Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Theoretical Aspects for the DET-Type Bioelectrocatalysis
3. Significance of Nanostructures of Electrodes for DET-Type Bioelectrocatalysis
3.1. Porous Carbon Electrodes
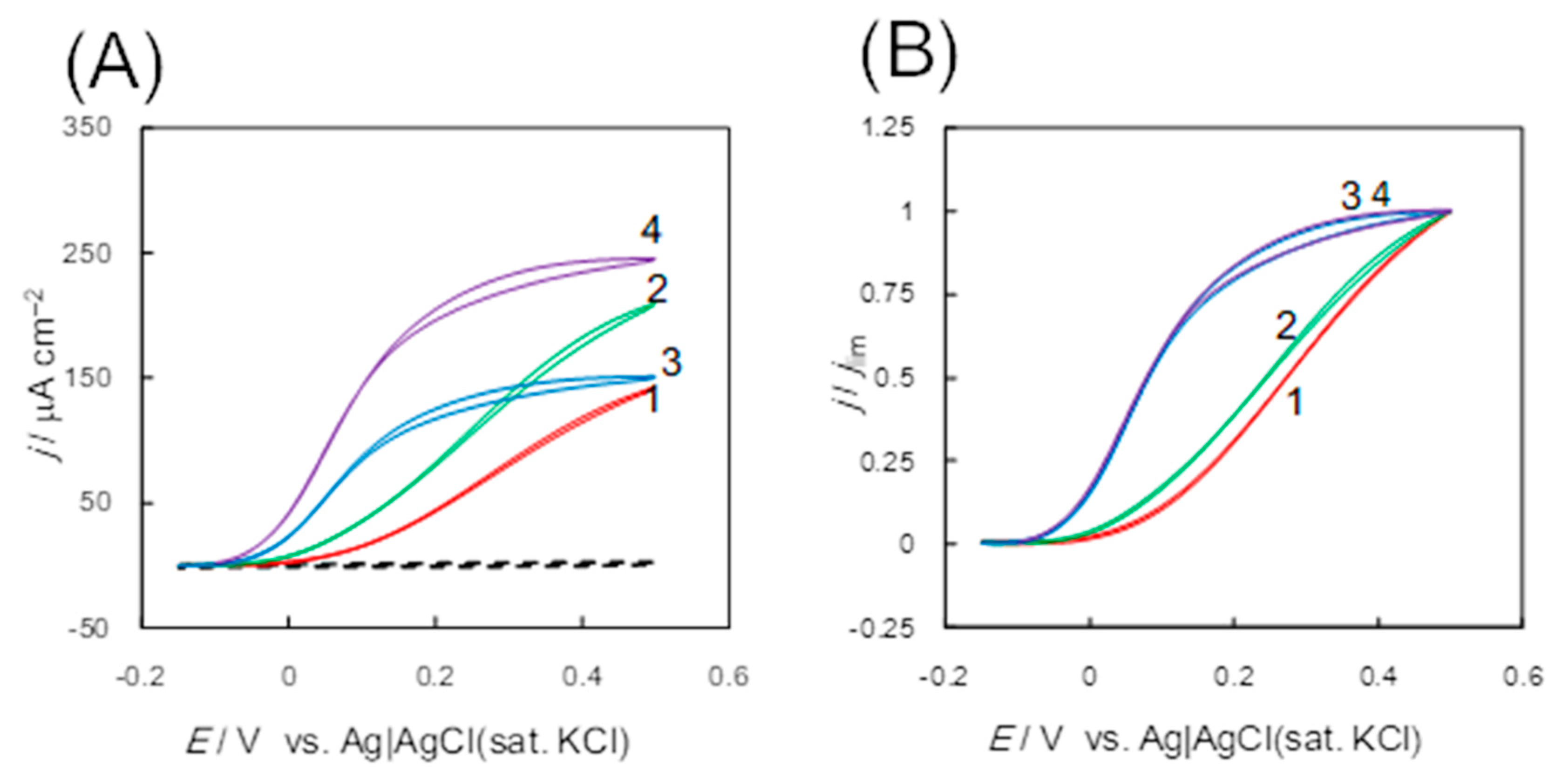
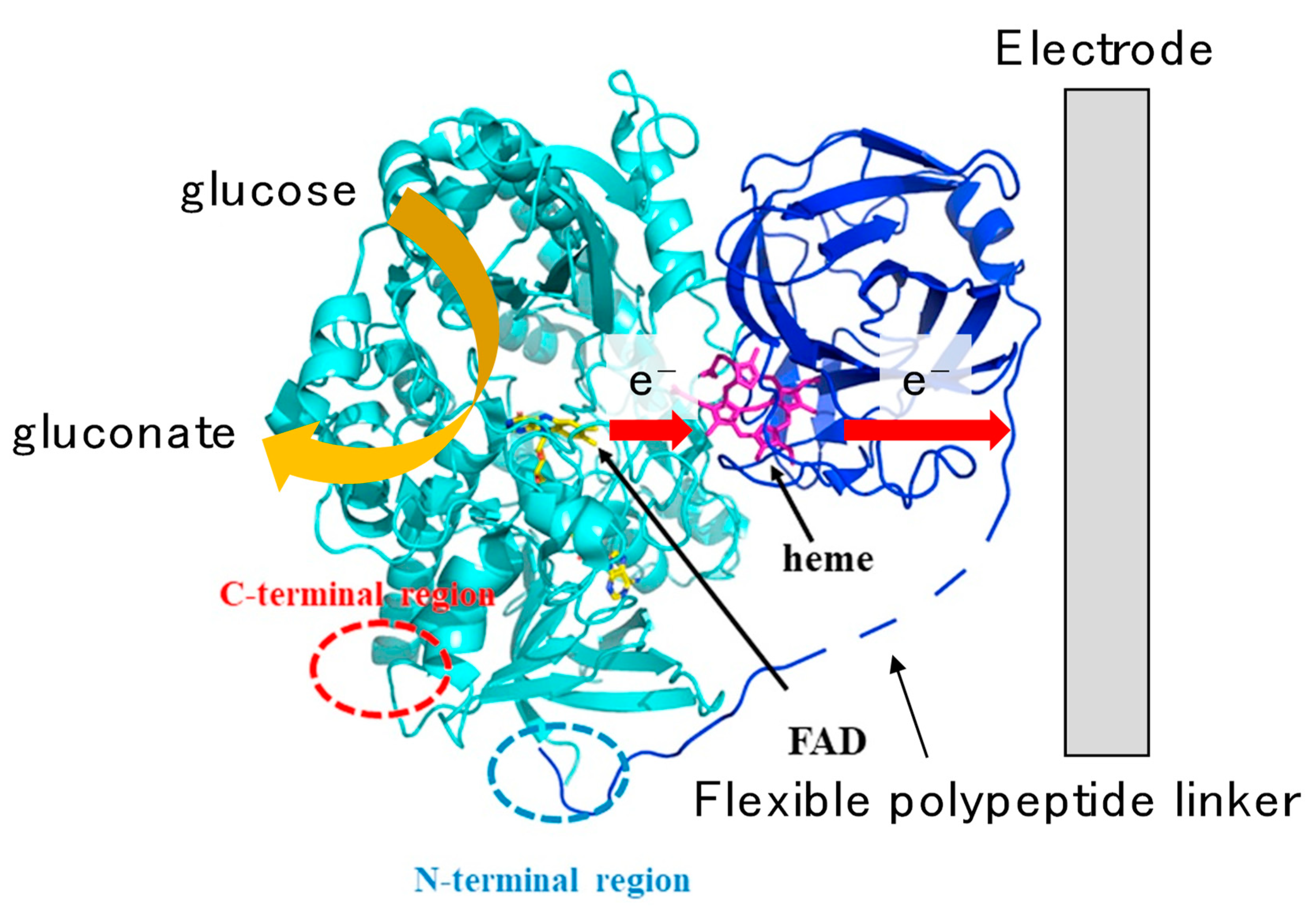
3.1.1. Fructose Dehydrogenase
3.1.2. Bilirubin Oxidase
3.1.3. Hydrogenase
3.1.4. Histamine Dehydrogenase
3.2. Nanoporous Metals and Aggregated Metal Nanoparticles
3.2.1. Hydrogenase
3.2.2. Bilirubin Oxidase
3.2.3. Fructose Dehydrogenase
4. Protein Engineering Methods for the Improvement of DET-Type Bioelectrocatalytic Performance
5. Interconversion of Redox Couples by Bidirectional DET-Type Bioelectrocatalysis
5.1. Hydrogenase
5.2. Formate Dehydrogenase
5.3. Ferredoxin-NADP+ Reductase
6. Conclusions
Funding
Conflicts of Interest
References
- Bartlett, P.N. Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Barton, S.C.; Gallaway, J.; Atanassov, P. Enzymatic biofuel cells for implantable and microscale devices. Chem. Rev. 2004, 104, 4867–4886. [Google Scholar] [CrossRef] [PubMed]
- Léger, C.; Bertrand, P. Direct electrochemistry of redox enzymes as tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Hirst, J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 14049–14054. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.T.; Minteer, S.D. Biofuel cells; enhanced enzymatic bioelectrocatalysis. Annu. Rev. Anal. Chem. 2012, 5, 157–179. [Google Scholar] [CrossRef]
- De Poulpiquet, A.; Ranava, D.; Monsalve, K.; Giudici-Orticoni, M.T.; Lojou, E. Biohydrogen for a new generation of H2/O2 biofuel cells: A sustainable energy perspective. ChemElectroChem 2014, 1, 1724–1750. [Google Scholar] [CrossRef]
- Mazurenko, I.; de Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Mano, N.; de Poulpiquet, A. O2 reduction in enzymatic biofuel cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef]
- Milton, R.D.; Minteer, S.D. Enzymatic bioelectrosynthetic ammonia production: Recent electrochemistry of nitrogenase, nitrate reductase, and nitrite reductase. ChemPlusChem 2017, 82, 513–521. [Google Scholar] [CrossRef]
- Fourmond, V.; Léger, C. Modelling the voltammetry of adsorbed enzymes and molecular catalysts. Curr. Opin. Electrochem. 2017, 1, 110–120. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E.; Willner, B. Electrical contact of redox enzyme layers associated with electrodes; routes to amperometric biosensors. Electroanalysis 1997, 9, 965–977. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L. Enzyme based amperometric biosensors. Curr. Opin. Electrochim. 2018, 10, 157–173. [Google Scholar]
- Krieg, T.; Sydow, A.; Schröder, U.; Schrader, J.; Holtmann, D. Reactor concepts for bioelectrochemical syntheses and energy conversion. Trends Biotechnol. 2014, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Shleev, S.; González-Arribas, E.; Falk, M. Biosupercapacitors. Curr. Opin. Electrochem. 2017, 5, 226–233. [Google Scholar] [CrossRef]
- Tsujimura, S.; Nakagawa, T.; Kano, K.; Ikeda, T. Kinetic study of direct bioelectrocatalysis of dioxygen reduction with bilirubin oxidase at carbon electrodes. Electrochemistry 2004, 72, 437–439. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundamentals and Applications, 2nd ed; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Léger, C.; Jones, A.K.; Albracht, S.P.J.; Armstrong, F.A. Effect of a dispersion of interfacial electron transfer rates on steady state catalytic electron transport in [NiFe]-hydrogenase and other enzymes. J. Phys. Chem. B 2002, 106, 13058–13063. [Google Scholar] [CrossRef]
- Xia, H.-Q.; Kitazumi, Y.; Shirai, O.; Kano, K. Enhanced direct electron transfer-type bioelectrocatalysis of bilirubin oxidase on negatively charged aromatic compound-modified carbon electrode. J. Electroanal. Chem. 2016, 763, 104–109. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Takeuchi, R.; Kitazumi, Y.; Shirai, O.; Kano, K. Significance of mesoporous electrodes for noncatalytic faradaic process of randomly oriented redox proteins. J. Phys. Chem. C 2016, 120, 26270–26277. [Google Scholar] [CrossRef]
- Sakai, K.; Xia, H.-Q.; Kitazumi, Y.; Shirai, O.; Kano, K. Assembly of direct-electron-transfer-type bioelectrodes with high performance. Electrochim. Acta 2018, 271, 305–311. [Google Scholar] [CrossRef]
- Kitazumi, Y.; Shirai, O.; Kano, K. Significance of nanostructures of an electrode surface in direct electron transfer-type bioelectrocatalysis of redox enzymes. ACS Symposium Series 1342. In Novel Catalyst Materials for Bioelectrochemical Systems: Fundamentals and Applications; Lakhveer Singh, L., Mahapatra, D.M., Liu, H., Eds.; American Chemical Society: Washington, DC, USA, 2020. [Google Scholar]
- Sakintuna, B.; Yürüm, Y. Templated porous carbons: A reviewarticle. Ind. Eng. Chem. Res. 2005, 44, 2893–2902. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J. Hyeon, Recent progress in the synthesis of porous carbon materials. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Ma, T.-Y.; Liu, L.; Yuan, Z.-Y. Direct synthesis of ordered mesoporous carbons. Chem. Soc. Rev. 2013, 42, 3977–4003. [Google Scholar] [CrossRef]
- Morishita, T.; Tsumura, T.; Toyoda, M.; Przepiórski, J.; Morawski, A.W.; Konno, H.; Inagaki, M. A review of the control of pore structure in MgO-templated nanoporous carbons. Carbon 2010, 48, 2690–2707. [Google Scholar] [CrossRef]
- Inagaki, M.; Toyoda, M.; Soneda, Y.; Tsujimura, S.; Morishita, T. Templated mesoporous carbons: Synthesis and applications. Carbon 2016, 107, 448–473. [Google Scholar] [CrossRef]
- Adachi, T.; Kaida, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Bioelectrocatalytic performance of d-fructose dehydrogenase. Bioelectrochemistry 2019, 129, 1–9. [Google Scholar] [CrossRef]
- Kawai, S.; Goda-Tsutsumi, M.; Yakushi, T.; Kano, K.; Matsushita, K. Heterologous overexpression and characterization of a flavoprotein-cytochrome c complex fructose dehydrogenase of Gluconobacter japonicus NBRC3260. Appl. Environ. Microbiol. 2013, 79, 1654–1660. [Google Scholar] [CrossRef]
- Tsujimura, S.; Nishina, A.; Hamano, Y.; Kano, K.; Shiraishi, S. Electrochemical reaction of fructose dehydrogenase on carbon cryogel electrodes with controlled pore sizes. Electrochem. Commun. 2010, 12, 446–449. [Google Scholar] [CrossRef]
- Xia, H.-Q.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Interaction between d-fructose dehydrogenase and methoxy-substituent-functionalized carbon surface to increase productive orientations. Electrochim. Acta 2016, 218, 41–46. [Google Scholar] [CrossRef]
- Kawai, S.; Yakushi, T.; Matsushita, K.; Kitazumi, Y.; Shirai, O.; Kano, K. The electron transfer pathway in direct electrochemical communication of fructose dehydrogenase with electrodes. Electrochem. Commun. 2014, 38, 28–31. [Google Scholar] [CrossRef]
- Funabashi, H.; Murata, K.; Tsujimura, S. Effect of pore size of MgO-templated carbon on the direct electrochemistry of d-fructose dehydrogenase. Electrochemistry 2015, 83, 372–375. [Google Scholar] [CrossRef]
- Kamitaka, Y.; Tsujimura, S.; Kataoka, K.; Sakurai, T.; Ikeda, T.; Kano, K. Effect of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 2007, 601, 119–124. [Google Scholar] [CrossRef]
- Lalaoui, N.; Le Goff, A.; Holzinger, M.; Consier, S. Fully oriented bilirubin oxidase on porphyrin functionalized carbon nanotube electrodes for electrocatalytic oxygen reduction. Chem. Eur. J. 2015, 21, 16868–16873. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Oyama, M.; Funabashi, H.; Ishii, S. Effects of pore size and surface properties of MgO-templated carbon on the performance of bilirubin oxidase-modified oxygen reduction reaction cathode. Electrochim. Acta 2019, 322, 134744. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Effects of mesoporous structures on direct electron transfer-type bioelectrocatalysis: Facts and simulation on a three-dimensional model of random orientation of enzymes. Electrochemistry 2017, 85, 82–87. [Google Scholar] [CrossRef]
- Funabashi, H.; Takeuchi, S.; Tsujimura, S. Hierarchical meso/macro-porous carbon fabricated from dual MgO templates for direct electron transfer enzymatic electrodes. Sci. Rep. 2017, 7, 45147. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Lojou, E. Hydrogenases as catalysts for fuel cells: Strategies for efficient immobilization at electrode interfaces. Electrochim. Acta 2011, 56, 10385–10397. [Google Scholar] [CrossRef]
- Mazurenko, I.; Clement, R.; Byrne-Kodjabachian, D.; de Poulpiquest, A.; Tsujimura, S.; Lojou, E. Pore size effect of MgO-templated carbon on enzymatic H2 oxidation by the hyperthermophilic hydrogenase from Aquifex aeolicus. J. Electroanal. Chem. 2018, 812, 221–226. [Google Scholar] [CrossRef]
- Alonso-Lomillo, M.A.; Rüdiger, O.; Maroto-Valiente, A.; Velez, M.; Rodríguez-Ramos, I.; Muñoz, F.J.; Fernández, V.M.; De Lacey, A.L. Hydrogenase-coated carbon nanotubes for efficient H2 oxidation. Nano Lett. 2007, 7, 1603–1608. [Google Scholar] [CrossRef]
- Fujieda, N.; Tsuse, N.; Satoh, A.; Ikeda, T.; Kano, K. Production of completely flavinylated histamine dehydrogenase, unique covalently bound flavin andiron-sulfur cluster containing enzyme of Nocardioides simplex in Escherichia coli, and its properties. Biosci. Biotechnol. Biochem. 2005, 69, 2459–2462. [Google Scholar] [CrossRef][Green Version]
- Reed, T.; Lushington, G.H.; Xia, Y.; Hirakawa, H.; Travis, D.M.; Mure, M.; Scott, E.E.; Limburg, J. Crystal structure of hystamine dehydrogenase from Nocardioides simplex. J. Biol. Chem. 2010, 285, 25782–25791. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, M.; Tsujimura, S.; Shirai, O.; Kano, K. Direct electrochemistry of histamine dehydrogenase from Nocardioides simplex. J. Electroanal. Chem. 2009, 625, 144–148. [Google Scholar] [CrossRef]
- Park, J.; Joo, J.; Kwon, S.G.; Jang, Y.; Hyeon, T. Synthesis of monodisperse spherical nanocrystals. Angew. Chem. Int. Ed. 2007, 46, 4630–4660. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Lee, Y.; Wang, C.; Yin, H.; Dai, S.; Sun, S. A facile synthesis of monodisperse Au nanoparticles and their catalysis of oxidation. Nano Res. 2008, 1, 229–234. [Google Scholar] [CrossRef]
- Takahashi, Y.; Wanibuchi, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using porous gold electrodes. J. Electroanal. Chem. 2019, 843, 47–53. [Google Scholar] [CrossRef]
- Gutiérrez-Sánchez, C.; Pita, M.; Vaz-Domínguez, C.; Shleev, S.; De Lacey, A.L. Gold nanoparticles as electronic bridges for laccase-based biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using thiol-modified gold nanoparticles on mesoporous carbon electrode. J. Electroanal. Chem. 2019, 832, 158–164. [Google Scholar] [CrossRef]
- Monsalve, K.; Roger, M.; Gutierrez-Sanchez, C.; Ilbert, M.; Nitsche, S.; Byrne-Kodjabachian, D.; Marchi, V.; Lojou, E. Hydrogen bioelectrooxidation on gold nanoparticle-based electrodes modified by Aquifex aeolicus hydrogenase: Application to hydrogen/oxygen enzymatic biofuel cells. Bioelectrochemistry 2015, 106, 47–55. [Google Scholar] [CrossRef]
- Pankratov, D.; Sundberg, R.; Suyatin, D.B.; Sotres, J.; Barrantes, A.; Ruzgas, T.; Maximov, I.; Montelius, L.; Shleev, S. The influence of nanoparticles on enzymatic bioelectrocatalysis. RSC Adv. 2014, 4, 38164–38168. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Belochapkine, S.; Magner, E. Nanoporous gold electrode with tuneable pore sizes for bioelectrochemical applications. Electroanalysis 2016, 28, 2415–2423. [Google Scholar] [CrossRef]
- Cracknell, J.A.; McNamera, T.P.; Lowe, E.D.; Blanford, C.F. Bilirubin oxidase from Myrothecium verrucaria: X-ray determination of the complete crystal structure and a rational surface modification for enhanced electrocatalytic O2 reduction. Dalton Trans. 2011, 40, 6668–6675. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Toyoda, M.; Sagara, K.; Takahashi, N.; Sato, A.; Kamitaka, Y.; Tsujimura, S.; Nakanishi, Y.; Sugiura, T.; Yamaguchi, S.; et al. X-ray analysis of bilirubin oxidase from Myrothecium verrucaria at 2.3 Å resolution using a twinned crystal. Acta Cryst. 2010, F66, 765–770. [Google Scholar]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The immobilization of fructose dehydrogenase on nanoporous gold electrodes for the detection of fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Mutation of heme c axial ligands in d-fructose dehydrogenase for investigation of electron transfer pathways and reduction of overpotential in direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2016, 67, 43–46. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a protein-engineered variant of d-fructose dehydrogenase for direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2017, 77, 112–115. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Protein-engineering improvement of direct electron transfer-type bioelectrocatalytic properties of d-fructose dehydrogenase. Electrochemistry 2019, 87, 47–51. [Google Scholar] [CrossRef]
- Kaida, Y.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Ultimate downsizing of d-fructose dehydrogenase for improving the performance of direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2019, 98, 101–105. [Google Scholar] [CrossRef]
- Tsuya, T.; Ferri, S.; Fujikawa, M.; Yamaoka, H.; Sode, K. Cloning and functional expression of glucose dehydrogenase complex of Burkholderia cepacia in Escherichia coli. J. Biotechnol. 2006, 123, 127–136. [Google Scholar] [CrossRef]
- Okuda-Shimazaki, J.; Loew, N.; Hirose, N.; Kojima, K.; Mori, K.; Tsugawa, W.; Sode, K. Construction and characterization of flavin adenine dinucleotide glucose dehydrogenase complex harboring a truncated electron transfer subunit. Electrochim. Acta 2018, 277, 276–286. [Google Scholar] [CrossRef]
- Yamashita, Y.; Ferri, S.; Huynh, M.L.; Shimizu, H.; Yamaoka, H.; Sode, K. Direct electron transfer type disposable sensor strip for glucose sensing employing an engineered FAD glucose dehydrogenase. Enzyme Microb. Technol. 2013, 52, 123–128. [Google Scholar] [CrossRef]
- Yamazaki, T.; Okuda-Shimazaki, J.; Sakata, C.; Tsuya, T.; Sode, K. Construction and characterization of direct electron transfer-type continuous glucose monitoring system employing thermostable glucose dehydrogenase complex. Anal. Lett. 2008, 41, 2363–2373. [Google Scholar] [CrossRef]
- Kakehi, N.; Yamazaki, T.; Tsugawa, W.; Sode, K. A novel wireless glucose sensor employing direct electron transfer principle based enzyme fuel cell. Biosens. Bioelectron. 2007, 22, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Loew, N.; Tsugawa, W.; Lin, C.; Probst, D.; La Belle, J.T.; Sode, K. The electrochemical behavior of a FAD dependent glucose dehydrogenase with direct electron transfer subunit by immobilization on self-assembled monolayers. Bioelectrochemistry 2017, 121, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, H.; Ferri, S.; Fujikawa, M.; Sode, K. Essential role of the small subunit of thermostable glucose dehydrogenase from Burkholderia cepacia. Biotechnol. Lett. 2004, 26, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yamazaki, T.; Yoshimatsu, K.; Kojima, K.; Tsugawa, W.; Ferri, S.; Sode, K. An Fe–S cluster in the conserved Cys-rich region in the catalytic subunit of FAD-dependent dehydrogenase complexes. Bioelectrochemistry 2016, 112, 178–183. [Google Scholar] [CrossRef]
- Yamashita, Y.; Suzuki, N.; Hirose, N.; Kojima, K.; Tsugawa, W. Mutagenesis study of the cytochrome c subunit responsible for the direct electron transfer-type catalytic activity of FAD-dependent glucose dehydrogenase. Int. J. Mol. Sci. 2018, 19, 931. [Google Scholar] [CrossRef]
- Yamaoka, H.; Yamashita, Y.; Ferri, S.; Sode, K. Site directed mutagenesis studies of FAD-dependent glucose dehydrogenase catalytic subunit of Burkholderia cepacia. Biotechnol. Lett. 2008, 30, 1967–1972. [Google Scholar] [CrossRef]
- Bollella, P.; Ludwig, R.; Gorton, L. Cellobiose dehydrogenase: Insights on the nanostructuration of electrodes forimproved development of biosensors and biofuel cells. Appl. Mater. Today 2017, 9, 319–332. [Google Scholar] [CrossRef]
- Okuda, J.; Sode, K. PQQ glucose dehydrogenase with novel electron transfer ability. Biochem. Biophys. Res. Commun. 2004, 314, 793–797. [Google Scholar] [CrossRef]
- Algov, I.; Grushka, J.; Zarivach, R.; Alfonta, L. Highly efficient flavin-adenine dinucleotide glucose dehydrogenase fused to a minimal cytochrome c domain. J. Am. Chem. Soc. 2017, 139, 17217–17220. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Okuda-Shimazaki, J.; Mori, K.; Kojima, K.; Tsygawa, W.; Ikebukuro, K.; Lin, C.-E.; Belle, J.L.; Yoshida, H.; Sode, K. Designer fungus FAD glucose dehydrogenase capable of direct electron transfer. Biosens. Bioelectron. 2019, 123, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, G.; Meharenna, Y.T.; Tsotsou, G.E.; Sadeghi, S.J.; Fairhead, M.; Giannini, S. Molecular Lego: Design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosens. Bioelectron. 2002, 17, 133–145. [Google Scholar] [CrossRef]
- Yuhashi, N.; Tomiyama, M.; Okuda, J.; Igarashi, S.; Ikebukuro, K.; Sode, K. Development of a novel glucose enzyme fuel cell system employing protein engineered PQQ glucose dehydrogenase. Biosens. Bioelectron. 2005, 20, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Igarashi, S.; Ferri, S.; Sode, K. Increasing stability of water-soluble PQQ glucose dehydrogenase by increasing hydrophobic interaction at dimeric interface. BMC Biochem. 2005, 6, 1. [Google Scholar] [CrossRef]
- Pereira, A.R.; Luz, R.A.S.; Lima, F.C.D.A.; Crespilho, F.N. Protein oligomerization based on brønsted acid reaction. ACS Catal. 2017, 7, 3082–3088. [Google Scholar] [CrossRef]
- Holland, J.T.; Lau, C.; Brozik, S.; Atanassov, P.; Banta, S. Engineering of glucose oxidase for direct electron transfer via site-specific gold nanoparticle conjugation. J. Am. Chem. Soc. 2011, 133, 19262–19265. [Google Scholar] [CrossRef]
- Ferapontova, E.; Schmengler, K.; Börchers, T.; Ruzgas, T.; Gorton, L. Effect of cysteine mutations on direct electron transfer of horseradish peroxidase on gold. Biosens. Bioelectron. 2002, 17, 953–963. [Google Scholar] [CrossRef]
- Güven, G.; Prodanovic, R.; Schwaneberg, U. Protein engineering−an option for enzymatic biofuel cell design. Electroanalysis 2010, 22, 765–775. [Google Scholar] [CrossRef]
- Eijsink, V.G.H.; Gaseidnes, S.; Borchert, T.V.; van den Burg, B. Directed evolution of enzyme stability. Biomol. Eng. 2005, 22, 21–30. [Google Scholar] [CrossRef]
- Wong, T.S.; Schwaneberg, U. Protein engineering in bioelectrocatalysis. Curr. Opin. Biotechnol. 2003, 14, 590–596. [Google Scholar] [CrossRef]
- Mate, D.M.; Gonzalez-Perez, D.; Falk, M.; Kittl, R.; Pita, M.; De Lacey, A.L.; Ludwig, R.; Shleev, S.; Alcalde, M. Blood tolerant laccase by directed evolution. Chem. Biol. 2013, 20, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Dementin, S.; Burlat, B.; Fourmond, V.; Leroux, F.; Liebgott, P.-P.; Hamdan, A.A.; Leger, C.; Rousset, M.; Guigliarelli, B.; Bertrand, P. Rates of intra-and intermolecular electron transfers in hydrogenase deduced from steady-state activity measurements. J. Am. Chem. Soc. 2011, 133, 10211–10221. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, L.; Idris, Z.; Vincent, K.A.; Lenz, O. Catalytic properties of the isolated diaphorase fragment of the NAD+-reducing [NiFe]-hydrogenase from Ralstonia eutropha. PLoS ONE 2011, 6, e25939. [Google Scholar] [CrossRef] [PubMed]
- Horch, M.; Lauterbach, L.; Lenz, O.; Hildebrandt, P.; Zebger, I. NAD(H)-coupled hydrogen cycling−structure–function relationships of bidirectional [NiFe] hydrogenases. FEBS Lett. 2012, 586, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Direct electron transfer-type four-way bioelectrocatalysis of CO2/formate and NAD+/NADH redox couples by tungsten-containing formate dehydrogenase adsorbed on gold nanoparticle-embedded mesoporous carbon electrodes modified with 4-mercaptopyridine. Electrochem. Commun. 2017, 84, 75–79. [Google Scholar] [CrossRef]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar] [CrossRef]
- Bassegoda, A.; Madden, C.; Wakerley, D.W.; Reisner, E.; Hirst, J. Reversible interconversion of CO2 and formate by a molybdenum-containing formate dehydrogenase. J. Am. Chem. Soc. 2014, 136, 15473–15476. [Google Scholar] [CrossRef]
- Hori, Y.; Vayenas, C.G.; White, R.E.; Gamboa-Aldeco, M.E. Modern Aspects of Electrochemistry; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a bioelectrochemical formate generating system from carbon dioxide and dihydrogen. Electrochem. Commun. 2018, 97, 73–76. [Google Scholar] [CrossRef]
- Siritanaratkul, B.; Megarity, C.F.; Roberts, T.G.; Samuels, T.O.M.; Winkler, M.; Warner, J.H.; Happe, T.; Armstrong, F.A. Transfer of photosynthetic NADP+/NADPH recycling activity to a porous metal oxide for highly specific, electrochemically-driven organic synthesis. Chem. Sci. 2017, 8, 4579–4586. [Google Scholar] [CrossRef]
- Wan, L.; Megarity, C.F.; Siritanaratkul, B.; Armstrong, F.A. A hydrogen fuel cell for rapid, enzyme-catalysed organic synthesis with continuous monitoring. Chem. Commun. 2018, 54, 972–975. [Google Scholar] [CrossRef] [PubMed]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. https://doi.org/10.3390/catal10020236
Adachi T, Kitazumi Y, Shirai O, Kano K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts. 2020; 10(2):236. https://doi.org/10.3390/catal10020236
Chicago/Turabian StyleAdachi, Taiki, Yuki Kitazumi, Osamu Shirai, and Kenji Kano. 2020. "Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes" Catalysts 10, no. 2: 236. https://doi.org/10.3390/catal10020236
APA StyleAdachi, T., Kitazumi, Y., Shirai, O., & Kano, K. (2020). Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts, 10(2), 236. https://doi.org/10.3390/catal10020236