res. 1995, 267, 65–77. [Google Scholar] [CrossRef]
- Isbell, H.S.; Frush, H.L. Reactions of carbhydrates with hydroperoxides 17. Mechanisms for hydroperoxide degradation of disaccharides and related-compounds. Carbohydr. Res. 1987, 161, 181–193. [Google Scholar] [CrossRef]
- Moreno, T.; Kouzaki, G.; Sasaki, M.; Goto, M.; Cocero, M.J. Uncatalysed wet oxidation of D-glucose with hydrogen peroxide and its combination with hydrothermal electrolysis. Carbohydr. Res. 2012, 349, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Vallieres, C.; Matlosz, M. A multisectioned porous electrode for synthesis of D-arabinose. J. Electrochem. Soc. 1999, 146, 2933–2939. [Google Scholar] [CrossRef]
- Jiricny, V.; Stanek, V. Productionof D-arabinose in a pilot-plant fluidized-bed electrochemical reactor. J. Appl. Electrochem. 1994, 24, 930–935. [Google Scholar] [CrossRef]
- Hourdin, G.L.; Germain, A.; Moreau, C.; Fajula, F. The catalysis of the Ruff oxidative degradation of aldonic acids by copper(II)-containing solids. J. Catal. 2002, 209, 217–224. [Google Scholar] [CrossRef]
- Gara, M.; Compton, R.G. Activity of carbon electrodes towards oxygen reduction in acid: A comparative study. New J. Chem. 2011, 35, 2647–2652. [Google Scholar] [CrossRef]
- Watson, V.J.; Delgado, C.N.; Logan, B.E. Improvement of activated carbons as oxygen reduction catalysts in neutral solutions by ammonia gas treatment and their performance in microbial fuel cells. J. Power Sources 2013, 242, 756–761. [Google Scholar] [CrossRef]
- Flexer, V.; Brun, N.; Destribats, M.; Backov, R.; Mano, N. A novel three-dimensional macrocellular carbonaceous biofuel cell. Phys. Chem. Chem. Phys. 2013, 15, 6437–6445. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Basséguy, R.; Délécouls-Servat, K.; Bergel, A. Glucose oxidase catalysed oxidation of glucose in a dialysis membrane electrochemical reactor (D-MER). Bioprocess Biosyst. Eng. 2004, 26, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Kumar, R.; Banerjee, S. Manufacture of gluconic acid: A review towards process intensification for green production. Chem. Eng. Process. 2016, 104, 160–171. [Google Scholar] [CrossRef]
- Parish, J.H. A Revolution in Biotechnology; Marx, J.L., Ed.; Cambridge University Press: New York, NY, USA, 1989; ISBN 0-521-32749-0. [Google Scholar]
- Sheldon, R.A. E factors, green chemistry and catalysis: An odyssey. Chem. Commun. 2008, 29, 3352–3365. [Google Scholar] [CrossRef]
- Jagdish Singh, N.V. Glucose oxides from Aspergillus niger: Production, characterization and immobilization for glucose oxidation. Adv. Appl. Sci. Res. 2013, 4, 250–257. [Google Scholar]
- Flocco, C.G.; Alvarez, M.A.; Giulietti, A.M. Peroxidase production in vitro by Armoracia lapathifolia (horseradish)-transformed root cultures: Effect of elicitation on level and profile of isoenzymes. Biotechnol. Appl. Biochem. 1998, 28, 33–38. [Google Scholar]
- Khan, G.F.; Ohwa, M.; Wernet, W. Design of a stable charge transfer complex electrode for a third-generation amperometric glucose sensor. Anal. Chem. 1996, 68, 2939–2945. [Google Scholar] [CrossRef]
- Pizzariello, A.; Stredansky, M.; Miertus, S. A glucose/hydrogen peroxide biofuel cell that uses oxidase and peroxidase as catalysts by composite bulk-modified bioelectrodes based on a solid binding matrix. Bioelectrochemistry 2002, 56, 99–105. [Google Scholar] [CrossRef]
- Merle, G.; Habrioux, A.; Servat, K.; Rolland, M.; Innocent, C.; Kokoh, K.B.; Tingry, S. Long-term activity of covalent grafted biocatalysts during intermittent use of a glucose/O-2 biofuel cell. Electrochim. Acta 2009, 54, 2998–3003. [Google Scholar] [CrossRef]
- Do, T.Q.N.; Varnicic, M.; Hanke-Rauschenbach, R.; Vidakovic-Koch, T.; Sundmacher, K. Mathematcal modeling of Porous Enzymatic Electrode with Direct Electron Transfer Mechanism. Electrochim. Acta 2014, 137, 616–626. [Google Scholar] [CrossRef]
- Genders, J.D.; Gopal, R.; Hartsough, D.M.; Kendall, P.M.; Long, W.J.; Mazur, D.J.; Zappi, G.D. Electrodialysis Methods for Purification and Recovery of Gluconic acid Derivatives. U.S. Patent 6541228B1, 1 April 2003. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flow Rate/mL min−1 | Conversion/% | Selectivity/% | Y:GA/% | Y:A/% | Y:FA/% |
|---|---|---|---|---|---|
| 10 and 14 | 49.0 | 83.7 | 41.5 | 3.0 | 5.0 + traces of other products |
| 5 | 57.0 | 96.5 | 55.0 | -Traces | 2.0 + traces of other products |
| 2 | 46.0 | 73.9 | 34.0 | 7.0 | 5.0 + traces of other products |
| Electrode | Ea/Ec/V (Time) | Overpotentials/V (Time) | Glucose Conversion/% | Selectivity/% | Y:GA/% | Y:A/% | Y:FA/% |
|---|---|---|---|---|---|---|---|
| ANODE: V-Gel/GOx-TTF | Ea = 0.2 (2 h) Ea = 0.15 (2 h) Ea = 0.1 (3 h) | 0.35 (2 h) 0.3 (2 h) 0.25 (3 h) | 35 | 100 | 34 | 0 | 1 |
| ANODE: V-Gel/GOx-TTF | Ea = 0.15 (2 h) Ea = 0.1 (2 h) Ea = 0.05 (3 h) | 0.30 (2 h) 0.25 (2 h) 0.20 (3 h) | 6 | 100 | 6 | 0 | 0 |
| CATHODE: V-PVDF/GOx-HRP | Ec = 0.05 (2 h) Ec = 0.0 V (2 h) Ec = −0.05 (3 h) | 0.50 (2 h) 0.55 (2 h) 0.60 (3 h) | 56 | 80 | 44 | 6 | 6 + traces of other products |
| CATHODE: V-PVDF/GOx-HRP | Ec = 0.15 (2 h) Ec = 0.1 (2 h) Ec = 0.05 (3 h) | 0.4 (2 h) 0.45 (2 h) 0.50 (3 h) | 43 | 70 | 29 | 7 | 7 + traces of other products |
| Reactor | Time/h | Cell Potential/V | Conversion/% | Selectivity/% | Y:GA/% | Y:A/% | Y:FA/% |
|---|---|---|---|---|---|---|---|
| 1 | 16 | U = −0.1 | 67 | 95.5 | 64 | -Traces | 3 + traces of other products |
| 2 | 21 | U = −0.1 (5.5 h) U = −0.05 (3.5 h) U = 0.0 (3 h) U = 0.05 (9 h) | 80 | 92 | 74 | 4 | 2 + traces of other products |
| System | Conditions | Cell Potential/V | Flow Rate/mL min−1 | Conversion/% | Selectivity/% |
|---|---|---|---|---|---|
| GOx-HRP based cathode | pH 6.00/22 °C | Ucell = 0.0 | 5 | 60 | 97 |
| BOD based cathode | pH 7.00/37 °C | Ucell = 0.0 | 5 | 28 | 100 |
| Indicators | Electroenzymatic | Fermentation | Electroenzymatic |
|---|---|---|---|
| STY, g h−1 dm−3 | 33.7–36.1 | 0.132–21 | 0.13–4.9 |
| Product titre, g dm−3 | 3.1 | 100–350 | 1.66–14.5 |
| tcycle, h | 8–20 | 20–100 | 3–12 |
| Reaction yield, % | 75 | 74–99.4 | 43–85 |
| Atom efficiency, % | 100 | 100 | 99 |
| E-factor/kg kg−1 | 9 | 0.06 [11] | N/A |
| References | This work | [45] | [17,18,19,20,44] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varničić, M.; Zasheva, I.N.; Haak, E.; Sundmacher, K.; Vidaković-Koch, T. Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid. Catalysts 2020, 10, 269. https://doi.org/10.3390/catal10030269
Varničić M, Zasheva IN, Haak E, Sundmacher K, Vidaković-Koch T. Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid. Catalysts. 2020; 10(3):269. https://doi.org/10.3390/catal10030269
Chicago/Turabian StyleVarničić, Miroslava, Iva N. Zasheva, Edgar Haak, Kai Sundmacher, and Tanja Vidaković-Koch. 2020. "Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid" Catalysts 10, no. 3: 269. https://doi.org/10.3390/catal10030269
APA StyleVarničić, M., Zasheva, I. N., Haak, E., Sundmacher, K., & Vidaković-Koch, T. (2020). Selectivity and Sustainability of Electroenzymatic Process for Glucose Conversion to Gluconic Acid. Catalysts, 10(3), 269. https://doi.org/10.3390/catal10030269