Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution

 , , , ,
, , , ,
Abstract
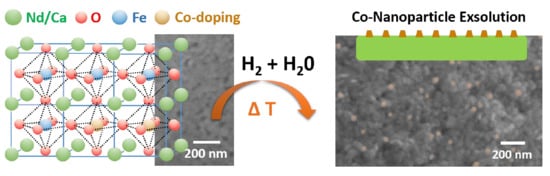
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
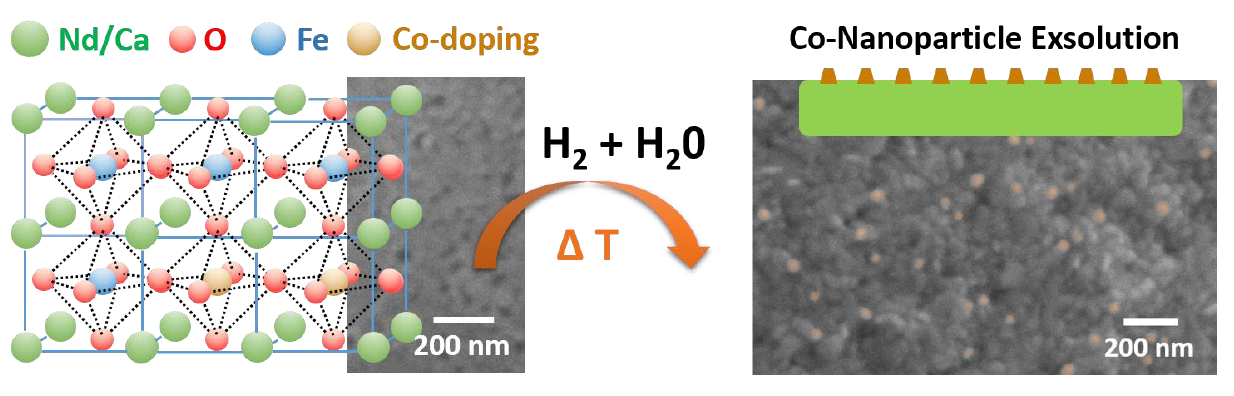
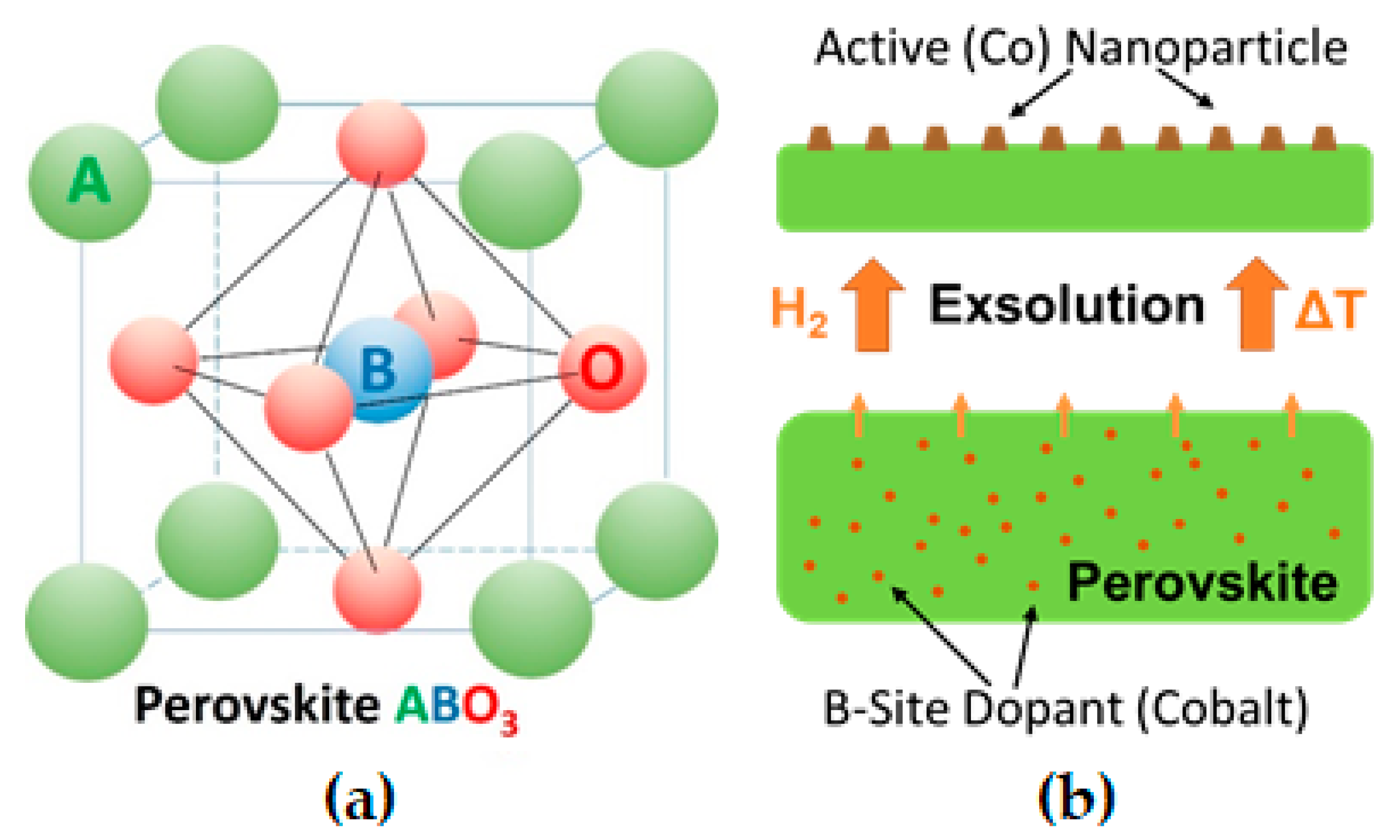
2.1. Materials Design
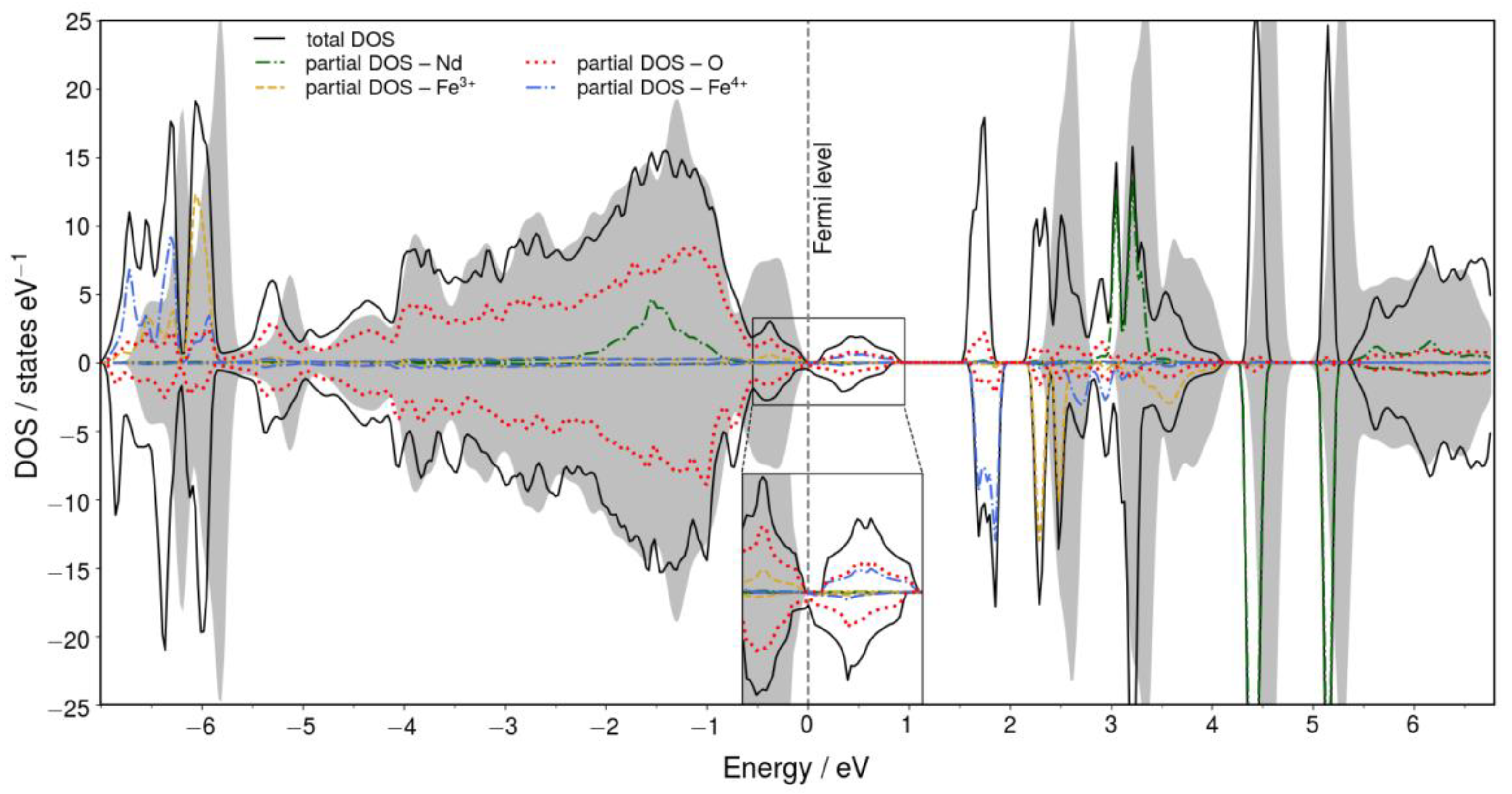
2.2. DFT Calculations
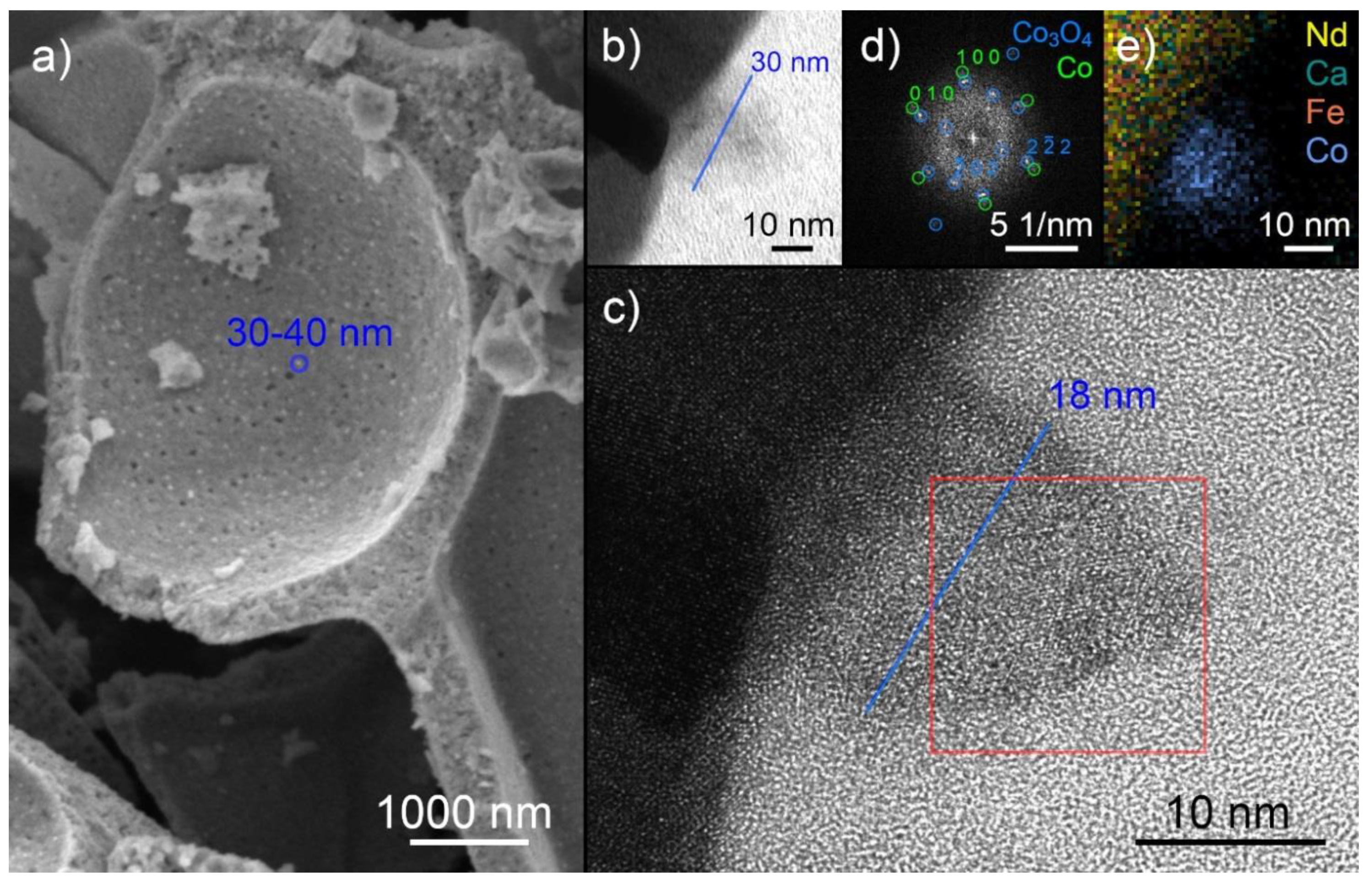
2.3. Exsolution Properties
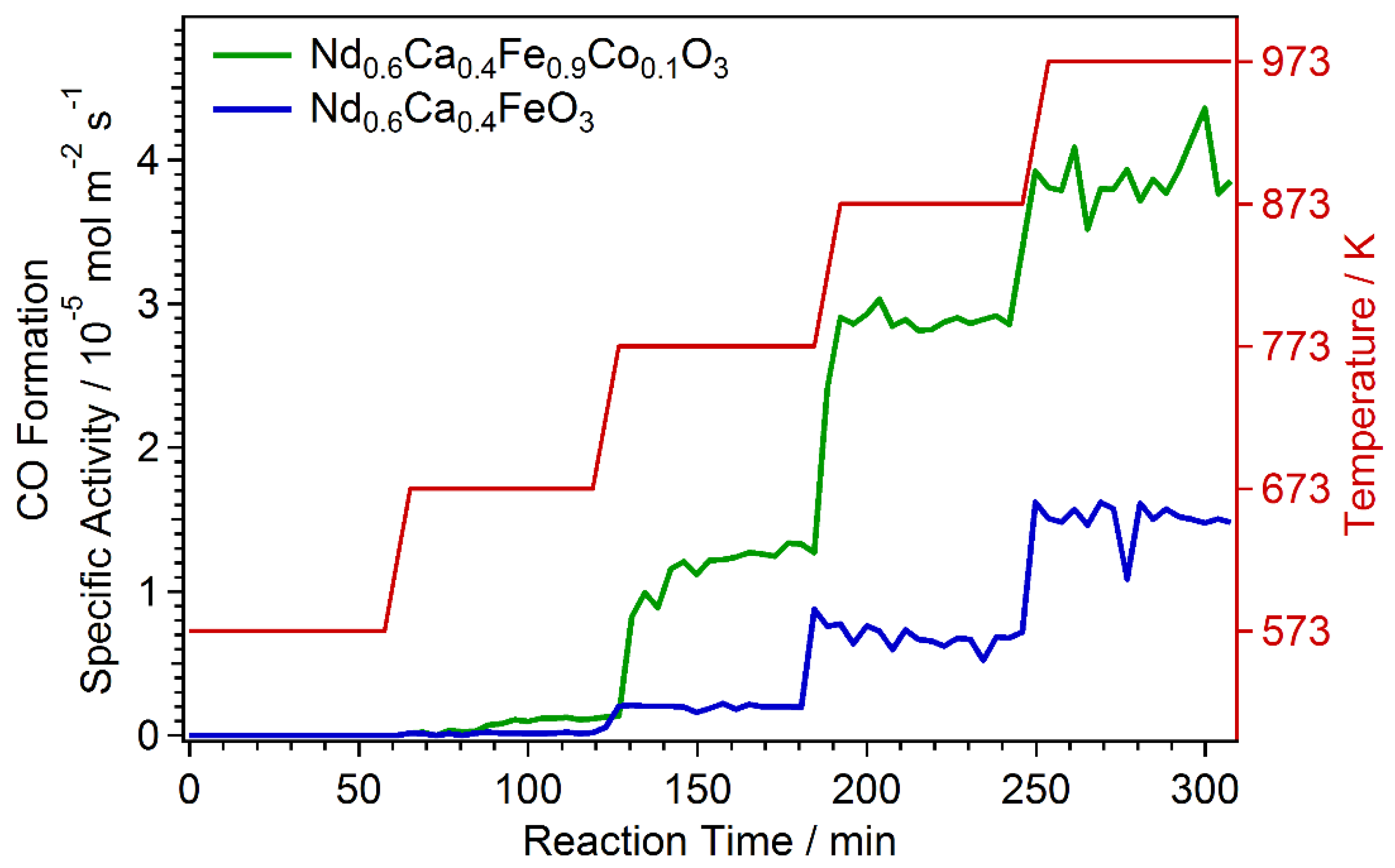
2.4. Catalytic Test Measurements
3. Materials and Methods
3.1. Synthesis of Novel Perovskites
3.2. Characterization Methods
3.3. DFT Calculations
3.4. Catalytic Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gorte, R.J.; Vohs, J.M. Nanostructured anodes for solid oxide fuel cells. Curr. Opin. Colloid Interface Sci. 2009, 14, 236–244. [Google Scholar] [CrossRef]
- Yates, J.T.; Campbell, C.T. Surface chemistry: Key to control and advance myriad technologies. Proc. Natl. Acad. Sci. USA 2011, 108, 911–916. [Google Scholar] [CrossRef]
- Rousseau, S.; Marie, O.; Bazin, P.; Daturi, M.; Verdier, S.; Harle, V. Investigation of Methanol Oxidation over Au/Catalysts Using Operando IR Spectroscopy: Determination of the Active Sites, Intermediate/Spectator Species, and Reaction Mechanism. J. Am. Chem. Soc. 2010, 132, 10832–10841. [Google Scholar] [CrossRef]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Menard, H.; Irvine, J.T.S. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef]
- Katz, M.B.; Zhang, S.Y.; Duan, Y.W.; Wang, H.J.; Fang, M.H.; Zhang, K.; Li, B.H.; Graham, G.W.; Pan, X.Q. Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: Bulk versus surface re-dispersion. J. Catal. 2012, 293, 145–148. [Google Scholar] [CrossRef]
- Tsekouras, G.; Neagu, D.; Irvine, J.T.S. Step-change in high temperature steam electrolysis performance of perovskite oxide cathodes with exsolution of B-site dopants. Energy Environ. Sci. 2013, 6, 256–266. [Google Scholar] [CrossRef]
- Kobsiriphat, W.; Madsen, B.D.; Wang, Y.; Shah, M.; Marks, L.D.; Barnett, S.A. Nickel- and Ruthenium-Doped Lanthanum Chromite Anodes: Effects of Nanoscale Metal Precipitation on Solid Oxide Fuel Cell Performance. J. Electrochem. Soc. 2010, 157, B279–B284. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Rameshan, R.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Fleig, J.; Kloetzer, B. Enhancing Electrochemical Water-Splitting Kinetics by Polarization-Driven Formation of Near-Surface Iron (0): An In Situ XPS Study on Perovskite-Type Electrodes. Angew. Chem. Int. Ed. 2015, 54, 2628. [Google Scholar] [CrossRef]
- Adijanto, L.; Padmanabhan, V.B.; Gorte, R.J.; Vohs, J.M. Polarization-Induced Hysteresis in CuCo-Doped Rare Earth Vanadates SOFC Anodes. J. Electrochem. Soc. 2012, 159, F751–F756. [Google Scholar] [CrossRef]
- Tanaka, H.; Uenishi, M.; Taniguchi, M.; Tan, I.; Narita, K.; Kimura, M.; Kaneko, K.; Nishihata, Y.; Mizuki, J. Intelligent catalyst having the self-regenerative function of Pd, Rh and Pt for automotive emissions control. Catal. Today 2006, 117, 321–328. [Google Scholar] [CrossRef]
- Katz, M.B.; Graham, G.W.; Duan, Y.W.; Liu, H.; Adamo, C.; Schlom, D.G.; Pan, X.Q. Self-Regeneration of Pd-LaFeO3 Catalysts: New Insight from Atomic-Resolution Electron Microscopy. J. Am. Chem. Soc. 2011, 133, 18090–18093. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Sun, Y.F.; Li, J.H.; Zeng, Y.M.; Amirkhiz, B.S.; Wang, M.N.; Behnamian, Y.; Luo, J.L. A-site deficient perovskite: The parent for in situ exsolution of highly active, regenerable nano-particles as SOFC anodes. J. Mater. Chem. A 2015, 3, 11048–11056. [Google Scholar] [CrossRef]
- Papargyriou, D.; Irvine, J.T.S. Nickel nanocatalyst exsolution from (La,Sr)(Cr,M,Ni)O3 (M=Mn,Fe) perovskites for the fuel oxidation layer of Oxygen Transport Membranes. Solid State Ion. 2016, 288, 120–123. [Google Scholar] [CrossRef]
- Zhao, Q.; Lorenz, H.; Turner, S.; Lebedev, O.I.; Van Tendeloo, G.; Rameshan, C.; Klotzer, B.; Konzett, J.; Penner, S. Catalytic characterization of pure SnO2 and GeO2 in methanol steam reforming. Appl. Catal. Gen. 2010, 375, 188–195. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.S.; Miller, D.N.; Menard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Burnat, D.; Kontic, R.; Holzer, L.; Steiger, P.; Ferri, D.; Heel, A. Smart material concept: Reversible microstructural self-regeneration for catalytic applications. J. Mater. Chem. A 2016, 4, 11939–11948. [Google Scholar] [CrossRef]
- Tanaka, H.; Taniguchi, M.; Uenishi, M.; Kajita, N.; Tan, I.; Nishihata, Y.; Mizuki, J.; Narita, K.; Kimura, M.; Kaneko, K. Self-regenerating Rh- and Pt-based perovskite catalysts for automotive-emissions control. Angew. Chem. Int. Ed. 2006, 45, 5998–6002. [Google Scholar] [CrossRef]
- Oh, T.S.; Rahani, E.K.; Neagu, D.; Irvine, J.T.S.; Shenoy, V.B.; Gorte, R.J.; Vohs, J.M. Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites during Exsolution. J. Phys. Chem. Lett. 2015, 6, 5106–5110. [Google Scholar] [CrossRef]
- Haag, J.M.; Barnett, S.A.; Richardson, J.W.; Poeppelmeier, K.R. Structural and Chemical Evolution of the SOFC Anode La0.30Sr0.70Fe0.70Cr0.30O3-δ upon Reduction and Oxidation: An in Situ Neutron Diffraction Study. Chem. Mater. 2010, 22, 3283–3289. [Google Scholar] [CrossRef]
- Gotsch, T.; Schlicker, L.; Bekheet, M.F.; Doran, A.; Grunbacher, M.; Praty, C.; Tada, M.; Matsui, H.; Ishiguro, N.; Gurlo, A.; et al. Structural investigations of La0.6Sr0.4FeO3-δ under reducing conditions: Kinetic and thermodynamic limitations for phase transformations and iron exsolution phenomena. RSC Adv. 2018, 8, 3120–3131. [Google Scholar] [CrossRef]
- Kwon, O.; Sengodan, S.; Kim, K.; Kim, G.; Jeong, H.Y.; Shin, J.; Ju, Y.W.; Han, J.W. Exsolution trends and co-segregation aspects of self-grown catalyst nanoparticles in perovskites. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Park, J.; Nam, S.Y.; Kim, K.J.; Choi, G.M.; Parkin, S.S.P.; Jang, H.M.; Irvine, J.T.S. Lattice strain-enhanced exsolution of nanoparticles in thin films. Nat. Commun. 2019, 10, 1471. [Google Scholar] [CrossRef] [PubMed]
- Gotsch, T.; Kopfle, N.; Grunbacher, M.; Bernardi, J.; Carbonio, E.A.; Havecker, M.; Knop-Gericke, A.; Bekheet, M.F.; Schlicker, L.; Doran, A.; et al. Crystallographic and electronic evolution of lanthanum strontium ferrite (La0.6Sr0.4FeO3-δ) thin film and bulk model systems during iron exsolution. Phys. Chem. Chem. Phys. 2019, 21, 3781–3794. [Google Scholar] [CrossRef]
- Arrive, C.; Delahaye, T.; Joubert, O.; Gauthier, G. Exsolution of nickel nanoparticles at the surface of a conducting titanate as potential hydrogen electrode material for solid oxide electrochemical cells. J. Power Sources 2013, 223, 341–348. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J.; Laskowski, R.; Tran, F.; Marks, L.D. WIEN2k: An Augmented Plave Wave plus Local Oribtals Program for Calculating Crystal Properties; Vienna University of Technology: Wien, Austria, 2018. [Google Scholar]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chueh, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Lee, W.; Han, J.W.; Chen, Y.; Cai, Z.; Yildiz, B. Cation Size Mismatch and Charge Interactions Drive Dopant Segregation at the Surfaces of Manganite Perovskites. J. Am. Chem. Soc. 2013, 135, 7909–7925. [Google Scholar] [CrossRef]
- Geller, S. Crystallographic Studies of Perovskite-like Compounds V: Relative Ionic Sizes. Acta Crystallogr. 1957, 10, 248–251. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, W.; Liu, P.R.; Zhao, H.J.; Chen, J.; Deng, J.X.; Xing, X.R. Improved conductivity of NdFeO3 through partial substitution of Nd by Ca: A theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 29097–29102. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Kubicek, M.; Goetsch, T.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Kloetzer, B.; et al. Surface Chemistry of Perovskite-Type Electrodes During High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 35847–35860. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ni, W.J.; Wang, J.; Zhong, Q.; Han, M.F.; Zhu, T.L. Exploration of Co-Fe alloy precipitation and electrochemical behavior hysteresis using Lanthanum and Cobalt co- substituted SrFeO3-δ SOFC anode. Electrochim. Acta 2018, 277, 226–234. [Google Scholar] [CrossRef]
- Gao, Y.; Lu, Z.H.; You, T.L.; Wang, J.; Xie, L.; He, J.Q.; Ciucci, F. Energetics of Nanoparticle Exsolution from Perovskite Oxides. J. Phys. Chem. Lett. 2018, 9, 3772–3778. [Google Scholar] [CrossRef]
- Kim, K.J.; Han, H.; Defferriere, T.; Yoon, D.; Na, S.; Kim, S.J.; Dayaghi, A.M.; Son, J.; Oh, T.S.; Jang, H.M.; et al. Facet-Dependent in Situ Growth of Nanoparticles in Epitaxial Thin Films: The Role of Interfacial Energy. J. Am. Chem. Soc. 2019, 141, 7509–7517. [Google Scholar] [CrossRef] [PubMed]
- Steiger, P.; Nachtegaal, M.; Krocher, O.; Ferri, D. Reversible Segregation of Ni in LaFe0.8Ni0.2O3±δ During Coke Removal. Chemcatchem 2018, 10, 4456–4464. [Google Scholar] [CrossRef]
- Joo, S.; Kwon, O.; Kim, K.; Kim, S.; Kim, H.; Shin, J.; Jeong, H.Y.; Sengodan, S.; Han, J.W.; Kim, G. Cation-swapped homogeneous nanoparticles in perovskite oxides for high power density. Nat. Commun. 2019, 10, 697. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to form a Capacitor. U.S. Patent 3,330,697, 11 July 1967. [Google Scholar]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]
- ICDD PDF-4+ 2019; International Centre for Diffraction Data: Newtown Square, PA, USA, 2018.
- Karsai, F.; Tran, F.; Blaha, P. On the importance of local orbitals using second energy derivatives for d and f electrons. Comput. Phys. Commun. 2017, 220, 230–238. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band Theory and Mott Insulators—Hubbard-U Instead of Stoner-I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef]
- Kraushofer, F.; Jakub, Z.; Bichler, M.; Hulva, J.; Drmota, P.; Weinold, M.; Schmid, M.; Setvin, M.; Diebold, U.; Blaha, P.; et al. Atomic-Scale Structure of the Hematite alpha-Fe2O3 (1(1)over-bar02) “R-Cut” Surface. J. Phys. Chem. C 2018, 122, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, F.; Sakuma, R.; Aryasetiawan, F. Ab initio calculations of the Hubbard U for the early lanthanides using the constrained random-phase approximation. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef]
- Wollan, E.O.; Koehler, W.C. Neutron Diffraction Study of the Magnetic Properties of the Series of Perovskite-Type Compounds [(1-x)La,xCa]MnO3. Phys. Rev. 1955, 100, 545–563. [Google Scholar] [CrossRef]
- Slawinski, W.; Przenioslo, R.; Sosnowska, I.; Suard, E. Spin reorientation and structural changes in NdFeO3. J. Phys. Condens. Matter 2005, 17, 4605–4614. [Google Scholar] [CrossRef]
- Bartolome, J.; Palacios, E.; Kuzmin, M.D.; Bartolome, F.; Sosnowska, I.; Przenioslo, R.; Sonntag, R.; Lukina, M.M. Single-crystal neutron diffraction study of Nd magnetic ordering in NdFeO3 at low temperature. Phys. Rev. B 1997, 55, 11432–11441. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Ishizawa, N. Synchrotron X-ray study of the electron density in RFeO3 (R. = Nd, Dy). Acta Crystallogr. Sect. B Struct. Sci. 1999, 55, 1–7. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindenthal, L.; Rameshan, R.; Summerer, H.; Ruh, T.; Popovic, J.; Nenning, A.; Löffler, S.; Opitz, A.K.; Blaha, P.; Rameshan, C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts 2020, 10, 268. https://doi.org/10.3390/catal10030268
Lindenthal L, Rameshan R, Summerer H, Ruh T, Popovic J, Nenning A, Löffler S, Opitz AK, Blaha P, Rameshan C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts. 2020; 10(3):268. https://doi.org/10.3390/catal10030268
Chicago/Turabian StyleLindenthal, Lorenz, Raffael Rameshan, Harald Summerer, Thomas Ruh, Janko Popovic, Andreas Nenning, Stefan Löffler, Alexander Karl Opitz, Peter Blaha, and Christoph Rameshan. 2020. "Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution" Catalysts 10, no. 3: 268. https://doi.org/10.3390/catal10030268
APA StyleLindenthal, L., Rameshan, R., Summerer, H., Ruh, T., Popovic, J., Nenning, A., Löffler, S., Opitz, A. K., Blaha, P., & Rameshan, C. (2020). Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts, 10(3), 268. https://doi.org/10.3390/catal10030268